Abstract
The infection of plants by pathogenic microbes and the subsequent establishment of disease involve substantial changes in the biochemistry and physiology of both partners. Analysis of genes that are expressed during these interactions represents a powerful strategy to obtain insights into the molecular events underlying these changes. Clubroot of canola (Brassica napus), caused by the obligate parasite Plasmodiophora brassicae, has considerable economic impact but has not been characterized extensively at the molecular genetic level. Here we have used suppression subtractive hybridization (SSH) and expressed sequence tag (EST) analysis to investigate gene expression during the early stages of colonization of canola roots by P. brassicae. A cDNA library was constructed by SSH which consisted of 797 clones that represented 439 unigenes. Thirty-two of these genes were demonstrated to be of a P. brassicae origin, and of these, 24 had not been previously reported. The remaining 407 genes, which were of a canola origin, were subjected to gene ontology and in silico analyses. Real-time PCR analysis of ten P. brassicae and seven canola genes indicated that seven of the former and five of the latter were upregulated at 7 days after infection, suggesting the importance of these genes in pathogenesis or host resistance.
Résumé
L'infection des plantes par des microbes pathogènes et l'établissement subséquent de la maladie impliquent des modifications considérables sur le plan de la biochimie et de la physiologie des deux partenaires. L'analyse des gènes exprimés durant ces interactions constitue une stratégie énergique permettant de faire la lumière sur les événements moléculaires qui sous-tendent ces modifications. La hernie du canola (Brassica napus), causée par le parasite obligatoire Plasmodiophora brassicae, entraîne de graves répercussions économiques, mais n'a pas fait l'objet de recherches génétiques approfondies à l'échelle moléculaire. Au cours de cette étude, nous avons utilisé l'hybridation soustractive-sélective (HSS) et l'analyse des étiquettes de séquence transcrite (EST) pour examiner l'expression génique durant les premiers stades de la colonisation des racines du canola par P. brassicae. Une banque d'ADNc, comprenant 797 clones qui représentaient 439 unigènes, a été créée par HSS. Nous avons montré que l'origine de 32 de ces gènes remonte à P. brassicae et, de ceux-ci, 24 n'avaient jamais été signalés. Les 407 autres gènes, dont l'origine remontait au canola, ont été soumis à des analyses d'ontologie génique et in silico. L'analyse par PCR en temps réel de 10 gènes de P. brassicae et de 7 gènes de canola a montré que 7 des premiers et 5 des seconds étaient régulés à la hausse 7 jours après l'infection, ce qui suggère l'importance de ces gènes dans le processus de la pathogenèse ou de la résistance de l'hôte.
Introduction
Clubroot, caused by Plasmodiophora brassicae Woronin, is one of the most destructive soilborne diseases of cruciferous crops worldwide and is an emerging threat to canola (Brassica napus L.) production in Canada (Hwang et al., Citation2012). The pathogen is an obligate parasite with a life cycle partitioned to three stages: survival in the soil as resting spores, primary infection of root hairs, and secondary infection of and development within the root cortex (Kageyama & Asano, Citation2009). The persistence of P. brassicae resting spores in the soil for up to 20 years (Wallenhammar, Citation1996) makes management of clubroot through cultural strategies, such as rotation out of susceptible crops, difficult to implement. On the other hand, breeding for resistance to the disease is hindered by a poor understanding of the molecular and cellular biology and biochemistry of P. brassicae and a lack of resistance resources. Improved knowledge of the mechanisms for clubroot pathogenesis could contribute to the development of novel sources of resistance and other control measures.
To date, information on the genomic makeup of P. brassicae and the molecular basis of host resistance/susceptibility is very limited, largely because of the nature of this organism as an obligate parasite. Graf et al. (Citation2004) isolated a few partial cDNAs that had no significant homology to GenBank sequences. Suppression subtractive hybridization (SSH) has been conducted between RNA from P. brassicae-infected and uninfected Arabidopsis tissue, and a total of 76 new P. brassicae gene sequences were identified (Bulman et al., Citation2006). Many of the sequences were predicted to contain signal peptides for translocation of the encoded proteins. Again using SSH, but starting with B. rapa rather than Arabidopsis clubroot galls, Sundelin et al. (Citation2011) isolated about 140 clones. Half of these clones, which represented 24 unisequences, originated from P. brassicae. Of these, 10 unisequences were newly characterized P. brassicae genes. By using next generation sequencing (NGS) technologies, Burki et al. (Citation2010) generated 27,333 contigs assembled from 454 reads from P. brassicae infected B. rapa. Among these contigs, 3167 were from P. brassicae. However, no further analysis was conducted to reveal their functions or relevance with disease development. A few genes have been postulated to be related to the pathogenicity of P. brassicae (Ito et al., Citation1999; Brodmann et al., Citation2002; Ando et al., Citation2006; Siemens et al., Citation2009), mainly based on the observation that the expression of the gene was correlated with a certain stage of disease development. Until now, only one gene, a serine protease (PRO1), has been heterologously expressed and proven experimentally to be important for resting spore germination (Feng et al., Citation2010).
Studies of pathogen-induced changes in host gene expression and metabolism have also been hampered by the difficulties associated with working with this obligate parasite. A few studies used transcriptomic and proteomic approaches to elucidate the mechanisms of P. brassicae infection and coordinate plant reactions. Using a full-genome Affymetrix chip, Siemens et al. (Citation2006) found that more than 1000 Arabidopsis genes were differentially expressed in P. brassica-infected roots at either 10 or 23 days after inoculation versus non-infected roots. Two proteomic studies, both using two-dimensional gel electrophoresis and quantitative image analysis in conjunction with mass spectrometry-based protein identification, illustrated changes in protein abundance at 4 day after inoculation (dai) in Arabidopsis (Devos et al., Citation2006), and at 0.5, 1, 2 and 3 dai in B. napus (Cao et al., Citation2008). The observed differential expression on mRNA or protein levels at the different stages of pathogenesis suggested that the corresponding genes might be involved in the plant response to the clubroot pathogen.
Functional genomic approaches provide powerful tools for identifying expressed genes. Among these techniques, expressed sequence tags (ESTs, Adams et al., Citation1991), serial analysis of gene expression (SAGE, Velculescu et al., Citation1995) and massively parallel signature sequencing (MPSS, Brenner et al., Citation2000) have been successfully employed. Due to its relative simplicity and inexpensive nature, single pass EST sequencing has been the most widely used method to characterize genes associated with cellular development, and biotic and abiotic stress in plant research. Although many EST clones lack the complete sequences of mRNAs, they can be very helpful to investigate the biological function of particular genes, especially in poorly annotated genomes. Additionally, it is possible to obtain full-length clones starting from ESTs. EST resources are also important for comparative transcriptomics because gene expression profiles can be obtained by analysing ESTs generated under varying conditions.
Recently, whole transcriptome sequencing using next-generation sequencing (NGS) technologies or RNA sequencing (RNA-seq) has started to be utilized to reveal the complex landscape of the transcriptome from various organisms at an unprecedented level of sensitivity and accuracy (Ozsolak & Milos, Citation2011). Compared with traditional EST sequencing and other high-throughput technologies such as microarrays, NGS and RNA-seq offer a near-complete snapshot of a transcriptome with a higher dynamic range of expression level. However, common NGS platforms including Illumina, SOLiD and 454 suffer from the disadvantage that the sequence reads are often very short (35–500 bp). As a consequence, high technologies on transcriptome assembly are required after NGS.
Despite the advantages of NGS and RNA-seq, EST cloning incorporated with SSH (Diatchenko et al., Citation1996) has been used to identify genes involved in host–pathogen interactions. This method reduces, by subtractive hybridization, the cloning of abundantly expressed housekeeping genes or genes commonly expressed in both control and treated plants, thereby normalizing expressed cDNA profiles during library construction. Moreover, it significantly enhances, by suppression PCR, the chances of cloning differentially expressed genes. This is particularly important because many pathogenesis-related genes are expressed at low levels, and can be limited to a particular tissue or cell type. This technique has been used to isolate plant and pathogen genes that are expressed during infection (Huang et al., Citation2007). Both of the two cDNA libraries of P. brassicae-infected plants, one using Arabidopsis (Bulman et al., Citation2006) and the other B. rapa (Sundelin et al., Citation2011), were constructed by using the SSH technique.
In the present study, construction of a SSH cDNA library followed by EST analysis was used to achieve a better insight into the genes expressed during the compatible interaction between a susceptible canola cultivar and P. brassicae. The library is the first one based on canola and derived from root samples at an early stage of infection (7 days after inoculation). This stage corresponds to when primary infection is still prevalent and secondary infection is becoming well-established. The results generated from the present study will serve as a foundation for further cloning and functional analysis of the genes related to clubroot pathogenesis.
Materials and methods
Chemicals and standard techniques
Chemicals were purchased from Fisher Scientific Canada (Ottawa, ON) unless otherwise specified. PCRs were conducted in an Eppendorf Mastercycler Thermal Cycler (Eppendorf, Hamburg, Germany). DNA extraction from agarose gels was performed with a Wizard SV Gel & PCR Cleanup Kit (Promega, Madison, WI). Other molecular techniques, if not specified, were performed according to the protocols described by Sambrook & Russell (Citation2001). The accessibility of all websites listed in this paper was verified on the day of submission.
Plant material and Plasmodiophora brassicae inoculum
Canola cultivar ‘Westar’ and P. brassicae strain Led09 (Feng et al., Citation2010) were used as host and pathogen, respectively. Seeds were surface-sterilized in 1% (w/v) sodium hypochlorite for 5 min, washed with distilled water, and germinated on moistened filter paper for 7 days. The resulting seedlings were used in inoculation experiments. Galls that developed on ‘Westar’ roots after artificial infection by Led09 in a greenhouse were collected and kept at −20 °C or −80 °C until required. These galls were used to prepare resting spores and for RNA extraction in real-time PCR analysis, respectively.
Preparation of resting spores
Galls were homogenized in 10% (w/v) sucrose in a blender. The slurry was passed through eight layers of cheesecloth and the suspension was centrifuged in a 50-mL tube at 50 ×g for 5 min. The supernatant was transferred into a new tube and centrifuged at 2000 ×g for 5 min. The resulting pellet had two distinct layers: the lower black layer consisted of soil particles, and the upper white to brownish layer had resting spores (Bryngelsson et al., Citation1988). The upper layer was suspended in 5 mL water by gentle pipetting and transferred into a new tube containing 40 mL water. After centrifugation at 2000 ×g for 5 min, the supernatant was discarded. Spores in the pellet were adjusted to 1 × 107 spores mL−1 and surface-disinfected with 1.0 μg mL−1 colistin sulphate and 1.0 μg mL−1 vancomycin hydrochloride (both from Sigma-Aldrich Canada, Oakville, ON) at 25 °C in the dark for 24 h. The spores were used as inoculum or for genomic DNA extraction after removal of the antibiotics by washing twice with 40 mL distilled water.
Plant inoculation
The prepared resting spores were used to inoculate Sunshine #4 potting mix (Sun Gro Horticulture, Vancouver, BC) at a concentration of 1 × 107 spores mL−1 soil. To ensure infection, roots of ‘Westar’ seedlings were immersed in a resting spore suspension (5 × 107 spores mL−1) for 5 min before transplanting into 15-cm-diameter pots filled with the inoculated soil. The pots were kept in trays in a growth chamber maintained at 24 °C/18 °C (day/night) with a 16 h photoperiod and 80% RH, and watered from the bottom every second day with tap water at pH 6.4 (adjusted with HCl).
Samples for RNA extraction
At 7 day after inoculation (dai), plants were dug up and their roots were rinsed in tap water to remove potting mix residues. After removal of the excess water with filter paper, a 2-cm long root segment was cut 2 cm below the hypocotyl from each plant. Ten such segments were randomly selected and their infection by P. brassicae was investigated with a Zeiss AXIO microscope (Carl Zeiss, Thornwood, NY). On each root segment, primary infection was assessed as the percentage of infected root hairs in 10 fields of view using the 10× objective lens. For secondary infection, the total number of secondary plasmodia in the entire root segment was counted.
Suppression subtractive hybridization (SSH)
Total RNA was extracted from 100 mg ground powder of root segments collected from healthy or inoculated plants at 7 dai using a NucleoSpin RNA II Kit (Clontech Laboratories, Mountain View, CA). Two micrograms of the obtained total RNA was used to generate double-stranded cDNA with a SMARTer PCR cDNA Synthesis Kit (Clontech Laboratories). SSH was performed by using a PCR-Select cDNA Subtraction Kit (Clontech Laboratories), with the protocol being followed starting from the RsaI Digestion step. Using the double-stranded cDNA from infected roots as tester and those from healthy roots as driver, subtractive hybridization and suppression PCR were performed. The PCR product was separated on a gel and the band at 250–4000 bp was cut and used to construct the cDNA library.
Cloning and plasmid DNA miniprep
The PCR products generated from the SSH were cloned using the pGEM-T Easy Vector System (Promega). Ampicillin resistant colonies were isolated by blue-white screening, cultured in LB broth, and preserved by freezing at −80 °C with 15% (w/v) glycerol. Plasmid DNA was extracted from selected colonies by the Alkaline Lysis with SDS protocol or by using PureYield Plasmid Miniprep Kit (Promega). Plasmid DNA concentrations were determined with a ND-1000 spectrophotometer (Thermo Scientific, Rockford, IL).
Sequencing
Using one of the M13 primers, sequencing PCRs were conducted in BigDye terminator reagent (ABI, Foster City, CA) following the manufacturer's instructions. PCR products were sequenced directly with an ABI PRISM 3100 automated DNA sequencer at the University of Alberta, Edmonton, AB. Since the cDNAs had been cut by RsaI (and were thus expected to be short) and only one-shot sequencing was conducted for each clone, the term ‘expressed sequence tag’ (EST) is used hereafter to refer to all of the generated sequences.
EST processing
All DNA sequences were checked for quality and then analysed by VecScreen (http://www.ncbi.nlm.nih.gov/VecScreen/VecScreen.html), which identified and removed vector and adaptor sequences. Manual searching and removing of the two nested PCR primer sequences, which were parts of the adaptors and had been introduced into the ESTs before SSH, was also conducted to complement the VecScreen analysis. The cleaned sequences were analysed with the CAP3 program with default parameters to obtain cluster contigs (http://deepc2.psi.iastate.edu/aat/cap/cap.html).
Basic local alignment search tool (Blast) searching
Identification of EST origin (canola or P. brassicae) was performed by similarity searches against the NCBI databases (http://blast.ncbi.nlm.nih.gov/Blast.cgi) using three approaches: BlastN against the non-redundant nucleotide collection (nr/nt) database, BlastN against the EST database (termed BlastN-EST hereafter) and BlastX against the non-redundant protein sequences (nr) database. Cut-off values were set up as ≤ 1e−10 for BlastN and BlastX, and ≤ 1e−40 for BlastN-EST. Best hits were recorded when their E-value was less than the cut-off values. If the E-values of all hits for a Blast search were higher than the cut-off value, the Blast result was recorded as ‘Non’ (no hit).
After confirmation by PCR (see below), the P. brassicae ESTs were subjected to the three Blast searches again but using the default parameters and the best hits were recorded. After functional annotations (see below), BlastX were conducted for selected canola ESTs against the Arabidopsis genome sequence (AtGDB) hosted at Plant Genome Database (http://www.plantgdb.org). Best hits with an E-value ≤1e−20 were recorded and compared with the up/downregulated genes identified by Siemens et al. (Citation2006).
PCR identification of Plasmodiophora brassicae clones
Based on the Blast results, 34 clones were subjected to PCR identification. Primers (Supplemental Table 1, available online) were designed based on the cDNA sequences by using the Primer3 program (http://frodo.wi.mit.edu/primer3) and synthesized by Integrated DNA Technologies (Coralville, IA). Genomic DNAs were extracted from P. brassicae resting spores and healthy canola roots collected from the prepared seedlings using a Plant DNA kit (Omega Bio-Tek, Norcross, GA). Amplification was performed with 40 cycles of denaturation at 94 °C for 30 s (4 min for the first cycle), annealing at 58 °C for 45 s and extension at 72 °C for 1 min.
Functional annotation by Blast2GO
Functional annotation was performed for all ESTs except those identified by PCR to be P. brassicae-originated by using Blast2GO version 2.2.3 (Conesa et al., Citation2005), following the standard procedure of BlastX for the unigenes dataset (parameters: nr database, high scoring segment pair cut-off length 33, report 20 hits, maximum E-value 1e−3), followed by mapping and annotation (parameters: E-value hit filter 1e−6, annotation cut-off 55, GO weight 5, HSP-hit coverage cut-off 20). GO terms were summarized according to their molecular functions, biological processes and cellular components. Enzyme mapping of the annotated sequences was performed with Direct GO to Enzyme Mapping and used to query the Kyoto Encyclopedia of Genes and Genomes (KEGG) to define the KEGG orthologs.
Gene expression analysis by real-time PCR
Total RNA was extracted from galls, uninfected and infected roots at 7 dai using the PureYield RNA Midiprep System (Promega). The total RNA was used to generate first strand cDNA with an iScript cDNA Synthesis Kit (Bio-Rad Canada, Mississauga, ON). Based on the obtained cDNA, real-time PCR was conducted to confirm the differential expression of ten selected P. brassicae genes and seven canola genes. Primers (Supplemental Table 2) were designed based on the EST sequences using the Primer3 server (http://frodo.wi.mit.edu/primer3). PCR was run in a final volume of 10 μL using 0.75 μM of each primer with the SYBR Green Core Reagents Kit (Life Technologies, Carlsbad, CA), following the manufacturer's instructions. The comparative CT method was used to analyse and present the data according to Schmittgen & Livak (Citation2008). The canola elongation factor 1α alpha gene (FJ529180) and the P. brassicae actin 1 gene (AAR88383) was used for normalization.
Results
Time-point of RNA isolation
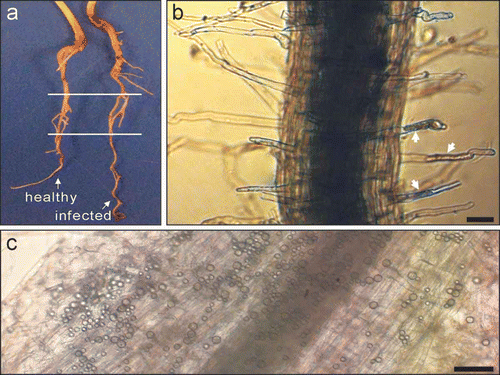
At 7 dai, there were no macroscopic differences between the inoculated and healthy plants (). Under the microscope, primary () and secondary () infections were commonly observed on inoculated plants. Based on ten 2-cm long root samples cut from the inoculated plants, the primary infection rate and the secondary plasmodium number per sample were 47.7± 18.1% and 83 ± 42%, respectively.
Fig. 1. Canola roots at 7 days after inoculation with Plasmodiophora brassicae. a, A healthy and an infected plant. The two horizontal lines define the segment of root used for RNA extraction. b, c, Primary infection (b) and secondary plasmodia (c) on the inoculated roots. Some of the root hairs with primary infections are indicated by arrows in (b). Bars = 50 μm.
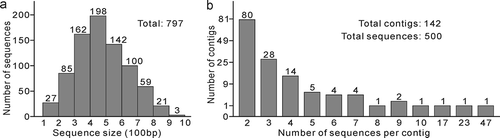
Characteristics of the cDNA library
From a single transformation, 831 colonies were selected and from which plasmid DNA was extracted. Using PCR with the M13 primers, a band larger than 250 bp was amplified from 797 clones. These clones were sequenced. Once the raw sequences were trimmed of vector, adaptor and poly-A sequences, the sizes of the cleaned sequences ranged from 103 to 945 bp with an average of 480 bp and two-thirds of the sequences were larger than 400 bp (). A total of 439 high-quality trimmed sequences (unisequences) were obtained after CAP3 assembling, revealing that the redundancy of the cDNA library is 44.9% (1–439/797). The 439 unisequences consisted of 297 singlets and 142 contigs with each contig containing 2–47 merged ESTs (). The 439 sequences had an average length of 516 bp and an average GC content of 44.2%.
Fig. 2. Sequence size distribution of the 797 ESTs (a) and distribution of ESTs in 142 contigs (b).
Origin of ESTs
The 439 unisequences were compared against known databases to infer their origins (canola or P. brassicae), based on significant sequence similarity to experimentally verified DNA or protein sequences. Homology searches performed by BlastN, BlastX and BlastN-EST revealed that most of the sequences shared similarities with known DNA or proteins (). Among the three Blast methods, BlastN-EST gave the most reliable results with respect to confirmation of plant-origin ESTs. Of the 439 sequences, 406 produced best hits from plant species, with 391 from Brassica species, 12 from Raphanus species, 1 from A. thaliana and 2 from non-cruciferous plants (C007 and C065). Since all of the crucifer-originated best hits identified by BlastN-EST had an E-value less than 1e−40, the corresponding ESTs were excluded in the PCR confirmation of P. brassicae-origin. The remaining 35 sequences () were subjected to PCR identification. These sequences covered all potential P. brassicae-origin sequences identified by BlastX and BlastN ().
Fig. 3. Identification of EST origin. a, Similarity search by Blast. Plant, P. brassicae and Others, the best hit of the sequence is from plant, Plasmodiophora brassicae or other organisms, respectively; Non, no hit. The red (online; horizontal in print) bar indicates clones subjected to PCR identification. All Blasts were conducted on 10–13 November 2011. b, PCR identification of EST origin using sequence-specific primers and DNA from resting spores of P. brassicae (left) or healthy canola root (right). C007-C136, contig 007 to contig 136; S007-S827, singlet 007 to singlet 827; M, Promega 100-bp DNA ladder.
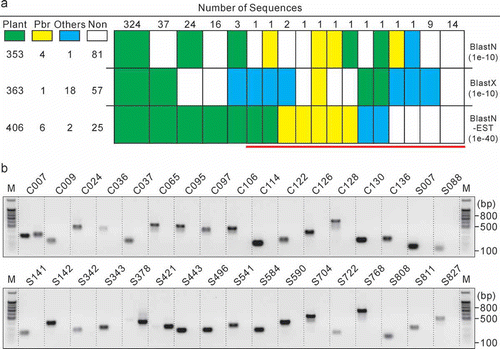
Among the 35 sequences that were predicted to be of non-plant origin, one sequence (C132) contained only a simple sequence repeat and no primer binding sites could be located by the Primer3 program. Thus, this sequence was excluded from the PCR analysis. At the annealing temperature of 58 °C, all of the 34 sequences were shown to be present in the DNA sample extracted from either healthy canola root or P. brassicae resting spores (). C007 was present in both healthy plants and resting spores and this was observed when the PCR was re-conducted with an annealing temperature at 60 °C, 62 °C or 64 °C (data not shown), indicating that the sequences in canola and P. brassicae share a high level of sequence identity. Since BlastN of C007 produced a best hit (GenBank accession number: AB526843) as P. brassicae small subunit ribosomal RNA with an E-value of 0.0 () and the entire 363 bp of C007 aligned to AB526843 with 100% identity, we believe that C007 is derived from P. brassicae. Primers for S378 and S421 produced bands from the healthy plant but not from P. brassicae (), indicating that these two sequences are derived from the host. Indeed, BlastN and BlastX of S378, and BlastX of S421, all produced a best hit with plant origin. All other sequences produced bands only from P. brassicae, indicating that they originated from the parasite. In summary, 32 sequences were found to be of P. brassicae origin (), among which 24 were novel and identified in the present study. The 32 P. brassicae sequences consisted of 15 contigs and 17 singlets. Since the 15 contigs were assembled from 141 clones, the proportion of P. brassicae clones in our library is 19.8% (158/797). The 24 new genes identified were submitted to GenBank under the accession numbers JK747433–JK747456.
Table 1. The best hits from Blast searches with the 32 Plasmodiophora brassicae clones found by suppression subtractive hybridization
Functional annotations
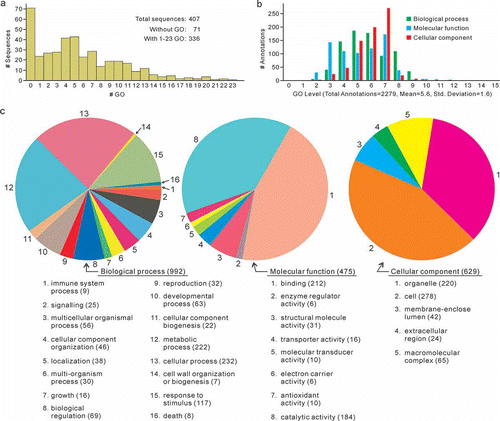
The 407 sequences not originating from P. brassicae were subjected to gene ontology (GO) annotation to interpret their function. When annotated by the Blast2GO program, most of the unisequences could be mapped and annotated. However, 38 sequences did not have significant matches from BlastX and therefore could not be mapped or annotated. Of the sequences with BlastX hits, nine could not be mapped and 24 could not be annotated. The remaining 336 (407–38–9–24) sequences were each assigned to at least one gene ontology (GO) term and several sequences had as many as 23 GO terms associated with them (). The gene ontologies were assigned to the annotated sequences according to biological process, molecular function and/or cellular component (). For gene products, the GO level indicates the specificity of the description of gene ontology terms. A higher level of GO therefore corresponds to a more specific description of gene products. The ESTs were distributed across many levels of GO annotations. A total of 2279 annotations were made with a mean level of 5.6 (). The GO level 2 was used in annotating the data and constructing pie charts (). A total of 992 GO terms were observed as a biological process. Among these, 232 (23.4%), 222 (22.4%) and 117 (11.8%) were related to cellular process, metabolic process and response to stimulus, respectively. In the category of molecular function, the vast majority of the 475 GO terms were involved in binding (212, 44.6%) and catalytic activities (184, 38.7%). Among the 629 GO terms in the cellular component category, 278 (44.2%) and 220 (35.0%) corresponded to cell parts and organelle, respectively.
Fig. 4. Functional annotation of ESTs conducted on 6–7 December 2011. a, Distribution of the number of annotated sequences versus the number of GO terms. b, The GO-level distribution of the annotated sequences. c, Pie charts of the second-level GO terms.
Out of 407 sequences, 106 were assigned with KEGG pathways based on enzyme codes generated by Blast2GO. Several KEGG pathways were represented by five or more ESTs (). The highest number of KEGG mappings was extracted from the methane metabolism pathway, with 16 sequences. BlastX were conducted for the 106 sequences against the Arabidopsis genome sequences. Seven sequences () produced best hits that were present in the lists of most up- or downregulated Arabidopsis genes after infection by P. brassicae (Siemens et al., Citation2006). These seven sequences were submitted to GenBank under the accession numbers JK7789863–JK789869.
Table 2. Number of genes involved into reconstruction of KEGG pathway maps (partial list, number of ESTs > 4)
Table 3. Canola clones with high sequence similarity to Arabidopsis genes that were up- or downregulated during clubroot pathogenesis
Expression of selected genes
The differential expression of ten and seven selected genes from P. brassicae and canola, respectively, was analysed by real-time PCR. The results were represented as the ratio between resting spores and in planta at 7 dai for P. brassicae genes, and between healthy and inoculated plants for canola genes (). Among the ten P. brassicae genes, seven were upregulated and three were downregulated at 7 dai. One gene, represented by clone C065, showed the most distinct upregulation. Furthermore, compared with the housekeeping gene, this gene was highly expressed both in resting spores and at the 7-dai infection stages. For the seven canola genes, five were upregulated and two were downregulated.
Table 4. Real-time PCR analysis of expression of selected genes
Discussion
The SSH procedure was used in the present study because, in theory, it allows the creation of a cDNA library enriched for sequences involved in the plant–pathogen interaction. Using PCR amplification and similarity searches, the results indicated that SSH was successful in creating a library with genes from both canola and P. brassicae at a time point when macroscopic symptoms were not visible but the pathogen was actively invading the root cortex. Indeed, both primary and secondary infections are prevalent at 7 dai on the same root (Feng et al., Citation2010; Feng et al., Citation2012b ) and P. brassicae actin gene transcripts could be readily detected by Northern hybridization (Feng et al., Citation2010). Since the materials and the time of sampling were different with the two previously constructed cDNA libraries (Bulman et al., Citation2006; Sundelin et al., Citation2011), genes identified from this library, in conjunction with those from other libraries, will provide a more comprehensive picture of gene expression during clubroot pathogenesis.
In this study, in order to determine the origin (canola or P. brassicae) of the assembled ESTs (unisequences) in the library derived from mixed tissues, a multifaceted sequence analysis approach was used based on three Blast searches. The results showed that BlastN-EST is the strongest method for determination of canola genes. It is better than BlastX due to the advantage that frame-shift artefacts arising from sequencing errors scarcely disturb the performed alignment and, on the other hand, it is better than BlastN versus other databases due to the fact that for canola more ESTs are available than other sequences. With the results from BlastN-EST as the major criteria, most plant genes were identified. Results from the PCR identification also supported the reliability of BlastN-EST, in which most of the sequences with non-plant best hits from BlastN-EST were demonstrated by PCR to be of P. brassicae origin.
Of the 32 identified P. brassicae genes, 17 had significant BlastX hits (E ≤ 1e−10) in the GenBank nr database. Of these 17 significant hits, 10 were either hypothetical or predicted proteins with unknown functions (). Of the 407 non-P. brassicae sequences, 336 were assigned to certain functions based on BlastX (). Thus, the SSH procedure was more effective at identifying host genes than pathogen genes. This is probably due to the higher ratio of host to pathogen mRNA, especially in our case in which canola roots at an early infection stage were used to generate the library. The relative biomass contributions from the host and pathogen were, therefore, highly unequal.
The percentage of pathogen clones in the current library was 19.8%, which is lower than for an Arabidopsis–P. brassicae library (Bulman et al., Citation2006) constructed at 42 dai (193/232 = 83%) and a B. rapa–P. brassicae library (Sundelin et al., Citation2011) constructed at 35 dai (69/140 = 49%) when large galls already formed, but higher than for SSH libraries conducted from other pathosystems at early infection stages, for example 4% (57/1345) for an Arabidopsis–Peronospora parasitica SSH library (Bittner-Eddy et al., Citation2003) and 1% (1/106) for a banana–Mycosphaerella fijiensis SSH library (Portal et al., Citation2011). The rate of redundancy for the SSH cDNA library was 44.9%. Because the library was not directional (constructed by TA cloning) and sequenced from only one direction, some degree of redundancy will not be detected, and more than one contig may correspond to the same transcript. For example, C027 and C050 share high similarity with alternative regions of AT4G11310 ().
Of the 32 P. brassicae genes identified, only eight were found to have P. brassicae-origin hits by the three Blasts. Using plant roots at later infection stages when plenty of P. brassicae resting spores had already formed, Sundelin et al. (Citation2011) identified 24 P. brassicae genes, of which 14 were also identified by Bulman et al. (Citation2006). Among the three most frequent genes in common in their libraries (PbsHSP1, PbGST1 and PbSUR2), only PbSUR2 was identified in the present study as a singlet (S808). The abundance of an EST can help understand the relative proportions of various transcripts. In the present library, the most abundant non-rRNA ESTs were C128 and C009 with 47 and 17 copies, respectively. Both of these represent P. brassicae genes and are not present in the two previously constructed libraries. All of these observations suggest that there are different gene expression patterns in early- and late-clubroot infection stages. Besides S808, another two genes were shared by the three libraries, PbsHSP2 (S142) and PbSUNK9 (C126), indicating that these three genes may play roles during different disease development stages.
Given the facts that very small numbers of P. brassicae sequences are available in the database and that P. brassicae is phylogenetically non-related to most of the model species, we were not surprised by the observation that most identified P. brassicae genes failed to produce any Blast hits. These sequences may represent novel genes that have not been previously characterized in other organisms, or are indeed unique to P. brassicae.
One interesting finding from the current study is that several canola genes share sequence similarity with Arabidopsis genes that were found to be up/downregulated during pathogenesis (). Siemens et al. (Citation2006) indicated that most known defence- or resistance-related genes were either not differentially expressed or downregulated during Arabidopsis clubroot development (23 dai vs. 10 dai). At 23 dai, resting spores were developed and the galls were forming (Siemens et al., Citation2006). Thus, the host genes involved in resistance/susceptibility may no longer be as active as during the early infection stage. Arabidopsis orthologs of the upregulated genes observed in the present study also showed higher expression at 10 dai in relation to 23 dai, and so did the downregulated genes. Given the perception that the two time points in the present study could be reversely correspondent to the two time points in Siemens et al. (Citation2006) in regard to gene expression, the regulation patterns of the seven canola genes are in consistency with their Arabidopsis orthologs. On the other hand, a new hypothesis, which states that P. brassicae opens the door to secondary infection by suppressing host resistance during primary infection, was recently proposed (Feng et al., Citation2012a ). This hypothesis is supported by the prevalence of downregulated genes identified by Siemens et al. (Citation2006) and in the current study. Under such a scenario, it can be postulated that the stage-dependently expressed genes identified in the current study must play important roles in the interaction between the plant and the pathogen. Compared with downregulated genes, more upregulated genes were identified from the ten P. brassicae genes. These upregulated genes could be important in early stages of clubroot pathogenesis.
Although the length and quality of the sequences are lower than alternative, less sensitive methods, the SSH method has the important advantage of detecting differentially expressed genes occurring at low transcript levels. It has been a useful strategy to identify host genes important for resistance. For example, in B. napus, genes encoding NAC-domain transcription factors have been isolated through subtractive expressed-sequence-tag analysis from the cDNA libraries of plants subjected to abiotic and biotic stresses (Hegedus et al., Citation2003). In the present study, more than 400 plant sequences were identified of which 336 represent genes with a predicted function. Among the 336 genes, a significant proportion is associated with response(s) to infection stimuli, which likely includes genes related to clubroot resistance.
The cDNA library presented in the present study provides limited yet useful insight into the transcriptional composition of both canola and P. brassicae. The EST sequences should enhance the effectiveness of molecular studies, especially for gene expression profiling and the design of microarrays. The genes represented can be further studied by various molecular techniques to reveal their functions in either P. brassicae pathogenicity or canola resistance. Additionally, the cluster and redundancy information should be useful for further subtraction of the most abundant transcripts included in the cDNA library, making further EST analysis more effective.
Supplementary material
Download PDF (156.7 KB)Acknowledgements
The authors acknowledge the funding from the Canola Agronomic Research Program (Alberta Canola Producers Commission, Manitoba Canola Growers Association, SaskCanola and the Canola Council of Canada), the Alberta Crop Industry Development Fund (ACIDF) and the Clubroot Risk Mitigation Initiative (AAFC/CCC).
References
- Adams , M.D. , Kelley , J.M. , Gocayne , J.D. , Dubnick , M. , Polymeropoulos , M.H. Xiao , H. 1991 . Links complementary DNA sequencing: expressed sequence tags and human genome project . Science , 252 : 1651 – 1656 .
- Ando , S. , Yamada , T. , Asano , T. , Kamachi , S. , Tsushima , S. , Hagio , T. and Tabei , Y. 2006 . Molecular cloning of PbSTKL1 gene from Plasmodiophora brassicae expressed during club root development . J. Phytopathol. , 154 : 185 – 189 .
- Bittner-Eddy , P.D. , Rehmany , A.P. , Birch , P. and Beynon , J.L. 2003 . Use of suppression subtractive hybridization to identify downy mildew genes expressed during infection of Arabidopsis thaliana . Mol. Plant Pathol. , 4 : 501 – 507 .
- Brenner , S. , Johnson , M. , Bridgham , J. , Golda , G. , Lloyd , D.H. Johnson , D. 2000 . Gene expression analysis by massively parallel signature sequencing (MPSS) on microbead arrays . Nat. Biotechnol. , 18 : 630 – 634 .
- Brodmann , D. , Schuller , A. , Ludwig-Müller , J. , Aeschbacher , R.A. , Wiemken , A. , Boller , T. and Wingler , A. 2002 . Induction of trehalase in Arabidopsis plants infected with the trehalose-producing pathogen Plasmodiophora brassicae . Mol. Plant Microbe Interact. , 15 : 693 – 700 .
- Bryngelsson , T. , Gustafsson , M. , Gréen , B. and Lind , C. 1988 . Uptake of host DNA by the parasitic fungus Plasmodiophora brassicae . Physiol. Mol. Plant Pathol. , 33 : 163 – 171 .
- Bulman , S. , Siemens , J. , Ridgway , H.J. , Eady , C. and Conner , A.J. 2006 . Identification of genes from the obligate intracellular plant pathogen, Plasmodiophora brassicae . FEMS Microbiol. Lett. , 264 : 198 – 204 .
- Burki , F. , Kudryavtsev , A. , Matz , M.V. , Aglyamova , G.V. , Bulman , S. , Fiers , M. , Keeling , P.J. and Pawlowski , J. 2010 . Evolution of Rhizaria: new insights from phylogenomic analysis of uncultivated protists . BMC Evol. Biol. , 10 : 377
- Cao , T. , Srivastava , S. , Rahman , M.H. , Kav , N.N.V. , Hotte , N. , Deyholos , M.K. and Strelkov , S.E. 2008 . Proteome-level changes in the roots of Brassica napus as a result of Plasmodiophora brassicae infection . Plant Sci. , 174 : 97 – 115 .
- Conesa , A. , Gotz , S. , Garcia-Gomez , J.M. , Terol , J. , Talon , M. and Robles , M. 2005 . Blast2GO: a universal tool for annotation, visualization and analysis in functional genomics research . Bioinformatics , 21 : 3674 – 3676 .
- Devos , S. , Laukens , K. , Deckers , P. , van der Straeten , D. , Beeckman , T. Inze , D. 2006 . A hormone and proteome approach to picturing the initial metabolic events during Plasmodiophora brassicae infection on Arabidopsis . Mol. Plant Microbe Interact. , 19 : 1431 – 1443 .
- Diatchenko , L. , Lau , Y.F.C. and Campbell , A.P. 1996 . Suppression subtractive hybridization: a method for generating differentially regulated or tissue-specific cDNA probes and libraries . Proc. Natl. Acad. Sci. USA , 93 : 6025 – 6030 .
- Feng , J. , Hwang , R. , Hwang , S.F. , Strelkov , S.E. , Gossen , B.D. , Zhou , Q. and Peng , G. 2010 . Molecular characterization of a serine protease Pro1 from Plasmodiophora brassicae that stimulates resting spore germination . Mol. Plant. Pathol. , 11 : 503 – 512 .
- Feng , J. , Hwang , S.F. and Strelkov , S.E. 2012a . Studies into primary and secondary infection processes by Plasmodiophora brassicae on canola . Plant Pathol , in press, DOI: 10.1111/j.1365–3059.2012.02612.x
- Feng , J. , Xiao , Q. , Hwang , S.F. , Strelkov , S.E. and Gossen , B.D. 2012b . Infection of canola by secondary zoospores of Plasmodiophora brassicae produced on a nonhost . Eur. J. Plant Pathol. , 132 : 309 – 315 .
- Graf , H. , Fähling , M. and Siemens , J. 2004 . Chromosome polymorphism of the obligate biotrophic parasite Plasmodiophora brassicae . J. Phytopathol. , 152 : 86 – 91 .
- Hegedus , D. , Yu , M. , Baldwin , D. , Gruber , M. , Sharpe , A. , Parkin , I. , Whitwill , S. and Lydiate , D. 2003 . Molecular characterization of Brassica napus NAC domain transcriptional activators induced in response to biotic and abiotic stress . Plant Mol. Biol. , 53 : 383 – 397 .
- Huang , X. , Li , Y. , Niu , Q. and Zhang , K. 2007 . Suppression subtractive hybridization (SSH) and its modifications on microbiological research . Appl. Microbiol. Biotechnol. , 76 : 753 – 760 .
- Hwang , S.F. , Strelkov , S.E. , Feng , J. , Gossen , B.D. and Howard , R.J. 2012 . Plasmodiophora brassicae: a review of an emerging pathogen of the Canadian canola (Brassica napus) crop . Mol. Plant Pathol. , 13 : 105 – 113 .
- Ito , S. , Ichinose , H. , Yanagi , C. , Tanaka , S. , Kameya-Iwaki , M. and Kishi , F. 1999 . Identification of an in planta-induced mRNA of Plasmodiophora brassicae . J. Phytopathol. , 147 : 79 – 82 .
- Kageyama , K. and Asano , T. 2009 . Life cycle of Plasmodiophora brassicae . J. Plant Growth Regul. , 28 : 203 – 211 .
- Ozsolak , F. and Milos , P.M. 2011 . RNA sequencing: advances, challenges and opportunities . Nat. Rev. Genet. , 12 : 87 – 98 .
- Portal , O. , Izquierdo , Y. , De Vleesschauwer , D. , Sánchez-Rodríguez , A. , Mendoza-Rodríguez , M. , Acosta-Suárez , M. , Ocaña , B. , Jiménez , E. and Höfte , M. 2011 . Analysis of expressed sequence tags derived from a compatible Mycosphaerella fijiensis–banana interaction . Plant Cell Rep. , 30 : 913 – 928 .
- Sambrook , J. and Russell , D.W. 2001 . Molecular Cloning: A Laboratory Manual , 3rd , Cold Spring Harbor , NY : Cold Spring Harbor Laboratory Press .
- Schmittgen , T.D. and Livak , K.J. 2008 . Analyzing real-time PCR data by the comparative C T method . Nat. Protoc. , 3 : 1101 – 1108 .
- Siemens , J. , Keller , I. , Sarx , J. , Kunz , S. , Schuller , A. , Nagel , W. , Schmülling , T. , Parniske , M. and Ludwig-Müller , J. 2006 . Transcriptome analysis of Arabidopsis clubroots indicate a key role for cytokinins in disease development . Mol. Plant Microbe Interact. , 19 : 480 – 494 .
- Siemens , J. , Graf , H. , Bulman , S. , In , O. and Ludwig-Müller , J. 2009 . Monitoring expression of selected Plasmodiophora brassicae genes during clubroot development in Arabidopsis thaliana . Plant Pathol. , 58 : 130 – 136 .
- Sundelin , T. , Jensen , D.F. and Lübeck , M. 2011 . Identification of expressed genes during infection of Chinese cabbage (Brassica rapa subsp. pekinensis) by Plasmodiophora brassicae . J. Eukaryot. Microbiol. , 58 : 310 – 314 .
- Velculescu , V.E. , Zhang , L. , Vogelstein , B. and Kinzler , K.W. 1995 . Serial analysis of gene expression . Science , 270 : 484 – 487 .
- Wallenhammar , A.C. 1996 . Prevalence of Plasmodiophora brassicae in a spring oilseed rape growing area in central Sweden and factors influencing soil infestation levels . Plant Pathol. , 45 : 710 – 719 .