Abstract
The released genome sequence of Ustilago maydis provided immense insight into this pathogen's genetic structure; however, thorough annotation of the genome requires data from many sources. Information from 4425 expressed sequence tags from the filamentous dikaryon provided an excellent resource for genome annotation. This allowed confirmation and correction of gene models, as well as the documentation of transcript structural features. The depth of coverage provided by the normalized dikaryon cDNA library contributed to the discovery of new candidate pathogenesis genes and enabled the identification of U. maydis antisense and noncoding RNAs. Candidate pathogenesis genes were identified based on their representation in the dikaryon library only or in the dikaryon and diploid cDNA libraries, followed by comparative analysis with the genome sequences of other plant pathogens. Six genes that were conserved only among pathogenic fungi and six genes unique to U. maydis were confirmed to be expressed in planta using reverse-transcriptase PCR. The transcript level of three of these genes varied during the transition from filamentous dikaryotic growth to the formation of the teliospore. This discovery provides representative genes that can be used to begin dissecting the control of gene expression during this transition in pathogenic development. Many of the newly identified U. maydis noncoding RNAs were differentially represented among cDNA libraries, suggesting potential functional roles in different U. maydis cell types. The deletion of one such ncRNA in a solopathogenic strain reduced virulence, revealing a new type of pathogenesis gene in fungi.
Résumé
La séquence publiée du génome d'Ustilago maydis a ouvert de larges perspectives quant à la structure génétique de cet agent pathogène. Toutefois, l'annotation complète du génome nécessite des données provenant de plusieurs sources. L'information relative à 4 425 étiquettes de séquence transcrites d'un dicaryon filamenteux a été essentielle à l'annotation du génome. Elle a permis de confirmer et de corriger les modèles des gènes ainsi que de documenter les transcriptions des éléments structuraux. L'ampleur de la banque d'ADNc normalisée de dicaryon a contribué à la découverte de nouveaux gènes de pathogénie candidats et a permis l'identification des ARN antisens et non codants d'U. maydis. Les gènes de pathogénie candidats ont été identifiés en fonction de leur représentation dans la banque de dicaryon uniquement ou dans les banques de dicaryon et d'ADNc diploïde, puis par des analyses comparatives de séquences génomiques d'autres phytopathogènes. L'expression, in planta, de six gènes de champignons pathogènes seulement et de six gènes propres à U. maydis a été confirmée par RT-PCR. Le niveau de transcription de trois de ces gènes a varié durant la phase de transition entre la croissance dicaryotique filamenteuse et la formation de téleutospores. Cette découverte fournit des gènes représentatifs qui peuvent être utilisés pour commencer à analyser le contrôle de l'expression génique durant cette phase de transition du développement de la pathogénie. Plusieurs des ARN non codants d'U. maydis nouvellement identifiés étaient différentiellement représentés dans les banques d'ADNc, ce qui suggère des rôles fonctionnels possibles chez différents types de cellules d'U. maydis. La délétion d'un tel ARN non codant chez une souche solopathogène a réduit la virulence, ce qui a révélé la présence d'un nouveau type de gène de pathogénie chez les champignons.
Introduction
Ustilago maydis (DC) Corda is the causal agent of common smut of corn. The hallmark of this disease is the formation of tumours on all aerial parts of the plant (Spellig et al., Citation1994; Horst et al., Citation2008). These tumours house diploid teliospores, which disperse, germinate and complete meiosis, resulting in the formation of non-pathogenic haploid sporidia (Banuett & Herskowitz, Citation1989). Compatible haploids fuse to form the pathogenic dikaryon, the primary infectious form of U. maydis (Banuett & Herskowitz, Citation1996). The dikaryon is an obligate parasite that invades the plant through direct penetration or passively through natural openings, such as stomata, or the silks of corn cobs (reviewed in Martínez-Espinoza et al., Citation2002). Upon penetration, the dikaryotic hyphae grow within and between plant cells, eliciting the formation of tumours (Snetselaar & Mims, Citation1992; Banuett & Herskowitz, Citation1996). Hyphal fragmentation occurs and diploid teliospores develop within tumour tissue, completing the disease cycle (Snetselaar & Mims, Citation1994; Banuett & Herskowitz, Citation1996). The formation of the pathogenic dikaryon is necessary for tumour formation and teliospore development, and is therefore essential for the continuation of the U. maydis life cycle.
Stable dikaryon formation is a unique feature of basidiomycetes (Anderson & Kohn, Citation2007); it is the primary foraging form and provides nutrients for fruiting body formation and sexual reproduction. The U. maydis dikaryon is dependent on the host plant for sustained growth and development (Kahmann & Kämper, Citation2004). However, dikaryon mycelial growth can be induced in the laboratory on medium containing charcoal (Day & Anagnostakis, Citation1971), allowing the study of this pathogenic cell type independent of the host. While the dikaryon is the primary pathogenic form of U. maydis, it is not the only one. In nature, a low percentage of teliospores germinate as solopathogenic diploids (Christensen, Citation1931; Holliday, Citation1961; Banuett & Herskowitz, Citation1989; Kojic et al., Citation2002). The diploid form can also be created in the laboratory by forced mating of complementary auxotrophs. These diploids are stable and can be induced to form filaments in the same manner as dikaryons (Holliday, Citation1974; Banuett & Herskowitz, Citation1989). The virulence of these filamentous growth forms has been compared (Banuett & Herskowitz, Citation1996; Babu et al., Citation2005) and it is generally accepted that they respond differently during growth within the plant (reviewed in Banuett, Citation2010). Culturing these pathogenic forms, independent of the host, provides an opportunity to compare the genes they express during growth and offers insight regarding the differential virulence of the diploid and dikaryon.
cDNA library creation and the generation of expressed sequence tags (ESTs) is a fast and efficient way of obtaining gene expression profiles for various tissues and pathogenic cell types (Adams et al., Citation1991; Verdun et al., Citation1998; Kessler et al., Citation2002). The availability of U. maydis cDNA sequences (Sacadura & Saville, Citation2003; Nugent et al., Citation2004; Ho et al., Citation2007) has facilitated the molecular characterization of genes and provided information for comparative genomics, microarray analysis, genome annotation and meiosis-specific gene discovery (Austin et al., Citation2004; Babu et al., Citation2005; Zahiri et al., Citation2005; Kämper et al., Citation2006; Donaldson & Saville, Citation2008). Obtaining sequences from full-length transcripts physically links the open reading frames (ORFs) to the untranslated regions (UTRs), in contrast to simply aligning sequences upstream of the start of translation. This has enabled accurate identification of transcriptional start sites and promoters (Doyle et al., Citation2011). Analysis of the filamentous dikaryon cDNA library provided information for further genome annotation and an opportunity to examine gene expression in the pathogenic form of U. maydis.
Unlike previous U. maydis cDNA libraries, the filamentous dikaryon cDNA library was normalized, allowing the impact of normalization on clone redundancy to be assessed. Normalization decreases redundancy that results from cloning abundant or intermediately expressed mRNAs, thereby increasing the frequency of rare mRNA representation in the cDNA library (Kessler et al., Citation2002). With the increased depth of gene coverage provided by normalization, the filamentous dikaryon cDNA library was used to improve U. maydis genome annotation. This included contributing to full-length cDNA analysis (Doyle et al., Citation2011), as well as identifying candidate pathogenesis genes and noncoding RNAs (ncRNAs). For the purpose of this study, ncRNAs are defined as regions of the genome that are transcribed but do not contain ORFs as annotated by the Munich Information centre for Protein Sequences (MIPS) U stilago m aydis database (MUMDB). Expression profiling of candidate genes supports previously unidentified transitions in gene expression during U. maydis pathogenesis, and the functional investigation of a ncRNA suggests that it acts, directly or indirectly, in pathogenic development. These significant findings support the importance of targeted and deeper transcriptome analysis in fungal pathogens.
Materials and methods
Strains, culture conditions and production of Ustilago maydis cell types
Compatible U. maydis haploid strains 518 (a2b2) and 521 (a1b1) were used for cDNA library creation. These strains, and D132 (a1a2 b1b2), originally isolated and described by Holliday (Citation1961), were obtained from Sally Leong (University of Wisconsin, Madison, WI, USA). For RT-PCR experiments, RNA was isolated from U. maydis strains FB1 (a1b1) and FB2 (a2b2), provided by Flora Banuett (Banuett & Herskowitz, Citation1989). Haploid cells and the forced diploid D132 were grown overnight in YEPS medium (1% yeast extract, 2% sucrose, 2% peptone; at 28–30 °C, 250 rpm). To induce filamentous growth on plates, overnight cultures were diluted with sterile dH2O to an OD600 of 1.5, and then equal volumes of compatible haploid cultures were combined. 10 μL aliquots of this mixture or of D132 were spotted on double complete medium (DCM) plates with charcoal and incubated at 28 °C (Day & Anagnostakis, Citation1971). Filamentous growth was monitored and 518 × 521 dikaryons were harvested 3–5 days after spotting, whereas FB1 × FB2 dikaryons and D132 diploids were harvested 4–6 days after spotting. To harvest budding diploid cells, D132 was spotted on DCM without charcoal and colonies were harvested 2 days after spotting. Budding haploid cells, FB1 and FB2 were spotted separately on DCM with charcoal and harvested 3 days after spotting. For RNA isolation leading to RT-PCR experiments, 20–30 colonies were harvested from plates, pooled and then frozen in liquid nitrogen prior to storage at −80 °C.
Teliospores were harvested from mature tumours of ‘Golden Bantam’ cobs infected with FB1 × FB2. Cob infection and teliospore harvesting were carried out essentially as described by Zahiri et al. (Citation2005). Differences include: overnight haploid cell cultures were diluted with sterile dH2O to a final OD600 of 1, equal volumes of compatible haploid cultures were combined and 6 mL of the mixture was injected down the cob silk shafts. Teliospores were vacuum dried and stored at 4 °C following isolation from the tumours.
RNA isolation and cDNA library construction
All procedures were carried out as suggested by the manufacturers, unless otherwise stated. 518 × 521 dikaryotic mycelia were frozen in liquid nitrogen and total RNA was extracted using TRIzol reagent (Life Technologies). Poly (A)+ RNA was purified from total RNA using a mRNA Purification Kit (Amersham BioSciences, now GE). 1.5 μg of poly (A)+ RNA was reverse-transcribed using the CDS-3M adapter (Evrogen). The cDNA was prepared using the Primer-Extension protocol of the Creator SMART cDNA library Construction Kit (Clontech) and purified using the QIAquick PCR Purification Kit (Qiagen). cDNA quality was checked by electrophoresis on a 1.5% agarose gel (1× TAE) followed by visualization through ethidium bromide staining. cDNA normalization was performed using the TRIMMER-DIRECT cDNA Normalization Kit (Evrogen). Normalized cDNA was unidirectionally cloned into the asymmetric SfiI restriction enzyme sites of the pDNR-LIB vector. Cloned cDNAs were transformed by electroporation into ElectroMax DH5α– Escherichia coli cells (Life Technologies). Transformants were plated on LB agar plates (1% tryptone, 0.5% yeast extract, 1% NaCl, 2% agar) containing 30 μg mL−1 chloramphenicol and, following overnight growth at 37 °C, individual colonies were picked and inoculated into 96-well microtitre plates containing LB medium supplemented with 30 μg mL−1 chloramphenicol, grown, and stored frozen at −80 °C in 15% glycerol. cDNA clones in this library have a ‘DIK’ designation.
RNA isolation for RT-PCR was conducted as follows. Tissue was ground in liquid nitrogen using a mortar and pestle. Ground tissue or vacuum-dried teliospores (400 mg) were resuspended in TRIzol reagent, and transferred into 2 mL screw-cap tubes containing Lysing Matrix C (MP Biomedicals). Cells were disrupted as described in Zahiri et al. (Citation2005). RNA isolation was carried out following the manufacturer's protocol for TRIzol reagent. RNA was precipitated using RNA precipitation solution (0.8 M disodium citrate/1.2 M NaCl) and isopropanol (Sambrook & Russell, Citation2001), washed with 75% ethanol and resuspended in diethylpyrocarbonate (DEPC) treated water. Approximately 50 μg of total RNA was treated with DNAseI (RNase-free, New England Biolabs), precipitated as above, resuspended in DEPC treated water and diluted to 100 ng μL−1. Genomic contamination was assessed by PCR using 200 ng of DNaseI treated total RNA, in a 25 μL reaction volume with AmpliTaq Gold (Applied Biosystems) reaction mix, and primers flanking the intron in um02491 (umGapd; Smith & Leong, Citation1990; ). Cycling conditions were 95 °C for 10 min, 35 cycles of 95 °C for 30 s, 63 °C for 15 s and 72 °C for 30 s, followed by a 72 °C hold for 10 min. Products were electrophoretically separated on a 2% agarose gel (1× TAE) and visualized by ethidium bromide staining. RNA quality was assessed, following glyoxal denaturation, by agarose gel electrophoresis (data not shown; Sambrook & Russell, Citation2001). Reverse transcription using the ABI TaqMan Gold RT-PCR kit (ABI) was carried out in a 20 μL reaction volume, using 400 ng of DNaseI treated total RNA, primed with oligo-d(T)16. The cycling conditions were 25 °C for 10 min, 50 °C for 30 min, and 95 °C for 10 min. cDNA was diluted eight-fold with sterile dH2O and 4 μL of this was used in each PCR reaction (see below for gene expression analysis).
Table 1. Primers used in this study
DNA sequencing
Prior to sequencing, plasmid DNA was isolated from E. coli cultures, grown for 16–18 h in 96-well plates containing 1 mL of LB supplemented with 30 μg mL−1 chloramphenicol per well, using Whatmann's 96-well Plasmid Miniprep System (VWR International) following the manufacturer's protocol. DNA quality was tested electrophoretically on 1% agarose gels (1× TAE) and visualized by ethidium bromide staining. Nucleotide sequences corresponding to the 5′ end of the mRNA were determined by Big Dye Terminator chemistry (ABI) using the protocol previously described by Ho et al. (Citation2007).
Sequence analysis
Raw sequence data were imported into the SeqManII module of Lasergene v.5.0 (DNASTAR). Default settings were used to trim sequences based on quality to remove ambiguous bases. FASTA files were exported from SeqManII. To remove contaminating vector sequences, a perl script was written to extract only the region of the EST that aligned to the U. maydis chromosome assembly file Umaydis_contig_nuc_140408.fas (Mewes et al., Citation2008), from MUMDB (http://mips.helmholtz-muenchen.de/genre/proj/ustilago), following a blastn with a cutoff of E < 1e−20. ESTs less than 150 nts were discarded.
Contig assembly and assignment of contigs to MUMDB genes
Ustilago maydis cDNA libraries were previously constructed from germinating (T11) and dormant (TDO) teliospores (Sacadura & Saville, Citation2003; Ho et al., Citation2007), from haploid cells grown in rich medium (HCM), under carbon starvation (MMC) and under nitrogen starvation (MMN) conditions (Ho et al., Citation2007) as well as forced diploids grown as filaments (D12; Nugent et al., Citation2004). ESTs from all cDNA libraries were assembled, with those from the filamentous dikaryon (DIK) cDNA library, into contigs and these contigs were assigned MUMDB genes as previously described by Ho et al. (Citation2007).
MUMDB Functional Catalogue (FunCat) category assignment
MUMDB genes identified in the EST contig assembly (see above) were functionally categorized using the October 2009 update of the MUMDB FunCat (Ruepp et al., Citation2004; Mewes et al., Citation2008). Only MUMDB genes with valid MUMDB ORF predictions were used in this analysis. The cell type associated with the ESTs used in the contig assembly and subsequent MUMDB gene identification was retained to identify putative differences in the functional classes of genes between: (1) the filamentous and non-filamentous cell types, and (2) the diploid and dikaryon filamentous cell types.
Comparison of the DIK cDNA library to other U. maydis cDNA libraries
Using the U. maydis EST contig assembly, the success of the cDNA library normalization was assessed. This was accomplished by comparing the number of ESTs corresponding to specific MUMDB genes in each of the cDNA libraries. In order to analyse transcripts expressed in the filamentous (pathogenic) cell types, a set of 981 non-redundant MUMDB genes, which overlapped contigs either unique to the DIK cDNA library or shared only between the DIK and D12 cDNA libraries, was generated.
Gene expression analysis, RT-PCR methods and gene selection
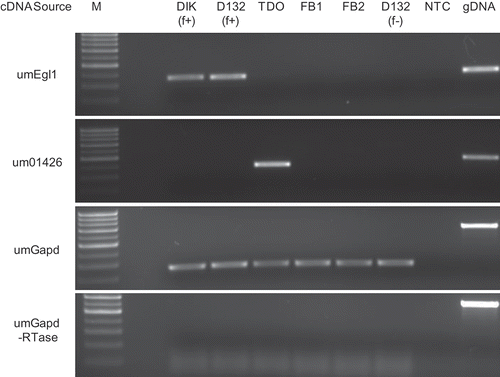
Primers for genes identified in this paper were designed using Primer3 (Rozen & Skaletsky, Citation2000), around introns (if applicable), targeting the 3′ end of the genes. Genes were selected based on their cDNA library distribution. Primer sequences are presented in . Cycling conditions for all PCRs were 95 °C for 10 min, 30 cycles of 95 °C for 30 s, 63 °C for 15 s and 72 °C for 30 s, followed by a 72 °C hold for 10 min. One quarter of the final product mixture was electrophoretically separated on a 2% agarose gel (1× TAE) and visualized by ethidium bromide staining. Quality control was conducted using the filament-specific gene um06332 (umEgl1; Schauwecker et al., Citation1995), the teliospore specific gene um01426 (Ho et al., Citation2007), and the constitutively expressed um02491 (umGapd). These quality screens were repeated following all PCRs to confirm consistent band migration and cDNA integrity.
Comparison of putative U. maydis filament-specific proteins with other fungi
Fungi sequenced using whole-genome shotgun (wgs) reads that were complete, or in assembly (with the exception of Schizosaccharomyces pombe: NCBI genome chromosomes), were selected from the NCBI Eukaryotic Genome Sequencing Projects database (http://www.ncbi.nlm.nih.gov/genomes/leuks.cgi). This set of fungi included ascomycetes, basidiomycetes and a single oomycete, which were categorized as non-pathogenic or pathogenic (). The oomycete, Phytophthora infestans, was included to represent plant pathogens with a divergent evolutionary path. Protein sequences for the 981 non-redundant MUMDB transcripts identified as being present only in the DIK cDNA library, or shared only between the DIK cDNA library and the D12 cDNA library, were used in tblastn searches against the individual wgs databases listed in and in blastp searches against the NCBI non-redundant database. The BLOSUM62 matrix and default parameters were used for these searches, with the exception that the ‘EXPECT’ threshold was changed to E < 1e−4. Results from the individual tblastn searches (April 2009) were combined to identify the presence (or absence) of proteins in the various fungi. To confirm if a given protein was U. maydis specific or pathogen specific the blastp results (conducted in August 2011) were analysed. Those proteins considered U. maydis specific were used in a blastp search against the non-redundant protein sequence database, and proteins considered pathogen specific were used in a blastp against fungi (taxid: 4751) in the same database. In these searches, the maximum number of aligned sequences returned was limited to 10 and opportunistic pathogenic fungi were considered pathogens. Following these screens, all proteins considered U. maydis-specific or pathogen-specific were screened against the Sporisorium reilianum genome sequence (http://mips.helmholtz-muenchen.de/genre/export/sites/default/sporisorium/Download/; Schirawski et al., Citation2010).
Table 2. Fungi used in tblastn searches
Creation of an Ustilago maydis ncRNA1 deletion strain
Deletion of ncRNA1 was performed by replacing the ncRNA1 locus with a hygromycin B resistance cassette following the PCR-based strategy originally described by Kämper (Citation2004). Plasmid pMF1-h (Brachmann et al., Citation2004) was obtained from Jörg Kämper (Karlsruhe Institute of Technology, Karlsruhe, Germany). Briefly, primers pB_Left-F and pB_Left_Rev_SfiI, and pB_Right-F and pB_Right_Rev_SfiI (see ) were used to amplify ∼1.1 kb flanking DNA regions to ncRNA1. The PCR products were purified using the QIAquick PCR Purification Kit (Qiagen). The purified flanking regions were digested with SfiI, and pMF1-h was digested with SfiI and BsaI. Digested flanking regions and the ∼2.7 kb fragment containing the hygromycin B resistance cassette were purified using the QIAquick Gel Purification Kit (Qiagen) and ligated using T4 DNA ligase (New England Biolabs). The ∼4.9 kb deletion construct was purified using the QIAquick Gel Purification Kit (Qiagen). The construct was amplified using primers pB_Nested_F and pB_Nested_R (), cloned into pCR-XL-TOPO (Life Technologies) yielding pncRNA1-HYG, and verified by sequencing. A ∼4.8 kb ncRNA1 deletion construct was amplified from pncRNA1-HYG using primers pB_Nested-F and pB_Nested-R and purified using the QIAquick Gel Purification Kit (Qiagen) prior to U. maydis transformation.
Ustilago maydis was transformed using a modification of the Yee (Citation1998) protocol. These modifications included combining ∼3 μg DNA with 30 μg heparin in a final volume of 20 μL (with dH2O) and incubating on ice for 5 min before adding it to 125 μL of protoplasts in STC (1 M sorbitol, 25 mM Tris-HCl, pH 7.5, 50 mM CaCl2). This mixture was incubated on ice for 10 min, then 100 μL of 50% polyethylene glycol solution (50% w/v PEG 3350, 25 mM Tris-HCl, pH 7.5, 50 mM CaCl2) was added and the incubation continued at room temperature (RT) for 20 min while rotating on a rotisserie. 1 mL SCS (50 mM sodium citrate, 1 M sorbitol, pH 5.8) was added and the mixture was centrifuged for 5 min (RT, 1000 × g). Following aspiration of the supernatant, 1 mL STC was added and the mixture was centrifuged for 5 min (RT, 600 × g). The supernatant was aspirated and protoplasts were resuspended in 200 μL DCM (with 100 mM glucose and 1 M sorbitol). The protoplasts were spread over DCM agar plates (with 1 M sorbitol and 250 μg mL−1 hygromycin B). After 7 days at 28 °C, colonies were transferred to fresh hygromycin B-containing YEPS agar plates to confirm resistance to antibiotic selection and to obtain single colonies. Resistant colonies were transferred to selection plates a second time, containing 250 μg mL−1 hygromycin B and 20 μg mL−1 phleomycin. Genomic DNA was isolated from putative transformants using the protocol described by Hoffman & Winston (Citation1987). To identify putative transformants and confirm transformation occurred at the ncRNA1 locus, PCRs were performed to amplify a region containing part of the hygromycin B cassette and an area of the flanking ncRNA1 locus, outside of the area of recombination. To conduct this screen, PCR was performed using primer pairs pB_Left-F and pMF1HygOUT-F, or pB_Right-R and pMF1HygOUT-R (). RNA was isolated from SG200 and putative transformants, and first-strand reverse transcription was carried out using a ncRNA1-specific primer (ncRNA1-FS). Loss of the ncRNA1 transcript was confirmed by the absence of RT-PCR amplification, using the primer pair ncRNA1-F and ncRNA1-R. Two independently generated deletion strains were used in the pathogenesis assay.
Plant growth conditions, in planta sampling and pathogenesis assay
‘Golden Bantam’ corn seeds (Ontario Seed Company) were planted (18 per 20 cm pot) in Sunshine Professional Growing Mix (Mix#1, Sungro Horticulture Canada). Germination and growth proceeded in a Conviron CMP 4030 growth chamber (18 h light, 70% RH, 30 °C; 6 h dark, 70% RH, 25 °C). For RT-PCR of in planta time-points, haploid strains (FB1, FB2) were grown overnight in YEPS medium (28 °C, 250 rpm) and adjusted to ∼106 cells mL−1. Equal numbers of compatible haploids were combined and ∼0.5 mL was injected into the base of seven-day-old seedlings. Stem samples were collected and pooled from 3–5 infected plants at 8 and 14 days post infection (DPI). For each pathogenesis assay, SG200 strains were grown overnight in YEPS medium (28 °C, 250 rpm), adjusted to ∼106 cells mL−1, and ∼0.5 mL was injected into the base of 45, seven-day-old seedlings. Symptoms were scored at 7, 10 and 14 DPI according to the 0–5 disease scoring scale described previously by Gold et al. (Citation1997). Disease ratings included: 0 = no symptoms; 1 = anthocyanin production and/or chlorosis; 2 = small leaf tumours; 3 = small stem tumours; 4 = large stem tumours; and 5 = plant death. Two separate pathogenesis assays were performed, consisting of a distinct ncRNA1 deletion mutant and a control (SG200).
Results
Filamentous dikaryon (DIK) cDNA library
During the isolation of total RNA from filamentous dikaryon cultures, only those with obvious white ‘fuz’ growth were chosen. This method maximized the representation of RNA from filaments. A total of 4425 ESTs were generated for the filamentous dikaryon (DIK) cDNA library with an average length of 522 nts. The sequences were submitted to dbEST at NCBI under accession numbers JK841280 to JK845704. Contigs were assembled and assigned MUMDB genes as previously described by Ho et al. (Citation2007). A total of 2640 non-redundant MUMDB genes were identified (Supplementary ).
Comparing the DIK cDNA library with previously constructed U. maydis cDNA libraries [germinating (T11) and dormant (TDO) teliospores (Sacadura & Saville, Citation2003; Ho et al., Citation2007), haploid cells grown in rich medium (HCM), under carbon starvation (MMC) and under nitrogen starvation (MMN) conditions (Ho et al., Citation2007) and forced diploids grown as filaments (D12; Nugent et al., Citation2004)] identified 596 unique DIK cDNA library genes (Supplementary ).
Approximately 2% of the DIK contigs were antisense to a MUMDB gene; these were identified as natural antisense transcripts (NATs; Supplementary ). Of these NATs, approximately 96% were unique to the DIK cDNA library. Of the total non-redundant NATs (nrNATs) identified to date in the U. maydis cDNA libraries, 18% (45/247) are uniquely found in the filamentous dikaryon cDNA library.
Aligning the ESTs with the genome sequence revealed that 58 noncoding RNAs are represented in the DIK cDNA library (). With this finding, the other U. maydis cDNA libraries were screened and the number of ncRNAs found in each was recorded ().
Table 3. EST representation among cDNA libraries and functional associations
DIK and cell type cDNA libraries
The set of 2640 DIK non-redundant MUMDB predicted ORFs was compared with cDNA libraries listed above to determine the number of genes represented in DIK and the cDNA library it was compared with. This analysis permitted sharing between cDNA libraries; therefore, ESTs were not solely found in the paired cDNA libraries (Supplementary ). With this limitation, the filamentous DIK cDNA library shared the highest representation of ORFs (51%) with the filamentous diploid cDNA library.
Fig. 1. RT-PCR for genes putatively identified as (a) dikaryon specific, (b) constitutively expressed. M: marker; DIK: dikaryon; D132: diploid; f+: filamentous; f-: non-filamentous; TDO: dormant teliospore; FB1, FB2: haploid cells; gDNA: genomic DNA; NTC: no template control.

Fig. 2. Controls for RT-PCR analysis. M: marker; DIK: dikaryon; D132: diploid; f+: filamentous; f-: non-filamentous; TDO: dormant teliospore; FB1, FB2: haploid cells; gDNA: genomic DNA; NTC: no template control.
The second assessment, of genes commonly represented in the DIK cDNA library and the other cDNA libraries, excluded sharing; this restriction allowed the number of genes represented only in the DIK cDNA library and the compared cDNA library to be determined. To represent this as a percentage, the number of shared genes was divided by the total non-redundant MUMDB genes for each individual cDNA library (data not shown). The DIK cDNA library shared the highest degree of unique overlap with the diploid cDNA library at 15%, followed by the dormant teliospore at 8%.
The expressed genes uniquely represented in a given cDNA library were screened to determine if they encoded secreted proteins (Mueller et al., Citation2008; http://mips.helmholtz-muenchen.de/genre/proj/ustilago/Search/index.html) or if they were associated with gene clusters as identified in Kämper et al. (Citation2006; ).
Impact of normalization
The DIK cDNA library was normalized and created with SMART technology. This normalization increased the likelihood of detecting rare mRNAs and provided an opportunity to compare normalized and non-normalized cDNA libraries with regards to clone redundancy (Kessler et al., Citation2002). shows a lower number of ESTs per ORF in the DIK cDNA library compared with the other cDNA libraries. To further assess the impact of normalization on the filamentous dikaryon cDNA library, the five genes with the highest EST representation in each cDNA library were examined (Supplementary ). The DIK cDNA library had the lowest redundancy at 1% relative to the total number of ESTs in its cDNA library.
cDNA library representation and transcript levels
EST presence in a given cDNA library is indicative of gene expression within the cell type used to create the cDNA library; however, cDNA libraries are not comprehensive. To assess if the transcripts of genes represented only in the DIK cDNA library or only in the DIK and D12 cDNA libraries, were present in other cell types, RT-PCR screens were carried out. These screens were limited to the five genes, in each category, that had the highest representation in the DIK cDNA library. The results are presented in , and Supplementary These figures emphasize the importance of using RT-PCR analysis to supplement cDNA library data. EST representation in a given cDNA library cannot be used as the sole resource for identifying differential gene expression between cell types. The RT-PCR results revealed differential transcription of one gene (um02095) and differential splicing of two others (um01821, um11032). Each of these genes has a different expression pattern in the dormant teliospore. The genes with differential splicing were reassessed by RT-PCR for 35 cycles, which more definitively showed the larger PCR products resulting from retained introns in the dormant teliospore.
EST libraries created from multiple cell types can also be used to identify genes that are present in all cell types, which may represent constitutively expressed genes. Eighty-three genes with representation in all cDNA libraries were identified (Supplementary ) and five were selected for RT-PCR analysis. Selection was based upon the genes having a similar level of representation in each cDNA library. The results of the RT-PCR indicated that all five genes were expressed in all cell types. Um00175 (related to translation initiation factor 3 subunit F) had a very similar level of expression in all cell types while there was minor variation in expression of four other genes ( b).
MUMDB Functional Catalogue category assignment of ESTs
ESTs were classified based on the MUMDB FunCat assignment of their corresponding ORF (Ruepp et al., Citation2004; Mewes et al., Citation2008). At the time of this analysis, 4475 of the 6786 MUMDB predicted ORFs were functionally classified. No gene was present more than once in a given functional category; however, a single gene may have more than one FunCat assignment and therefore may be represented more than once in this analysis.
Functional annotation can highlight differences in genes expressed in different cell types (Ho et al., Citation2007). FunCat assignment was performed for each cDNA library, listed above, to identify differences in category representation based on cell type and cDNA library construction. The normalized DIK cDNA library had the highest percentage of unique ORFs at 23%, relative to the other six cDNA libraries used in this study (). The number of genes with a FunCat assignment was similar across all cDNA libraries, with percentages ranging from 73–79% (data not shown). The percentage of genes that were unique to each cDNA library and had a FunCat assignment ranged from 50–67% (data not shown). All cDNA libraries had high representation in the ‘protein with binding function or cofactor requirement (structural or catalytic)’ and ‘metabolism’ categories (Supplementary ).
Fig. 3. FunCat distribution for genes unique to cell type groupings.
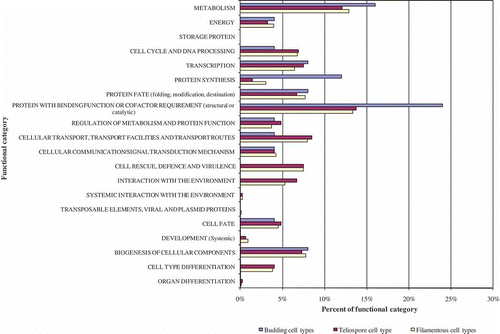
To examine the filamentous (pathogenic) cell type relative to the teliospore and non-pathogenic budding haploid, cDNA libraries were grouped based on growth form. Groupings included genes only represented in the filamentous cell types DIK and D12, genes represented only in the budding cell types HCM, MMN and MMC, and genes represented only in the teliospore (TDO; ).
Fig. 4. RT-PCR assessing U. maydis transcript levels in different cell types and during in planta pathogenic development. Genes putatively identified as (a) Ustilago maydis specific, or (b) pathogen specific. (c) The genes ncRNA1 and um12316. (d) RT-PCR controls. M: marker; DIK: dikaryon; D132: diploid; f+: filamentous; TDO: dormant teliospore; FB1, FB2: haploid cells; DPI: days post infection; gDNA: genomic DNA; NTC: no template control.

Filamentous diploid and dikaryon cell types elicit symptoms of infection in planta that suggest a variation in virulence (Banuett & Herskowitz, Citation1996; Babu et al., Citation2005; Banuett, Citation2010). This could be a consequence of expressing different genes in these cell types. The FunCat category distribution was compared between the DIK, D12 and the filamentous cell type groupings and revealed no substantial differences ( and Supplementary ).
DIK cDNA library representation and gene presence in other fungi
A total of 981 genes represented only in the dikaryon library or in the DIK and D12 libraries (Supplementary ) were compiled for use in comparisons with other fungal DNA sequences in the NCBI databases (). These comparisons identified six ORFs found only in U. maydis (one ORF was predicted in MUMDB but not found in the NCBI database) and six conserved only among fungal pathogens (). Four of the 12 genes were represented in U. maydis dikaryon cDNA libraries created and utilized for the initial genome annotation (Kämper et al., Citation2006); however, analysis of these cDNA libraries has not been reported. RT-PCR was carried out on all 12 genes. Haploid cells were grown on DCM with charcoal to account for any impact charcoal may have on gene expression (Babu et al., Citation2005). The transcript levels in the dikaryon and diploid growth forms were compared with the EST representation in the respective libraries ( a, b). RT-PCR results indicated that um01330, um03327 and um01479 had higher transcript levels in the diploid relative to the dikaryon, a result that contrasts with these genes not being represented in the diploid EST library (Supplementary ). Higher expression levels were found for um06485 in the filamentous cell types relative to the other cell types. Each of the 12 genes are expressed during in planta growth. Um05787 transcripts were not detected at 8 DPI or in the dormant teliospore but they were detected at 14 DPI. Um02341 is detected at 8 and 14 DPI yet it is present at a low level, in the dormant teliospore.
Fig. 5. Deletion of ncRNA1 attenuates pathogenicity. (a) Schematic of the ncRNA1 locus. Regions of U. maydis (top) and S. reilianum (bottom) chromosomes (chr) are represented by dashed black lines. A region of S. reilianum chr05 not shown is represented by diagonal black double-lines. Annotated ORFs are represented by pentagons; putative 3′ UTRs surrounding the ncRNA1 deletion by red and orange vertical lines; intergenic regions by solid black lines; introns by solid coloured lines and the noncoding transcript (ncRNA1) by a green arrow. The region removed from ΔncRNA1 strains is highlighted in grey. Putative orthologues shared between U. maydis and S. reilianum are indicated by solid lines of the same colour. (b) RT-PCR examining the effect of ΔncRNA1 on select transcript levels. RNA was isolated from ΔncRNA1 #6 and ΔncRNA1 #7 with independent SG200 controls (#1 and #2). Transcript-specific primers were used to generate cDNA for um02150, ncRNA1, um02151 and um12316 with an internal umGapd-specific primer to control for relative amounts of RNA. ‘No reverse transcriptase’ and ‘no template’ controls were also performed (data not shown). (c) Pathogenicity of ΔncRNA1 U. maydis strains. Infection with independent ΔncRNA1 strains and their corresponding control (SG200) were performed in parallel. Seven-day-old maize seedlings (n = 45) were infected and the symptoms were scored 14 DPI. Separate infection assays (R1 and R2, respectively) differed slightly in the manifestation of disease symptoms. Bars represent the percentage of infected plants and the disease symptoms are depicted in the legend. The disease index (DI) for each strain is shown below the bar graph.
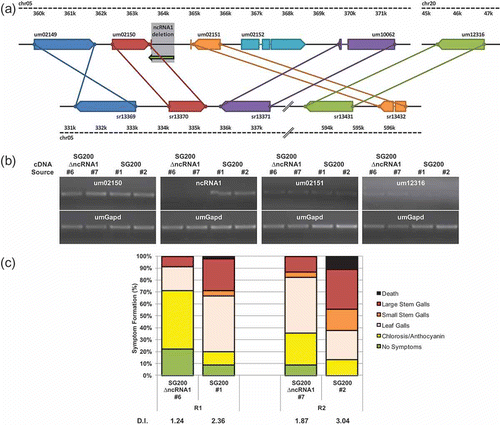
Table 4. Classification of filament-specific DIK represented genes assessed by tblastn and blastp analysis
Ustilago maydis ncRNA1 deletion
Examination of ESTs from the DIK and TDO cDNA libraries identified a ncRNA transcribed from the intergenic region separating the 3′ ends of um02150 and um02151 on chromosome 5 ( a). Complete sequence determination of cloned cDNAs (TDO_20_B10, TDO_42_A07 and DIK_46_F04) revealed that all three represented polyadenylated transcripts that ranged in length from 646 to 675 nts. Contig assembly identified a noncoding transcript, 807 nts in length, named ncRNA1 ( a). NcRNA1 is complementary (antisense) to 65 nts of the um02150 3′UTR. Blastn to the U. maydis genome revealed sequence similarity between ncRNA1 (nt 226–464) and the 3′ end of the um12316 ORF (nt 1280–1512) on chromosome 20. The similarity extends 35 nts into the putative 3′ UTR of um12316. NcRNA1 and the um12316 locus are identical at 209/275 nts. NcRNA1 does not contain an open reading frame, the longest stretch of codons is 54 (ATG to stop, including three cysteine codons). The coded putative peptide would not contain an N-terminal secretion signal, as determined by a search with Signal-P (http://www.cbs.dtu.dk/services/SignalP/). The longest codon stretch upstream of ncRNA1 is 31 (ATG to stop). The region of U. maydis chromosome 5 containing ncRNA1 is rearranged relative to the corresponding region of the closely related S. reilianum. Many genes surrounding ncRNA1 on U. maydis chr05 have a similar arrangement on S. reilianum chr05; however, um02152 has no orthologue in S. reilianum, and um02151 is adjacent to the um12316 orthologue, at a different locus on S. reilianum chr05 ( a). There is no evidence of an orthologue to ncRNA1 in the S. reilianum genome. This difference between U. maydis and S. reilianum is interesting given that these two fungi infect the same host (Zea mays), yet they yield different disease phenotypes.
RT-PCR results indicated that ncRNA1 had higher transcript levels in the filamentous diploid and teliospore, relative to other cell types. Higher transcript levels were found for um12316 in the filamentous cell types relative to the other cell types. Additionally, ncRNA1 was detected in planta, at 8 and 14 DPI, while um12316 transcripts were not detected at 8 DPI or in the dormant teliospore but they were detected at 14 DPI ( c).
Combined, these observations made ncRNA1 a candidate for deletion analysis. Analysis of fully sequenced cDNAs (Doyle et al., Citation2011) facilitated the deletion of ncRNA1 without affecting the adjacent 3′ UTRs corresponding to um02150 and um02151 ( a). Two independent U. maydis ncRNA1 deletion strains were created in the SG200 background. Neither contained the ncRNA1 transcript, and in both, the transcript levels of the flanking genes um02150 and um02151 were not distinguishable from wild-type SG200. However, RT-PCR results suggest transcript levels of um12316 are higher in the mutants relative to wild-type SG200 ( b). Pathogenesis of the two ΔncRNA1 U. maydis strains was assessed at 14 DPI in two independent assays. Deletion of ncRNA1 led to a measurable decrease in virulence, relative to wild-type SG200 infections ( c).
Discussion
The released genome sequence of U. maydis (Kämper et al., Citation2006) provided immense insight into this pathogen's genetic structure, including the identification of gene clusters involved in pathogenesis. However, thorough annotation of the genome requires data from many sources. Sequence information from expressed transcripts is an excellent resource for genome annotation, allowing the confirmation or correction of gene models, the accurate identification of intron/exon boundaries, as well as the identification of transcript structural features (Doyle et al., Citation2011). cDNA cloning and long read lengths ensure that sequences of the ORFs and UTRs are physically linked. Beyond this, analysis of the dikaryon cDNA library allowed an assessment of the limitations of normalization, a comparison of transcript representation in various cell types, the detection of potential constitutively expressed genes, the discovery of noncoding RNAs, and the identification of new candidate pathogenesis genes.
The dikaryon cDNA library was normalized during its creation in an attempt to maximize gene identification (Ozsolak & Milos, Citation2011). Before using the data in comparative analyses, the attributes of normalization were assessed. The DIK cDNA library contained fewer ESTs per ORF represented than any of the non-normalized cDNA libraries (). It also had the highest percentage of unique ORFs () and the lowest redundancy rate for the five genes with the highest representation (Supplementary ). These attributes indicate that clone redundancy was reduced by normalization. Some genes represented in the DIK cDNA library were not found in any other cDNA library and RT-PCR results indicate that genes with low transcript levels in dikaryons are captured in the normalized DIK library (). These transcripts may be at higher levels in other cell types (), yet they were not captured in non-normalized EST libraries. RT-PCR results for a set of genes uniquely represented in the dikaryon cDNA library revealed that these genes were expressed in other cell types ( a). Kessler et al. (Citation2002) found similar differences when comparing normalized and non-normalized cDNA libraries of Aspergillus fumigatus and Yang et al. (Citation2009) found that normalized wheat cDNA libraries increased the identification of novel genes. It has been suggested that normalization can lead to preferential amplification of shorter sequences, which in turn reduces the amount of information obtained per transcript (Kessler et al., Citation2002). Analysis by Doyle et al. (Citation2011) of U. maydis cDNA libraries indicated that cDNAs created using SMART technology, which includes the normalized DIK cDNA library, do not differ in clone length from the non-normalized cDNA libraries created using the Cap-trapper method. It can be concluded from this that normalization, by the methods utilized here, increases the representation of transcripts and does not inhibit the creation of U. maydis full-length cDNA clones.
Comparing all available U. maydis EST libraries (DIK, HCM, MMC, MMN, TDO, T11 and D12), 83 common genes were identified (Supplementary ), refining the list of core genes identified by Ho et al. (Citation2007) and providing the opportunity to screen for candidate constitutively expressed genes. The inclusion of constitutively expressed genes when assessing variation in gene expression across cell types or treatment conditions is necessary to account for variation in the amount of starting material used in each sample and to provide a reference for relative expression levels (Guénin et al., Citation2009). RT-PCR indicated um00175, a gene related to translation initiation factor 3 subunit F, had similar transcript levels across all U. maydis cell types assayed ( b). Eukaryotic translation initiation factor 3 subunit F has also been identified as a potential housekeeping gene in humans (Eisenberg & Levanon, Citation2003).
A major morphological transition in the U. maydis life cycle is the fusion of compatible non-pathogenic budding haploid cells to form the pathogenic filamentous dikaryon (Banuett, Citation2010). Haploid cells induced to fuse and form dikaryons on plates are not 100% converted. Therefore, dikaryotic filaments induced in the laboratory and used for RNA isolation contain some haploid cells. These haploid cells differ from those grown on plates in pure culture in that they are grown on medium containing charcoal (Babu et al., Citation2005), and the presence of cells with a compatible mating type triggers pheromone production. This indicates that differences in transcript levels detected in the DIK cDNA library, relative to the haploid cDNA libraries, may reflect changes in expression resulting from the presence of pheromones, or the formation of the dikaryon. In support of the latter, RT-PCR analysis revealed that the transcript of um06485, represented in the DIK library, was present at a higher level in the filamentous dikaryon and diploid, relative to the non-pathogenic haploid cells ( a). Acknowledging this limitation, we sought to gain knowledge of potential functional differences between U. maydis cell types by comparing FunCats of genes represented in each of the cDNA libraries. A high level of genes classified in the FunCat category ‘protein synthesis’ were present in the non-filamentous haploid U. maydis growth forms compared with the filamentous forms. This difference may reflect cytoplasmic content differences. In contrast to unstressed haploid cells that divide by budding, the dikaryon growing on plates has active cytoplasm only in the apical cell (Banuett et al., Citation2008). Therefore, the amount of active cytoplasm per amount of biomass may be less in filamentous growth forms on plates than haploid cells in culture. A low representation of genes involved in protein synthesis was also noted in the filamentous growth form of Penicillium chrysogenum (van den Berg et al., Citation2008) and in the pathogenic growth phase of Fusarium oxysporum relative to its vegetative non-pathogenic growth phase (Iida et al., Citation2006). The FunCat categories ‘cell rescue defence and virulence’ and ‘interaction with the environment’ were highly represented in the U. maydis filamentous cell types relative to the non-pathogenic haploid forms. This suggests an increase in genes that allow the fungus to sense and protect itself from the environment. Similarly, Morton et al. (Citation2011) found an increase in genes that allowed Aspergillus fumigatus to survive the defensive responses of the host during infection of human dendritic cells. Genes found in the FunCat category ‘cell type differentiation’ were higher in the filamentous cell types of U. maydis relative to the haploid cell types. This representation may reflect preparation for the complex environmental interaction that occurs during the development of filaments on and in the host. The FunCat category ‘cell type differentiation’ is also represented at a higher level in the development of the perithecium, a highly specialized and complex cell type in the ascomycete Gibberella zeae (Qi et al., Citation2006).
The ability to sense changes that occur within its host is critical to the proliferation and survival of a pathogenic fungus (Bahn et al., Citation2006). In U. maydis, the filamentous dikaryon is the primary pathogenic growth form; however, the rarely formed diploid is also pathogenic (Christensen, Citation1931; Holliday, Citation1961; Kojic et al., Citation2002). These two forms differ in nucleus status and have been noted to differ in their virulence on some corn varieties (Banuett & Herskowitz, Citation1996; Babu et al., Citation2005; Banuett, Citation2010). The nuclear state differences and this differential response to the host are consistent with the differences in gene expression observed by Babu et al. (Citation2005). In the analysis of the DIK and D12 cDNA libraries, no differences in functional categories of genes expressed were detected, yet the genes expressed were clearly different. This suggests that distinct genes are expressed in the filamentous diploid and dikaryon; however, these genes carry out similar functions.
Candidate pathogenesis gene selection, for this paper, was conducted as an alternative to previous gene cluster analysis (Kämper et al., Citation2006). The dikaryon and the diploid are pathogenic forms, and therefore it was hypothesized that there were uncharacterized pathogenesis genes among those represented only in the dikaryon cDNA library or in both the dikaryon and filamentous diploid cDNA libraries. This subset of ESTs represented 981 genes that were used in further comparisons. Among these genes, those conserved in other pathogenic fungi, but not in non-pathogenic fungi, would more likely be involved in pathogenesis. Pairwise comparisons identified six U. maydis proteins, which were conserved among pathogenic fungi, as well as six proteins found only in U. maydis (). RT-PCR analysis revealed that expression profiles for the 12 genes varied between cell types; however, all genes were expressed at some stage during in planta infection. The presence of these transcripts in infected corn tissue supports their potential role in pathogenesis.
None of the 12 genes have been deleted to assess a possible role in pathogenesis; however, rbf1 and the b mating type genes are required for the transcription of um10093. Rbf1 also controls the expression of um05787 (Zarnack et al., Citation2008). The b genes and rbf1 control the transition to pathogenic growth in U. maydis; therefore, it is possible that um10093 and um05787 have a role in pathogenesis (Heimel et al., Citation2010). Another gene, um05614, has a GCN5-related N-acetyltransferase (GNAT) domain. Proteins with GNAT domains appear to be over-represented in pathogenic fungi (Seidl et al., Citation2011). GNAT containing proteins may acetylate histones or ribosomal proteins (Yu et al., Citation2006), suggesting that um05614 could alter gene expression at one or more levels during filamentous growth and/or pathogenesis. Interestingly, this gene was conserved among distantly related pathogenic fungi but not found in the closely related S. reilianum. Therefore, it is possible that it could play a role in the distinct pathogenic phenotype exhibited by S. reilianum and U. maydis in their common host. These identified genes will be investigated to determine if they have a role in pathogenesis.
Ustilago maydis pathogenic development proceeds with transitions from filamentous dikaryotic growth, to karyogamy, hyphal fragmentation and the formation of the teliospore. At approximately 8 DPI, hyphal fragmentation produces what has been described as worm-like cells (Banuett & Herskowitz, Citation1996). At 14 DPI, cells undergo rounding and various stages of teliospore formation are present. The differences in transcript presence at 8 and 14 DPI may indicate differences in gene expression between worm-like and post worm-like U. maydis growth forms. Following the cells rounding up teliospores mature and enter dormancy. Therefore, differences in transcript levels between 14 DPI and dormant teliospores may reflect gene expression changes associated with teliospore maturation. Transcripts of um05787 are not detected at 8 DPI or in the dormant teliospore but are present at 14 DPI; whereas, transcripts of um02341 and um06485 are present at 8 DPI and 14 DPI but low or absent in the dormant teliospore ( a, b). Um05787, um02341 and um06485 may be useable as reporter genes denoting the transitions discussed above. This possibility will be investigated in future studies.
The in-depth analysis of the DIK cDNA library presented here highlights the importance of cDNA library data and its usefulness in multiple analyses. The DIK cDNA library aided in the confirmation of 742 gene models (101 with introns), as well as the identification of 25 gene models that require editing, 45 antisense transcripts (Doyle et al., Citation2011) and 58 noncoding RNAs. With the identification of ncRNAs in the DIK cDNA library, the other cDNA libraries were reanalysed and ncRNAs were found to be represented in all (). NcRNAs can be transcribed from intergenic regions, or from regions overlapping ORFs on the same strand (sense), or the opposite strand (antisense). While the functions of most eukaryotic ncRNAs are unknown, it has been determined that some long ncRNAs (> 200 nt), which are distinct from the short interfering RNAs (20–31 nts), have the ability to regulate gene expression at the transcriptional and post-transcriptional levels using a variety of mechanisms. NcRNA transcription can lead to transcriptional interference, chromatin remodelling and histone modifications, or when bound to proteins, ncRNAs can alter the protein's activity and localization (reviewed in Wilusz et al., Citation2009). A higher number of ncRNAs were discovered in the U. maydis DIK cDNA library than in the comparable, but not normalized, filamentous diploid cDNA library. Representation was higher still in the nitrogen stressed haploid cell (MMN) and dormant teliospore (TDO) cDNA libraries (). There were also differences in the representation of antisense RNAs among these libraries. The widespread representation of noncoding and antisense RNAs in U. maydis cDNA libraries suggests that they may provide core functions in cell metabolism or development.
One possibility is that some of the ncRNAs are actually coding RNAs and that they encode small peptides. Effectors, small cysteine rich proteins that influence the host to facilitate pathogenesis, have been identified in several fungal plant pathogens (reviewed in Stergiopoulos & de Wit, Citation2009; Ali & Bakkeren, Citation2011). NcRNA1 could code for an immature peptide of 54 aa. However, no unprocessed effectors are this small in size. Effectors usually have a secretion signal, for example the U. maydis effector Pep1 has a 26 aa signal that is cleaved from the protein during secretion (Doehlemann et al., Citation2009). The 54 aa sequence in ncRNA1 does not have a signal peptide cleavage site, indicating the lack of a recognizable secretion sequence. While it is theoretically possible that the ncRNA1 peptide is translated and secreted by some undescribed mechanism, the characteristics of the potential 54 aa coding region in ncRNA1 do not support its role as an effector protein. This, in turn, suggests that the ncRNA1 is in fact a noncoding RNA.
RT-PCR confirmed that ncRNA1 levels are higher in the dormant teliospore and filamentous diploid, relative to other cell types. It also indicated that ncRNA1 is expressed during in planta U. maydis growth ( c). This situation is not unique to U. maydis. A large number of uncharacterized ncRNAs in the oomycete P. infestans exhibit peak expression during biotrophic growth in infected potatoes, suggesting a role for ncRNAs in pathogenesis (Avrova et al., Citation2007). Similarly, a ncRNA in Aspergillus flavus exhibits temperature-dependent expression. This ncRNA is antisense to aflD and is thought to regulate the expression of the aflatoxin (AF) gene cluster, possibly through chromatin remodelling (Smith et al., Citation2008). Deletion of ncRNA1 in U. maydis resulted in strains that are indistinguishable from wild-type except that they have decreased virulence compared with wild-type ( c) and the deletion of ncRNA1 led to higher transcript levels for um12316, which codes for a secreted protein (Mueller et al., Citation2008). The mechanism linking the deletion and altered transcription level has not been determined. It is possible that ncRNA1 deletion directly impacts pathogenesis or that the resulting alteration in um12316 expression leads to reduced pathogenesis. Support for a possible direct role of ncRNA1 may come through comparison with other fungi.
Like U. maydis, Saccharomyces cerevisiae lacks functional RNA interference machinery (Laurie et al., Citation2008; Drinnenberg et al., Citation2009). In S. cerevisiae, functional roles for ncRNA in regulating gene expression at the transcriptional and post-transcriptional level include transcriptional interference and chromatin remodelling (reviewed in Berretta & Morillon, Citation2009; Harrison et al., Citation2009; Gullerova & Proudfoot, Citation2010). If ncRNA1 directly alters pathogenesis, it could do so by interacting with the 3′ UTR of um02150, which it overlaps. In S. cerevisiae, antisense RNAs overlap the ORF of the sense partner, so if this is the mechanism by which ncRNA1 functions, it is distinct from S. cerevisiae. Another possibility is that ncRNA1 is a functional ncRNA that interacts with a currently unidentified protein affecting its stability or localization. In S. pombe, a 500 nt ncRNA, transcribed from the Sme2 locus (named meiRNA), binds MEI2, affecting its cellular localization. The nuclear MEI2–meiRNA complex is required for premeiotic DNA replication and the initiation of meiosis I (reviewed in Yamamoto, Citation2010). Transcript analysis revealed levels of um02150 and um02151 were unchanged, while um12316 levels were higher in the ΔncRNA1 strains, relative to wild-type ( b). It is possible that differences in um12316 transcript levels are an indirect result of the physiology of the deletion strain, or that ncRNA1 directly affects um12316 transcript levels. Since ncRNA1 shares sequence similarity with um12316 on the sense strand, if there were an interaction in trans between ncRNA1 and the um12316 locus, it would most likely occur through RNA–DNA interactions, possibly facilitating chromatin remodelling. This mechanism would be distinct from the chromatin remodelling in S. cerevisiae, since in this yeast, remodelling is associated with cis-orientated antisense or sense transcripts.
Conclusions
Creation of a normalized cDNA library from filamentous dikaryon RNA allowed improved U. maydis genome annotation through the confirmation of many gene models and the correction of others. It also facilitated the identification of antisense transcripts and noncoding RNAs. Comparing the normalized and non-normalized U. maydis cDNA libraries led to the conclusion that normalization reduces the ability to identify genes that are differentially expressed, while increasing the frequency of detecting unique transcripts. Focusing on the filamentous pathogenic dikaryon enabled FunCat comparison of expressed genes in pathogenic and non-pathogenic cell types, as well as between pathogenic cell types that differ in their response to growth in the host. Six pathogen-specific and six U. maydis-specific candidate pathogenesis genes were identified. Transcript profiles for these candidate genes indicated all were expressed during pathogenic development and that three were differentially expressed during the transition from filamentous dikaryotic growth to the formation of dormant teliospores. This identifies previously undiscovered shifts in gene expression associated with important developmental transitions in U. maydis pathogenesis. In-depth analysis of the DIK cDNA library also identified ncRNAs that were differentially represented among cDNA libraries. Deletion of one such ncRNA, ncRNA1, led to a reduction in pathogenesis. This identifies a new class of pathogenesis genes in fungi. The analysis of the dikaryon cDNA library reveals potential pathogenesis genes that were not detected by other means. This supports the use of targeted in-depth transcriptome analysis to uncover novel details of pathogenesis by smut fungi.
tcjp_a_697077_sup_27778942.pdf
Download PDF (1.2 MB)Acknowledgements
We would like to acknowledge Natalie Gabovic and Dr Shaowu Meng for their assistance with cDNA library creation. We also acknowledge the Natural Sciences and Engineering Research Council (NSERC) of Canada for research funding and the Ontario Graduate Scholarship programme for student support.
Notes
*The first two authors contributed equally to this study.
References
- Adams , M.D. , Kelley , J.M. , Gocayne , J.D. , Dubnick , M. , Polmeropoulos , M.H. Xiao , H. 1991 . Complementary DNA sequencing: expressed sequence tags and human genome project . Science , 252 : 1651 – 1656 .
- Ali , S. and Bakkeren , G. 2011 . Fungal and oomycete effectors – strategies to subdue a host . Can. J. Plant Pathol. , 33 : 425 – 446 .
- Anderson , J. B. and Kohn , L. M. 2007 . “ Dikaryons, diploids, and evolution ” . In Sex in Fungi: Molecular Determination and Evolutionary Implications , Edited by: Heitman , J. , Kronstad , J.W. , Taylor , J. and Casselton , L. 333 – 348 . Washington , DC : ASM Press .
- Austin , R. , Provart , N.J. , Sacadura , N.T. , Nugent , K.G. , Babu , M. and Saville , B.J. 2004 . A comparative genomic analysis of ESTs from . Ustilago maydis. Funct. Integr. Genomics , 4 : 207 – 218 .
- Avrova , A.O. , Whisson , S.C. , Pritchard , L. , Venter , E. , De Luca , S. Hein , I. 2007 . A novel non-protein-coding infection-specific gene family is clustered throughout the genome of Phytophthora infestans. Microbiology . 153 : 747 – 759 .
- Babu , M.R. , Choffe , K. and Saville , B.J. 2005 . Differential gene expression in filamentous cells of . Ustilago maydis. Curr. Genet. , 47 : 316 – 333 .
- Bahn , Y. , Kojima , K. , Cox , G.M. and Heitman , J. 2006 . A unique fungal two-component system regulates stress responses, drug sensitivity, sexual development, and virulence of . Cryptococcus neoformans. Mol. Biol. Cell , 17 : 3122 – 3135 .
- Banuett , F. 2010 . “ Ustilago maydis and maize: a delightful interaction ” . In Cellular and Molecular Biology of Filamentous Fungi , Edited by: Borkovich , K.A. and Ebbole , D.J. 622 – 644 . Washington , DC : ASM Press .
- Banuett , F. and Herskowitz , I. 1989 . Different a alleles are necessary for maintenance of filamentous growth but not for meiosis . Proc. Natl. Acad. Sci. USA , 86 : 5878 – 5882 .
- Banuett , F. and Herskowitz , I. 1996 . Discrete developmental stages during teliospore formation in the corn smut fungus . Ustilago maydis. Development , 122 : 2965 – 2976 .
- Banuett , F. , Quintanilla Jr , R.H. and Reynaga-Peña , C.G. 2008 . The machinery for cell polarity, cell morphogenesis, and the cytoskeleton in the Basidiomycete fungus Ustilago maydis – A survey of the genome sequence . Fungal Genet. Biol. , 45 : S3 – S14 .
- Berretta , J. and Morillon , A. 2009 . Pervasive transcription constitutes a new level of eukaryotic genome regulation . EMBO Rep. , 10 : 973 – 982 .
- Brachmann , A. , König , J. , Julius , C. and Feldbrügge , M. 2004 . A reverse genetic approach for generating gene replacement mutants . Ustilago maydis. Mol. Genet. Gen. , 272 : 216 – 226 .
- Christensen , J.J. 1931 . Studies on the genetics of . Ustilago zeae. Phytopathology , 4 : 129 – 188 .
- Day , P.R. and Anagnostakis , S.L. 1971 . Corn smut dikaryon in culture . Nat. New Biol. , 231 : 19 – 20 .
- Doehlemann , G. , van der Linde , K. , Assmann , D. , Schwammbach , D. , Hof , A. Mohanty , A. 2009 . Pep1, a secreted effector protein of Ustilago maydis, is required for successful invasion of plant cells . PLoS Path. , 5 : e1000290
- Donaldson , M.E. and Saville , B.J. 2008 . Bioinformatic identification of Ustilago maydis meiosis genes . Fungal Genet. Biol. , 45 : S47 – S53 .
- Doyle , C.E. , Donaldson , M.E. , Morrison , E.N. and Saville , B.J. 2011 . Ustilago maydis transcript features identified through full-length cDNA analysis . Mol. Genet. Gen. , 286 : 143 – 159 .
- Drinnenberg , I.A. , Weinberg , D.E. , Xie , K.T. , Mower , J.P. , Wolfe , K.H. Fink , G.R. 2009 . RNAi in budding yeast . Science , 326 : 544 – 550 .
- Eisenberg , E. and Levanon , E.Y. 2003 . Human housekeeping genes are compact . Trends Genet. , 19 : 362 – 365 .
- Gold , S.E. , Brogdon , S.M. , Mayorga , M.E. and Kronstad , J.W. 1997 . The Ustilago maydis regulatory subunit of a cAMP-dependent protein kinase is required for gall formation in maize . Plant Cell , 9 : 1585 – 1594 .
- Guénin , S. , Mauriat , M. , Pelloux , J. , Van Wuytswinkel , O. , Bellini , C. and Gutierrez , L. 2009 . Normalization of qRT-PCR data: the necessity of adopting a systematic, experimental conditions-specific, validation of references . J. Exp. Bot. , 60 : 487 – 493 .
- Gullerova , M. and Proudfoot , N.J. 2010 . Transcriptional interference and gene orientation in yeast: noncoding RNA connections . Cold Spring Harbor Symposium , 75 : 299 – 311 .
- Harrison , B.R. , Yazgan , O. and Krebs , J.E. 2009 . Life without RNAi: noncoding RNAs and their functions in Saccharomyces cerevisiae . Biochem. Cell Biol. , 87 : 767 – 779 .
- Heimel , K. , Scherer , M. , Vranes , M. , Wahl , R. , Pothiratana , C. Schuler , D. 2010 . The transcription factor Rbf1 is the master regulator for b-mating type controlled pathogenic development . Ustilago maydis. PLoS Pathog. , 6 : e1001035
- Ho , E.C. , Cahill , M.H. and Saville , B.J. 2007 . Gene discovery and transcript analyses in the corn smut pathogen Ustilago maydis: expressed sequence tag and genome sequence comparison . BMC Genomics , 8 : 334
- Hoffman , C.S. and Winston , F. 1987 . A ten-minute DNA preparation from yeast efficiently releases autonomous plasmids for transformation of . Escherichia coli. Gene , 57 : 267 – 272 .
- Holliday , R. 1961 . The genetics of Ustilago maydis . Genet. Res. , 2 : 204 – 230 .
- Holliday , R. 1974 . “ Ustilago maydis ” . In Handbook of Genetics. First , Edited by: King , R.C. 575 – 595 . New York : Plenum .
- Horst , R.J. , Engelsdorf , T. , Sonnewald , U. and Voll , L.M. 2008 . Infection of maize leaves with Ustilago maydis prevents establishment of C4 photosynthesis . J. Plant Physiol. , 165 : 19 – 28 .
- Iida , Y. , Ohara , T. and Tsuge , T. 2006 . Identification of genes up-regulated during conidiation of Fusarium oxysporum through expressed sequence tag analysis . Fungal Genet. Biol. , 43 : 179 – 189 .
- Kahmann , R. and Kämper , J. 2004 . Ustilago maydis: how its biology relates to pathogenic development . New Phytol. , 164 : 31 – 42 .
- Kämper , J. 2004 . A PCR-based system for highly efficient generation of gene replacement mutants in Ustilago maydis . Mol. Genet. Gen. , 271 : 103 – 110 .
- Kämper , J. , Kahmann , R. , Bölker , M. , Ma , L.J. , Brefort , T. Saville , B.J. 2006 . Insights from the genome of the biotrophic fungal plant pathogen Ustilago maydis . Nature , 444 : 97 – 101 .
- Kessler , M.M. , Willins , D.A. , Zeng , Q. , Del Mastro , R.G. , Cook , R. Doucette-Stamm , L. 2002 . The use of direct cDNA selection to rapidly and effectively identify genes in the fungus Aspergillus fumigatus . Fungal Genet. Biol. , 36 : 59 – 70 .
- Kojic , M. , Kostrub , C.F. , Buchman , A.R. and Holloman , W.K. 2002 . BRCA2 homolog required for proficiency in DNA repair, recombination, and genome stability in Ustilago maydis . Mol. Cell , 10 : 683 – 691 .
- Lakshminarayan , M.I. , Anantharaman , V. , Wolf , M.Y. and Aravind , L. 2008 . Comparative genomics of transcription factors and chromatin proteins in parasitic protists and other eukaryotes . Int. J. Parasitol. , 38 : 1 – 31 .
- Laurie , J.D. , Linning , R. and Bakkeren , G. 2008 . Hallmarks of RNA silencing are found in the smut fungus Ustilago hordei but not in its close relative Ustilago maydis . Curr. Genet. , 53 : 49 – 58 .
- Martínez-Espinoza , A.D. , García-Pedrajas , M.D. and Gold , S.E. 2002 . The Ustilaginales as plant pests and model systems . Fungal Genet. Biol. , 35 : 1 – 20 .
- Mewes , H.W. , Dietmann , S. , Frishman , D. , Gregory , R. , Mannhaupt , G. Mayer , K.F. 2008 . MIPS: analysis and annotation of genome information in 2007 . Nucleic Acids Res. , 36 : D196 – D201 .
- Morton , C.O. , Varga , J.J. , Hornbach , A. , Mezger , M. , Sennefelder , H. Kneitz , S. 2011 . The temporal dynamics of differential gene expression in Aspergillus fumigatus interacting with human immature dendritic cells in vitro . PloS One , 6 : e1606
- Mueller , O. , Kahmann , R. , Aguilar , G. , Trejo-Aguilar , B. , Wu , A. and de Vries , R.P. 2008 . The secretome of the maize pathogen . Ustilago maydis. Fungal Genet. Biol. , 45 : S63 – 70 .
- Nugent , K.G. , Choffe , K. and Saville , B.J. 2004 . Gene expression during Ustilago maydis diploid filamentous growth: EST library creation and analyses . Fungal Genet. Biol. , 41 : 349 – 360 .
- Ozsolak , F. and Milos , P.M. 2011 . RNA sequencing: advances, challenges and opportunities . Nat. Rev. Genet. , 12 : 87 – 98 .
- Qi , W. , Kwon , C. and Trail , F. 2006 . Microarray analysis of transcript accumulation during perithecium development in the filamentous fungus Gibberella zeae (anamorph Fusarium graminearum) . Mol. Genet. Gen. , 276 : 87 – 100 .
- Rozen , S. and Skaletsky , H.J. 2000 . “ Primer3 on the WWW for general users and for biologist programmers ” . In Bioinformatics Methods and Protocols: Methods in Molecular Biology , Edited by: Krawetz , S. and Misener , S. 365 – 386 . Totowa , NJ : Humana Press .
- Ruepp , A. , Zollner , A. , Maier , D. , Albermann , K. , Hani , J. Mokrejs , M. 2004 . The FunCat, a functional annotation scheme for systematic classification of proteins from whole genomes . Nucleic Acids Res. , 32 : 5539 – 5545 .
- Sacadura , N.T. and Saville , B.J. 2003 . Gene expression and EST analyses of Ustilago maydis germinating teliospores . Fungal Genet. Biol. , 40 : 47 – 64 .
- Sambrook , J. and Russell , D.W. 2001 . Molecular Cloning: A Laboratory Manual , 3rd , Cold Spring Harbor , NY : Cold Spring Harbor Laboratory .
- Schauwecker , F. , Wanner , G. and Kahmann , R. 1995 . Filament-specific expression of a cellulose gene in the dimorphic fungus Ustilago maydis . Biol. Chem. H.-S. , 376 : 617 – 625 .
- Schirawski , J. , Mannhaupt , G. , Münch , K. , Brefort , T. , Schipper , K. Doehlemann , G. 2010 . Pathogenicity determinants in smut fungi revealed by genome comparison . Science , 330 : 1546 – 1548 .
- Seidl , M.F. , Van den Ackerveken , G. , Govers , F. and Snel , B. 2011 . A domain-centric analysis of Oomycete plant pathogen genomes reveals unique protein organization . Plant Physiol. , 155 : 628 – 644 .
- Smith , C.A. , Robertson , D. , Yates , B. , Nielsen , D.M. , Brown , D. Dean , R.A. 2008 . The effect of temperature on Natural Antisense Transcript (NAT) expression . Aspergillus flavus. Curr. Genet. , 54 : 241 – 269 .
- Smith , T.L. and Leong , S.A. 1990 . Isolation and characterization of an Ustilago maydis glyceraldehyde-3-phosphate dehydrogenase-encoding gene . Gene , 93 : 111 – 117 .
- Snetselaar , K.M. and Mims , C.W. 1992 . Sporidial fusion and infection of maize seedlings by the smut fungus Ustilago maydis . Mycologia , 84 : 193 – 203 .
- Snetselaar , K.M. and Mims , C.W. 1994 . Light and electron microscopy of Ustilago maydis hyphae in maize . Mycol. Res. , 98 : 347 – 355 .
- Spellig , T. , Regenfelder , E. and Reichmann , M. 1994 . Control of mating and development in Ustilago maydis. Antonie van Leeuwenhoek . 65 : 191 – 197 .
- Stergiopoulos , I. and de Wit , P.J.G.M. 2009 . Fungal effector proteins . Annu. Rev. Phytopathol. , 47 : 233 – 263 .
- van den Berg , M.A. , Albang , R. , Albermann , K. , Badger , J.H. , Daran , J.M. Driessen , A.J.M. 2008 . Genome sequencing and analysis of the filamentous fungus Penicillium chrysogenum . Nat. Biotechnol. , 26 : 1161 – 1168 .
- Verdun , R.E. , Di Paulo , N. , Urmenya , T.P. , Rondinelli , E. , Frasch , A.C. and Sanchez , D.O. 1998 . Gene discovery through expressed sequence tag sequencing in Trypanosoma cruzi . Infect. Immun. , 66 : 5393 – 5398 .
- Wilusz , J.E. , Sunwoo , H. and Spector , D.L. 2009 . Long noncoding RNAs: functional surprises from the RNA world . Gene Dev. , 23 : 1494 – 1504 .
- Yamamoto , M. 2010 . The selective elimination of messenger RNA underlies the mitosis–meiosis switch in fission yeast . Proc. Jpn. Acad. Ser. B Phys. Biol. Sci. , 86 : 788 – 797 .
- Yang , D. , Tang , Z.H. , Zhang , L.P. , Zhao , C.P. and Zheng , Y.L. 2009 . Construction, characterization, and expressed sequence tag (EST) analysis of normalized cDNA library of thermo-photoperiod-sensitive genic male sterile (TPGMS) wheat from spike developmental stages . Plant Mol. Biol. Rep. , 27 : 117 – 125 .
- Yee , A.R. 1998 . Specificity at the b mating type locus of Ustilago maydis , Vancouver , , Canada : PhD Thesis. The University of British Columbia .
- Yu , M. , de Carvalho , L.P.S. , Sun , G. and Blanchard , J.S. 2006 . Activity-based substrate profiling for Gcn5-related N-acetyltransferases: the use of chloroacetyl-Coenzyme A to identify protein substrates . J. Am. Chem. Soc. , 128 : 15356 – 15357 .
- Zahiri , A.R. , Babu , M.R. and Saville , B.J. 2005 . Differential gene expression during teliospore germination . Ustilago maydis. Mol. Genet. Gen. , 273 : 394 – 403 .
- Zarnack , K. 2006 . Die Auswirkung der posttranslationellen Aktivierung des Transkriptionsfaktors Prf1 auf die Pheromonantwort in Ustilago maydis , Marburg , , Germany : PhD Thesis, Philipps- University .
- Zarnack , K. , Eichhorn , H. , Kahmann , R. and Feldbrügge , M. 2008 . Pheromone-regulated target genes respond differentially to MAPK phosphorylation of transcription factor Prf1 . Mol. Microbiol. , 69 : 1041 – 1053 .