
Abstract
The Stewart’s wilt pathogen (Pantoea stewartii subsp. stewartii; Pss) of sweet corn is classified as a quarantine bacterium, requiring certification of grain shipments made to over 60 countries. This study reports the development and validation of the first multi-gene array for accurate and specific differentiation of Pss from Pantoea stewartii subsp. indologenes (Psi) and other Pantoea species using DNA, or RNA after generation of cDNA. The technique consisted of multiplex (16S rRNA, leuS, gyrB, rpoB, cpsD) PCR amplifications using universal and specific primers in a single reaction, followed by the hybridization of the digoxigenin-labelled amplicons to 22 specific oligonucleotide probes (19- to 24-mers) immobilized on a nylon membrane. The sensitivity of the array (10 fg of DNA) compared favourably with TaqMan real-time PCR. The specificity and reliability of the array was tested on 65 bacterial strains consisting of Pantoea species, closely and distantly related bacterial genera. Specific detection of Pss in infected corn leaves and seed homogenates was also validated using growth chamber-inoculated plants. The reported multi-gene DNA array could be a reliable tool for routine and specific detection of Pss.
Résumé
L’agent pathogène (Pantoea stewartii subsp. stewartii; PSS) de flétrissement de Stewart du maïs sucré est une bactérie de quarantaine, qui exigent la certification des grains avant l’expédition vers plus de 60 pays. Cette étude indique le développement et la validation de la première macropuce multi-gènique pour la détection et la différenciation spécifique de Pss de sous-espèce Pantoea stewartii subsp. indologenes (Psi) et d’autres espèces de Pantoea à l’aide de l’ADN, ou l’ARN après génération d’ADNc. La technique consiste l’amplification par PCR en multiplex (16S ARNr, leuS, gyrB, rpoB, cpsD) à l’aide des amorces universel et spécifiques dans une seule réaction, suivie de l’hybridation des sondes marquées avec digoxigénine sur 22 (19- à 24-mers) oligonucléotides spécifiques immobilisée sur une membrane de nylon. La sensibilité de la macropuce (10 fg d’ADN) se compare favorablement avec TaqMan PCR en temps réel. La spécificité et la fiabilité de la macropuce ont été evaluées avec 65 souches bactériennes composées des espèces de Pantoea et d’autres genres bactériens. La détection spécifique de Pss dans les feuilles de maïs infectés et d’homogénats de semences a également été validée à l’aide de plantes inoculées en chambre de croissance. Cette macropuce multi-gènique d’ADN pourrait être un outil fiable pour la détection courante et spécifique de Pss.
Introduction
Rapid and accurate identification/detection of pathogens responsible for specific plant diseases are prerequisites for selection of reliable management strategies to mitigate economic losses to growers. This is particularly challenging for bacterial plant pathogens with subspecies-level taxonomic classification, such as Pantoea stewartii subsp. stewartii (Smith 1898) Mergaert et al. (Citation1993), comb. nov. [Pss; syn. Erwinia stewartii (Mergaert et al. Citation1993)], the causal agent of the Stewart’s vascular wilt of corn. Stewart’s wilt is a serious bacterial disease of sweet corn in Canada and the USA (Stewart Citation1897; Tambong et al. Citation2008), but has not established in Europe according to the European and Mediterranean Plant Protection Organization (EPPO). Pss overwinters in mature corn flea beetles (Chaetocnema pulicaria F. E. Melsheimer, 1847), which spread the pathogen. Also, the pathogen can be distributed by seed transmission at very low frequencies (Block et al. Citation1999; Michener et al. Citation2002; Cook et al. Citation2005). Several countries have phytosanitary restrictions for seedborne Pss on corn seed and thus, require shipments to be certified Pss-free (Tambong et al. Citation2008; Wensing et al. Citation2010). A certification method should be able to differentiate Pss from its closely related subspecies, P. stewartii subsp. indologenes (Psi), which can be challenging using currently available molecular approaches. Errors in identification/detection of a quarantine plant pathogen such as Pss can have serious economic and/or political implications. Traditional techniques for detection/identification and certification of corn seeds, e.g. field inspection of plants to be harvested as seeds and enzyme-linked immunosorbent assay (ELISA) (Lamka et al. Citation1991), are time-consuming and relatively insensitive, especially on grains under long-term storage. This is a concern especially for countries or regions where the pathogen has not yet been reported.
Several PCR-based detection techniques, such as PCR-coupled ligase chain reaction (Wilson et al. Citation1994), nested primers (Blakemore et al. Citation1999), ‘miniprimer’ PCR (Xu et al. Citation2010), specific primers derived from hrpS, cpsDE and 16S rRNA intergenic (Coplin et al. Citation2002) or pstS-glmS region (Wensing et al. Citation2010), as well as TaqMan real-time PCR (Tambong et al. Citation2008; Wensing et al. Citation2010) have been developed. These assays, however, cannot be used alone to reliably differentiate the subspecies of P. stewartii (Tambong et al. Citation2008; Wensing et al. Citation2010). A ‘miniprimer’ PCR assay developed by our group (Xu et al. Citation2010) and a stepdown conventional PCR assay (Gehring et al. Citation2014) are reported to differentiate Pss from Psi in pure cultures but their effectiveness in accurate detection of the pathogen in infected corn leaves or seeds are yet to be demonstrated.
Membrane-based DNA array technology has been employed to detect and identify various microorganisms (Tambong et al. Citation2006; Tung et al. Citation2007; Ko et al. Citation2008; Lin et al. Citation2010; Chen et al. Citation2013), and has proved to be a sensitive, accurate and high-throughput tool (Tambong et al. Citation2006; Lin et al. Citation2010; Chen et al. Citation2013) with enormous multiplexing possibilities compared with other PCR-based assays. So far, all membrane-based arrays designed for the detection and identification of bacteria used only a single gene or DNA fragment; and as such, are prone to non-specific reactions, especially when environmental samples are used given the high number of different bacterial populations present. In addition, none of the reported bacterial arrays could be used for the detection of DNA as well as RNA; or for subspecies-level differentiation of plant pathogenic bacteria.
The objectives of this study were to sequence and analyse 16S rRNA, a pathogenicity-related cpsD (wceI) and three bacterial house-keeping genes (rpoB, leuS and gyrB) for the development of a multigene oligonucleotide array for rapid differentiation of Pss from Psi and other Pantoea species in pure cultures and plant samples. leuS encodes leucyl-tRNA synthetase involved in translation, rpoB encodes for the β subunit of RNA polymerase, and gyrB is the structural gene for DNA gyrase β subunit. The array was also used to detect the RNA of the target genes after cDNA generation. This is, probably, the first report of an array that is demonstrated to detect DNA as well as RNA of a phytobacterial pathogen.
Materials and methods
Bacterial strains and DNA extraction
A total of 65 Pantoea and non-Pantoea bacterial strains including all publicly available type strains of the genus Pantoea were used in this study (). Type strains of the genus Pantoea were obtained from the Belgian Coordinated Collections (BCCM/LMG; Ghent, Belgium). Pantoea and the majority of non-Pantoea bacterial strains were cultured in Luria–Bertani (LB: 10 g L−1 tryptone, 5 g L−1 yeast extract, 10 g L−1 NaCl) or nutrient broth (NB: 5 g L−1 peptone, 1 g L−1 meat extract, 5 g L−1 NaCl, pH 7.0 ± 0.2) and incubated at 28°C. Purified DNA, pure cultures and seeded homogenates from P. stewartii DOAB 21 (Tambong et al. Citation2008), isolated from infected sweet corn plants in Canada, were used to optimize the DNA array hybridization. The same strain was used for growth chamber inoculations. Strains of Pectobacterium spp. were cultured in tryptone yeast extract liquid media (Schaad et al. Citation1999) and Clavibacter michiganensis ssp. sepedonicus in nutrient broth supplemented with yeast extract (Smid et al. Citation1995). Stock bacterial cultures were maintained on the same medium supplemented with 25% w/v glycerol at −80°C. Genomic DNA of the bacterial strains was purified using Wizard SV Genomic DNA purification system Kit (Promega) following the manufacturer’s instructions.
PCR amplification and DNA sequencing of multiple genes
Partial sequences of 16S rRNA, cpsD (pathogenicity-related) and three core protein-coding genes (leuS, rpoB and gyrB) were generated for all the required strains not available in public databases. The primer pairs used in this study are listed in Table S1. Primers were synthesized by Invitrogen Inc. (Carlsbad, CA, USA). PCR amplifications were performed in a thermal cycler (Biometra) using 10 ng of bacterial DNA (1 µL), 1 µL of 10× PCR buffer, 0.75 µL of 2 mM deoxynucleoside triphosphates (dNTPs), 0.08 µL of each primer (20 µM), 0.1 µL of Titanium Taq DNA polymerase (5 U µL−1; BD/Biosciences/Clontech), and 6.99 mL of MilliQ PCR water (Fermentas). Sequencing was done as previously described (Tambong et al. Citation2006) using ABI BigDye Terminator chemistry v3.1 (Applied Biosystems) and run on an ABI 3130xl automated sequencer (Applied Biosytems/Hitachi). Sequences were edited in SEQMAN (DNASTAR).
Phylogenetic analysis using maximum likelihood
The gyrB, leuS and rpoB sequences were imported into MEGA5 (Tamura et al. Citation2011) for final editing, taking into account corresponding amino acids and checked for correct in-frame reading using RevTrans version1.4 (Wernersson & Pedersen Citation2003). gyrB-leuS-rpoB concatenated Sequences were aligned using a Linux version of MUSCLE (Edgar Citation2004). MEGA5 (Tamura et al. Citation2011) was used to compute Maximum likelihood (ML) phylogeny using the general time reversible (GTR) substitution model, selected on the basis of the Akaike information criterion implemented in jMODELTEST version 2.1.1 (Darriba et al. Citation2012) with 1000 bootstrap replications. ML was executed with the SPR and NNI tree improvement algorithms. Bootstrap values ≥70% were indicated on the phylogenetic tree. 16S rRNA and cps were not included in the analysis since these genes are highly conserved (Tambong et al. Citation2014).
Design and spotting of oligonucleotides
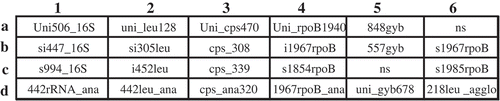
Twenty-two oligonucleotides (19- to 24-mers) () with unique polymorphisms (Tambong et al. Citation2006; Lin et al. Citation2010) were designed from multiple genes (16S rRNA, leuS, rpoB, gyrB and cpsD) for specific detection/differentiation of subspecies of Pantoea stewartii. The designed oligonucleotide were evaluated in silico for specificity using Basic Local Alignment Sequence Tool (BLAST, Altschul et al. Citation1990). Oligo Calculator version 3.26 (http://www.basic.northwestern.edu/biotools/OligoCalc.html) was used to determine the change in Gibbs free energy (dG) and melting temperature (Tm) based on the nearest neighbour kinetics for optimal hybridization. The selected oligonucleotides included 8 Pantoea stewartii (s994_16S, si305leu, cps_308, cps_339, 848gyb, 557gyb, uni_gyb678, s1967rpoB and s1985rpoB)-, 1 subspecies stewartii (s1854rpoB)-, 2 subspecies indologenes- (i452leu and i1967rpoB), 8 group (442rRNA_ana, uni_leu128, 442leu_ana, Uni_cps470, 1967rpoB_ana, uni_gyb678, s1985rpoB and 218leu_agglo)-, and 1 Pantoea genus (Uni_rpoB1940) – specific oligonucleotides. A 16S rRNA bacteria-specific oligonucleotide (Uni506_16S) was designed and spotted to serve as positive control and for normalization of hybridization signals. The oligonucleotides () were synthesized by Invitrogen (Canada) with a C6 5ʹ amino group, which acts as a covalent linker to the membrane. The layout of the oligonucleotides on the array is shown in . Amino-terminated oligonucleotides were diluted to 40 µM in sodium bicarbonate buffer (0.4 M, pH 8.0) in a sterile 384-well microplate and spotted with a VP 384S 1µL-slot pins multi-Blot Replicator (V&P Scientific Inc., San Diego, CA) in double rows of 6 on strips (6 by 8 cm) of Immunodyne ABC membranes (PALL Europe Limited, Portsmouth, England). Quadruplicates of the same oligonucleotides were spotted by using the 180° offset printing guide hole on the library copier (V&P Scientific Inc.). The spotted membranes were air dried for 10 min and transferred into blocking solution (2× SSC [1× SSC: 0.15 M NaCl plus 0.015 M sodium citrate], pH 7.0, amended with 5 g L−1 casein (BDH Biochemical, Poole, UK) and 0.05% (v/v) Tween 20) with agitation for 15 min. Membranes were washed in 2× SSC for 30 min and maintained in 2× SSC for short-term storage.
Table 1. Putative housekeeping genes, location on the array, code and oligonucleotide sequences evaluated in this study.
Fig. 1 Layout of multilocus oligonucleotides spotted on array. Uni506_16S was a positive control used to normalize hybridization intensity; NS, not spotted, negative control. 1, 16S rRNA; 2, leuS, 3, cpsD; 4, rpoB; 5, gyrB; 6, rpoB (b6 & c6) and leuS (d6). The corresponding sequences of all oligonucleotides are listed in .
Preparation of multiplex digoxigenin-labelled amplicons by PCR and hybridization
Gradient PCR was performed to determine optimal conditions for multiplex PCR amplifications of five distinct genes (16S rRNA, leuS, rpoB, gyrB and cpsD) in a single reaction. Simultaneous multiplex PCR amplifications and digoxigenin labelling were done in 40 µL reaction mixtures containing Titanium DNA Taq polymerase (4U; Clontech, Mountain View, CA), 10 ng of template DNA, digoxigenin-labelled dUTP, and other standard reaction substrates as reported previously (Tambong et al. Citation2006). Multiplex PCR amplifications were carried out in a Biometra thermocycler (Goettingen, Germany) with initial denaturation at 95°C for 3 min, followed by 40 cycles of 95°C for 45 s, 65°C for 45 s, and 72°C for 90 s and a final extension at 72°C for 8 min.
Hybridizations were done at 54°C as previously described (Tambong et al. Citation2006). Digoxigenin was detected by chemiluminescence following the protocol from the manufacturer by using anti-digoxigenin–alkaline phosphatase conjugate and the substrate CDP-Star (Roche Diagnostics GmbH, Laval, PQ). Replicate hybridizations were done to confirm the results and the specificity of the oligonucleotides. Chemiluminograms were scanned in colour at 600 ppi with HP Scanjet 5590 scanner and saved as .tiff file. ImageJ software (Schneider et al. Citation2012) was used to compute the hybridization signal (in grey values) minus the signal of the surrounding background for each spot. Mean hybridization signals of the quadruplicate spots of each oligonucleotide were calculated. Hybridization signal intensities, expressed in grey values, were normalized by calculating the ratio of the mean signal of each oligonucleotide to that of the Uni506_16S (positive control). Normalized ratios <0.05 (5%) were considered as background signals based on distribution analysis (data not shown) that predicted a cut-off value corresponding to a ratio of 0.1 (10% of the signal of Uni506_16S; positive control). Signals with normalized values of ≤0.1 were considered background signals because the spots were not visible without computer enhancement.
Sensitivity and specificity of the array on pure bacterial cultures
Sensitivity is defined as the minimum amount of DNA required to produce detectable hybridization signals (normalized signals of greater than 0.1) for all oligonucleotides required for reliable identification/detection of P. stewartii subsp. stewartii. Purified genomic DNA from Pss DOAB 21 was serially diluted 10-fold from 10 ng to 1 fg in autoclaved milliQ water and subjected to multiplex PCR amplifications as indicated above. All reactions were performed in triplicate.
Specificity of the assay was evaluated on genomic DNA of 65 strains including P. stewartii (23) and closely (24) and distantly (18) related bacterial species. The DNA of each strain was diluted to 10 ng and multiplex PCR and array hybridizations were performed as indicated above. Each strain was run in duplicate and repeated once.
Direct detection of Pss in growth chamber-inoculated and naturally infected corn samples
Sweet corn seeds ‘Earlivee’ were sown in commercial potting mix (1:1:1 soil: vermiculate: peat moss) and grown at 28°C and 16 h daylight in a growth chamber. Plants were inoculated as previously reported (Tambong et al. Citation2008). Briefly, the lowest leaf of 14-day-old plants was cut close to the whorl region and inoculated by infusion of 10 µL of a suspension (106 CFU) of P. stewartii strain DOAB 21. Two weeks after inoculation, 40 mg of material from leaves with disease symptoms or healthy controls were sliced and placed into a 1.5-mL tube containing 200 µL of autoclaved milliQ water and vortexed. The suspension was microcentrifuged briefly to remove debris and l µL subjected to digoxigenin-labelling multiplex PCR followed by hybridization on the array. Corn seeds harvested from plants inoculated with P. stewartii strain DOAB 21 were homogenized as previously described (Tambong et al. Citation2008). Total DNA was extracted using the MO BIO Soil Kit (Carlsbad, CA) and processed for hybridization as indicated above. Dilution plating on LB medium coupled with sequencing was used to verify the presence of P. stewartii in infected corn parts. Average bacterial counts in leaf tissue and corn homogenates were 107 and 105 cfu mL−1.
RNA extraction and cDNA macroarray analysis for detection of viable Pss and Psi
Total RNAs were extracted from 2 strains of Pss (DOAB 049 and DOAB 050) and 2 Psi strains (LMG 2632 and LMG 2630) using NucleoSpin RNAII Kit (Clontech, USA) following the manufacturer’s instructions. The extracted RNA was eluted from the column with 60 µL of RNase-free H2O after centrifugation at 11 000 g for 1 min. DNA-free kit (Life Technologies, USA) was then used to remove any genomic DNA contamination according to the manufacturer’s instructions. The quality, integrity and concentration of the extracted RNA were determined spectrophotometrically using Bioanalyzer (Agilent Technologies, USA). The cDNAs were synthesized using the EcoDry Premix Oligo dT (Clontech CA, USA) according to the manufacturer’s instructions. To enrich the synthesis of cDNAs of genes being targeted (gyrB, leuS and rpoB) and to obtain long sizes of cDNA fragments, 0.95 µM of the respective forward primer (Table S1) was added into reactions containing 300 ng of total RNA.
To check the expression of the 3 targeted genes (data not shown), real-time PCR assays were performed using SYBR Advantage qPCR Premix kit (Clontech, USA) in a total reaction volume of 10 µL, containing 5 µL of SYBR premix, 0.1 µL of each 20 µM primer pairs (gyrB3/B4 for gyrB gene, leuS3/S4 for leuS gene and rpoBjt112/jt748 for rpoB gene), 1 µL of 1/10th dilution of cDNA, and 3.8 µL of PCR H2O. The Real-time RT-PCR conditions were 95°C for 30 s, followed by 94°C, 5 s, 62°C, 30 s, and 72°C, 1.5 min for 40 cycles. A melting curve analysis was performed from 70°C to 95°C with 1°C increments and held for 30 s in a Chromo 4 real-time PCR machine and monitored by Opticon3.1 software as reported previously (Tambong et al. Citation2008).
To obtain DIG-labelled hybridization probes, cDNA DIG-PCR was performed in a 15-µL volume containing 1.5 µL 10× Titanium buffer (Clontech, USA), 0.5 µL of 2 mM dNTP (including 0.7 mM Digoxigenin-11-dUTP, Roche, Canada), 0.19 µL of each primer (gyrB3/B4, leuS3/S4 and rpoBjt112/jt748), 1.5 µL of 1/10th diluted cDNA, 0.15 µL of Titanium Taq DNA polymerase and 11.16 µL of PCR grade H2O. PCR was carried out in a Tprofessional PCR machine (Biometra, Germany) with the following cycling conditions: 95°C for 30 s, 94°C, 5 s, 62°C, 30 s, 72°C, 90 s for 36 cycles with a final extension at 72°C for 5 min. Two microlitres of the cDNA DIG-PCR amplicons were used to hybridize on the array as indicated above.
Sequence accession numbers
The majority of sequences used in this study are published in Tambong et al. (Citation2014) with GenBank numbers KF482571–KF482607, KF482608–KF482644 and KF482719–KF482755 for gyrB, leuS and rpoB, respectively. Twenty-one additional sequences of 7 P. stewartii subsp. indologenes strains were generated and deposited in GenBank with accession numbers KT863449–KT863455, KT863456–KT863462 and KT863463–KT863469 for the respective genes.
Results
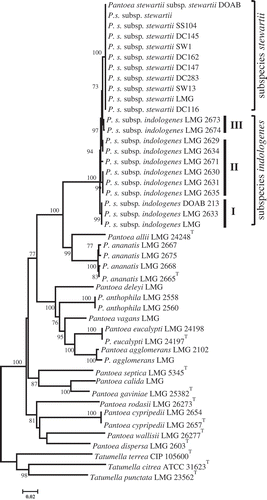
Pantoea phylogeny based on leuS-gyrB-rpoB concatenated gene
The expected phylogenetic associations were observed on the tree derived from concatenated gene sequences (). The tree inferred with concatenated sequences confirmed the phylogenetic affiliations of the core Pantoea subgroups (P. stewartii, P. ananatis, P. agglomerans and P. dispersa). Based on leuS-gyrB-rpoB concatenated sequences, Pantoea phylogeny showed that Pss strains are homogeneous while Psi strains are heterogeneous based on the alleles studied (). The Psi subcluster containing the type strain, LMG 2632, regrouped two strains that are genotypically similar. Another Psi subcluster contained strains LMG 2630, LMG 2631 and LMG 2635, while all the other Psi strains clustered distinctly either in pairs or as singletons. Phylogenetic discrimination of subspecies of P. stewartii using the selected genes suggests that these loci could be useful in the development of reliable molecular diagnostic tools.
Fig. 2 Maximum likelihood inferred phylogenetic tree of Pantoea species from gyrB-leuS-rpoB concatenated sequences. Pantoea stewartii subsp. stewartii strains seem to be homogeneous while those of P. stewartii subsp. indologenes are heterogeneous. General time reversible substitution model was used with 1000 bootstrap replicates. Tatumella spp. used as outgroup.
Verification of designed oligonucleotides by BLAST
The specificity of the 22 selected oligonucleotides was evaluated by BLAST to all publicly available GenBank entries. Figure S1 summarizes the BLAST results consisting of 3300 GenBank hits. The oligonucleotide Uni506_16S, spotted to serve as a positive control, had a total hit of 1790 entries consisting of Pantoea, Pectobacterium, Erwinia and Tatumella genera. Oligonucleotide si447_16S exhibited a 100% coverage and identity with 34 Pss GenBank entries. A universal oligonucleotide Uni_1940rpoB, derived from rpoB gene, showed a 100% identity with 1230 Pantoea, Pectobacteria, Erwinia and Dikeya GenBank entries. Two gyrB-derived (557gyb, 848gyb) and leuS-derived (si305leu) oligonucleotides exhibited a perfect match with, respectively, 11 and 6 P. stewartii GenBank entries. The oligonucleotide s1854rpoB showed a perfect match with 8 P. stewartii subsp. stewartii entries while i425leu exhibited a 100% identity to only a GenBank entry of Psi LMG 2632. A BLAST of the oligonucleotide i1967rpoB had a single hit with 100% sequence identity to Psi LMG 2630.
Development of a membrane-based array and validation for specific detection of Pss
Twenty-two specific oligonucleotides, 19 to 24 bases long, were designed from 16S rRNA, 3 core housekeeping genes (leuS, rpoB and gyrB) and a pathogenicity-related gene, cpsD (wceI). Four oligonucleotides (Uni506_16S, si447_16S, s994_16S and 442rRNA_ana) were designed from the 16S rRNA. Oligonucleotide Uni506_16S (; location a1) was designed as a universal bacteria target and used as a positive control for normalization of hybridization signals. Oligonucleotide s994_16S was spotted to detect all the subspecies of P. stewartii while 442leu_ana () was designed for specific detection of the P. ananatis subgroup (P. ananatis and P. allii). Two oligonucleotides (848gyb and 557gyb) (; locations b4 and c4) were designed from gyrB gene sequences to specifically target strains of P. stewartii. At least one oligonucleotide was designed for each subspecies of P. stewartii. Oligonucleotide i452leu (location c2; ) was spotted for specific detection of Psi subcluster I as shown in while oligonucleotide s1854rpoB (, location c4) was spotted to detect only strains of Pss. An oligonucleotide (i1967rpoB; ; location b4) was spotted to detect strains of Psi subcluster II (). Oligonucleotide 442leu_ana targeted the P. ananatis subgroup (P. ananatis and P. allii). Also, a leuS-derived oligonucleotide (218leuS_agglo; ) was spotted to target strains of P. agglomerans.
Of the 4 oligonucleotides designed from the cps gene, Uni_cps470 was spotted to detect P. stewartii and P. ananatis subgroups, 2 (cps_308 and cps_339; ) targeted only P. stewartii strains while cps_ana320 was designed to target strains of P. ananatis. Two blank spots, at random locations (a6 and c5), serve as background negative controls.
Validation of array for specific detection of Pss
Hybridization patterns of amplicons of 65 Pantoea and non-Pantoea bacterial strains were highly reproducible (). Hybridization signals of the positive control oligonucleotide (Un506_16S) were used for data normalization for each membrane. The normalized hybridization signals of the oligonucleotides ranged from 0 to 1.2 (Fig. S2). The oligonucleotide Uni506_16S exhibited detectable hybridization signals with the digoxigenin-labelled amplicons of all the strains of Pantoea species (Fig. 7). All 11 Pss strains exhibited consistent and positive detection signals (ratios between 0.7–1.2) with 14 out of the 22 oligonucleotides spotted (). The 14 oligonucleotides are derived from all the 5 genes used. This underscores a multigene detection system for reliable identification of Pss. The oligonucleotide s1854rpoB exhibited positive and detectable hybridization signals with all the strains of Pss but not with those of Psi (, ). shows a representative chemiluminogram of the hybridization signals of Pss strains. One oligonucleotide, i452leu, reliably and consistently differentiated the subcluster I strains of Psi (LMG 2632T, DOAB 213 and LMG 2633; ) from all the other Psi (subcluster II; ) and Pss strains (). The 6 Psi strains of subcluster II () had specific and consistent hybridization signals with oligonucleotide i1967rpoB (, ). Psi strains of subcluster III (LMG 2673 and LMG 2674; ) did not show detectable hybridization signals with either i425leu or i1967rpoB. Reliable detection and differentiation of Psi strains of subcluster III were achieved with positive hybridization signals of 13 oligonucleotides (Fig. 7). This is consistent with the phylogenetic association of these strains.
Fig. 3 Summary of hybridization patterns of multiplex digoxigenin-labelled PCR amplicons of Pantoea stewartii and other bacterial strains to an array of subspecies-, species- and group-specific oligonucleotides on nylon membranes. Chemiluminograms were scanned at 600 dpi using HP ScanJet 5590 scanner, and grey scale values of each dark spot, computed with ImageJ software and normalized using oligonucleotide Uni506_16S. Normalized signals are indicated by the following symbols: □, <0.1 (not detected); ▪, 0.15–0.39; ◘, 0.40–0.79; ■, >0.80. The locations of the oligonucleotides given at the top correspond to their locations on the membranes in the array. Oligonucleotides are presented in . The type strains were obtained from BCCM/LMG bacterial collection, Belgium or the Collection of Plant associated Bacteria (CFBP), France. Strains with prefixes SW or DC were kindly provided by Dr David Coplin.
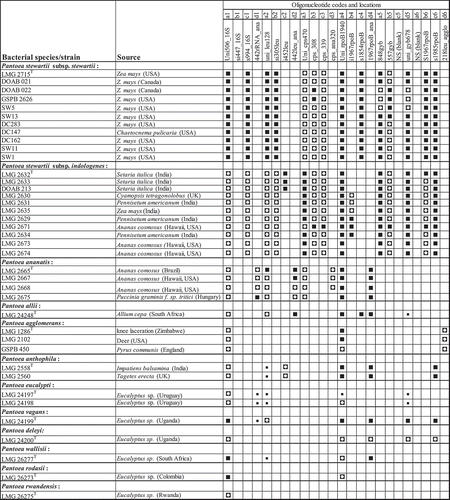
Fig. 3 (Continued.)
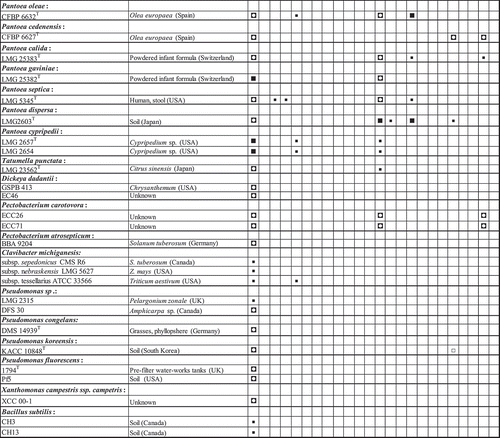
Closely related species such as P. ananatis LMG 2665T (, ), P. agglomerans LMG 1286T (, ) and P. allii LMG 24248T (, ) exhibited only 9, 3 and 6 positive reactions, respectively. One of the positive hybridization signals of P. allii is a cross-reaction with the oligonucleotide s1854rpoB spotted to detect Pss. Oligonucleotide 442leu_ana, designed to detect P. ananatis and P. allii, gave positive signals with all the strains of these two species () while oligonucleotide 218leu_agglo was specific for detection of P. agglomerans strains (). Only 3 or 1 positive reactions were observed for, respectively, Pectobacterium caratovora ECC26 (, ) or Dickeya dadantii GSPB 413, former members of the Erwinia group. While most of the designed oligonucleotides showed positive reactions, the oligonucleotide si447_16S, designed to detect all P. stewartii, did not show detectable positive reaction with any of the targeted strains.
Hybridizations were performed with PCR amplicons derived from serial dilutions of genomic DNA of P. stewartii DOAB 21 (data not shown) to determine the detection limit of the array, that is, the least amount of DNA at which all the expected 14 oligonucleotides exhibited detectable hybridization signals for reliable detection/identification of Pss strains. The array exhibited consistent positive hybridization signals with multiple genes down to the 10-fg level of DNA.
Potential use of the oligonucleotide array to detect Pss in infected corn plant tissues
Direct processing of Pss-inoculated leaf samples followed by hybridization of the digoxigenin-labelled PCR amplicons on the array consistently reproduced typical hybridization patterns of Pss but not with samples from non-inoculated control plants (Fig. 7). Non-inoculated plants exhibited a positive hybridization signal with only the bacterial-specific oligonucleotide, Uni506_16S (), indicating that the phyllosphere of the corn leaves harbour other micro-organisms. As expected, all the 14 oligonucleotides required for the reliable detection/identification of Pss exhibited positive hybridization reactions (). In addition, Pss was detected in seed homogenates derived from corn plants inoculated with the bacteria but not seeds derived from healthy controls and non-inoculated plants (data not shown). The detection of Pss on inoculated corn plants was validated by TaqMan real-time PCR and isolation and DNA sequencing where applicable.
Fig. 4 Hybridization profiles of 18 Pantoea species and subspecies, Pectobacterium and Dickeya (former Erwinia members) and Tatumella punctata. Note specificity of detection of Pantoea stewartii subsp. stewartii LMG 2715T and differentiation from Pantoea stewartii subsp. indologenes based on oligonucleotide s1854rpoB (location c4).
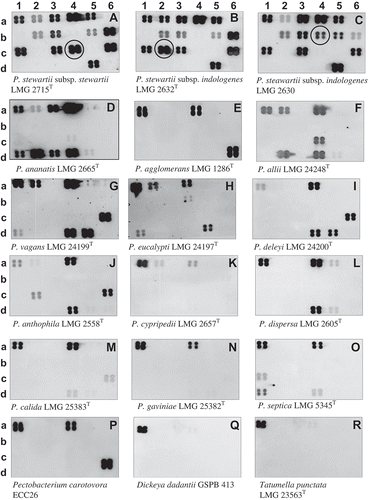
Fig. 5 Chemiluminograms showing specific detection of Pantoea stewartii subsp. stewartii DOAB 021 on corn plants infected or not under growth chamber conditions. a, non-infected plant, and b, infected plant. Leaf lesions (40 mg) were processed as described in Tambong et al. (Citation2008). No DNA extraction was performed. Four plants per treatments were analysed with consistent hybridization pattern obtained. Row numbers represent the different genes used: 1, 16S rRNA; 2, leuS; 3, cps; 4, rpoB; 5, gyrB; 6, mix: rpoB (b6 & c6) and leuS (d6) spotted on the membrane-based array.
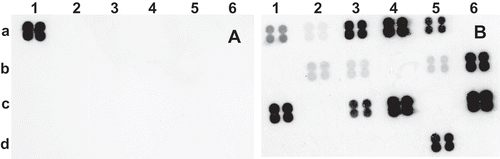
Fig. 6 Alignment of rpoB-derived oligonucleotide (s1854rpoB) sequences showing nucleotide mismatches and specificity to P. stewartii subsp. stewartii compared with other strains of Pantoea and Tatumella. Note nucleotide mismatches of oligonucleotide s1854rpoB are almost at the centre for effective differentiation.

Pss RNA detection by the array
The cDNAs generated were enriched and digoxigenin-labelled and hybridized to the same array for detection of 16S rDNA, rpoB, gyrB and leuS. The cps gene was not targeted and as such served as a control for specificity. Hybridization of the labelled cDNAs on the array gave the expected reactions with 12 oligonucleotides derived from the 4 genes (Fig. S3). The cps gene that was not enriched or targeted as well as the spotted negative controls gave no detectable hybridization signal, thus confirming the specificity of the array.
Discussion
This study describes the development and validation of a multi-gene membrane-based DNA array for reliable subspecies-level identification and differentiation of P. stewartii subsp. stewartii. This is the first oligonucleotide array with demonstrated reliability in differentiating subspecies of P. stewartii in pure cultures and infected plant parts using DNA or RNA after cDNA generation. Subspecies-level identification of P. stewartii strains is important since only Pss causes Stewart’s wilt of corn (Tambong et al. Citation2008; Gehring et al. Citation2014) and the presence of Psi in samples could result in false positives with significant economic impact to corn producers.
The Stewart’s wilt pathogen of corn is endemic to the Americas and more than 60 countries have phytosanitary restrictions for seedborne Pss on corn seed, requiring shipments to be Pss-free (Tambong et al. Citation2008). ELISA-based assays are largely used for commercial certification but due to their high detection limits, some infective populations might be missed (Tambong et al. Citation2008; Wensing et al. Citation2010). Several PCR-based detection techniques have been developed (Wilson et al. Citation1994; Blakemore et al. Citation1999; Tambong et al. Citation2008; Wensing et al. Citation2010). However, none of the previous assays could reliably differentiate subspecies of P. stewartii (Tambong et al. Citation2008; Wensing et al. Citation2010). We previously developed a ‘miniprimer’ PCR assay that is based on banding patterns to differentiate both subspecies in pure cultures (Xu et al. Citation2010). Also, Gehring et al. (Citation2014) reported a stepdown PCR assay based on single nucleotide polymorphisms (SNPs) in galE and recA for subspecies differentiation. Both assays reliably detected Pss and Psi in pure cultures but there are no data reported for their use in Pss detection in infected corn leaves or seeds. In addition, previous methods have not been reported to detect RNA of the target pathogen after cDNA generation.
Membrane-based DNA array technology has been employed in the detection and identification of various microorganisms (Tambong et al. Citation2006; Tung et al. Citation2007; Ko et al. Citation2008; Lin et al. Citation2010; Chen et al. Citation2013), and has proven to be a sensitive, accurate and high-throughput tool (Tambong et al. Citation2006; Lin et al. Citation2010; Chen et al. Citation2013) with enormous multiplexing possibilities compared with other PCR-based assays. Most membrane-based arrays reported to date for bacterial detection/identification are based on either a single genomic region or gene, e.g. 16S-23S rRNA intergenic spacer (Ko et al. Citation2008; Lin et al. Citation2010), or gyrB (Fukushima et al. Citation2003). The oligonucleotide array developed in our study used 3 discriminating housekeeping genes (leuS, rpoB, gyrB), a pathogenicity-related gene (cpsD) and 16S rRNA. This is, perhaps, the first multi-gene membrane-based array developed for detection/identification of plant pathogenic bacteria of agricultural significance and for differentiation among related subspecies. The use of multiple target regions is reported to increase the reliability and confidence in accurate detection and identification of Phytophthora species, a group of Oomycetes (Chen et al. Citation2013). DNA-based arrays could be powerful forensic tools but possess some inherent biological limitations of detecting dead cells in environmental samples. Given that the reported array could be used for detection of RNA after cDNA production, it could be instrumental in confirming, in cases of positive DNA detection, the viability of Pss being detected. None of the previous reported arrays and other molecular assays have been demonstrated to detect RNAs of the target pathogens.
The differentiation of Pss from Psi strains is based on the specificity of the oligonucleotide s1854rpoB that exhibits two critical nucleotide mismatches, almost at the centre of the oligonucleotide (). This is consistent with previous work (Kawasaki & Chehab Citation1994) that the discriminatory specificity is greatest when mismatches are in the centre of the oligonucleotide. However, the base composition of the entire immobilized fragment could also greatly affect its hybridizing potential and stability (Tambong et al. Citation2006). While the oligonucleotide s1854rpoB was designed to detect only Pss strains, it exhibited cross-reaction with P. allii. This single cross-reactivity should not affect the specificity of the array in accurate identification of Pss strains. The complete hybridization pattern of P. allii (7 positive reactions) is significantly different from that of Pss (14 positive reactions).
The reasons why one oligonucleotide (si447_16S; location b1) did not show detectable hybridization signals with any subspecies/species are unknown. This could be partly attributed to the base composition of the oligonucleotides and stringent hybridization conditions. Perhaps the oligonucleotide si447_16S might be situated within strong secondary structures that do not open up under the hybridization conditions used. Lievens et al. (Citation2005) and Tambong et al. (Citation2006) reported similar results when amplicons generated from DNA of Verticillium nubilum or Pythium spp. did not hybridize to any of the oligonucleotides designed for Verticillium or Pythium detection. Better prediction models for hybridizations that include potential secondary structures of the template should reduce this problem.
Another advantage of the current array is the high sensitivity. The assay showed a detection limit of 10 fg of DNA which compares favourably with that of real-time PCR (1000 fg) reported previously (Panicker & Bej Citation2005; Tambong et al. Citation2008). The high sensitivity of the array could be related to the use of digoxigenin in probe labelling. Because of the high sensitivity of the array, it could have application in routine certification of corn seed shipments as well as in forensic investigations.
In conclusion, the results reported here show that the oligonucleotide array designed and tested could be useful for the identification and detection of Pss in pure cultures and infected corn leaves and seeds, as well as reliably differentiating Pss from Psi. This array could become a tool for rapid detection of the pathogen in field samples and for reliable certification of corn shipments destined for export. Given that this array can be used to detect or identify several species in a single reaction mixture, specific oligonucleotides of all the different Pantoea species could be designed, spotted and validated for use as a routine diagnostic tool in medical or plant disease laboratories.
Supplemental Material
Download MS Word (5.8 MB)Acknowledgements
This research was funded by Agriculture and Agri-food Canada through projects # 1800 and #1136. I am thankful to R. Xu and R. Assabgui for technical assistance with multi-locus DNA sequencing, and Dr D. Coplin for kindly providing most of the Pss strains. Thanks to M. Nejjari and all summer students who worked on this project.
Supplemental material
Supplemental data for this article can be accessed here: http://dx.doi.org/10.1080/07060661.2015.1113442
References
- Altschul SF, Gish W, Miller W, Myers EW, Lipman DJ. 1990. Basic local alignment search tool. J Mol Biol. 215:403–410.
- Blakemore EJA, Law JR, Reeves JC. 1999. PCR identification of Erwinia stewartii and its comparison with two other methods. Seed Sci Technol. 27:385–396.
- Block CC, Hill JH, McGee DC. 1999. Relationship between late-season severity of Stewart’s bacterial wilt and seed infection in corn. Plant Dis. 83:527–530.
- Chen W, Djama ZR, Coffey MD, Martin FN, Bilodeau GJ, Radmer L, Denton G, Levesque CA. 2013. Membrane-based oligonucleotide array developed from multiple markers for the detection of many Phytophthora species. Phytopathology. 103:43–54.
- Cook KA, Weinzier RA, Pataky JK, Esker PD, Nutter JFW. 2005. Population densities of corn flea beetle (Coleoptera: Chrysomelidae) and incidence of Stewart’s wilt in sweet corn. J Econ Entomol. 98:673–682.
- Coplin DL, Majerczak DR, Zhang Y, Kim W-S, Jock S, Geider K. 2002. Identification of Pantoea stewartii subsp. stewartii by PCR and strain differentiation by PFGE. Plant Dis. 86:304–311.
- Darriba D, Taboada GL, Doallo R, Posada D. 2012. jmodeltest 2: more models, new heuristics and parallel computing. Nat Methods. 9:772.
- Edgar RC. 2004. Muscle: a multiple sequence alignment method with reduced time and space complexity. BMC Bioinformatics. 5:113–115.
- Fukushima M, Kakinuma K, Hayashi H, Nagai H, Ito K, Kawaguchi R. 2003. Detection and identification of Mycobacterium species isolates by DNA microarray. J Clin Microbiol. 41:2605–2615.
- Gehring I, Wensing A, Gernold M, Wiedemann W, Coplin DL, Geider K. 2014. Molecular differentiation of Pantoea stewartii subsp. indologenes from subspecies stewartii and identification of new isolates from corn seeds. J Appl Microbiol. 116:1553–1562.
- Kawasaki ES, Chehab FF. 1994. Analysis of gene sequences by hybridization of PCR-amplified DNA to covalently bound oligonucleotide probes. The reverse dot blot method. Meth Mol Biol. 28:225–236.
- Ko W-C, Lee N-Y, Su SC, Dijkshoorn L, Vaneechoutte M, Wang L-R, Yan -J-J, Chang TC. 2008. Oligonucleotide array-based identification of species in the Acinetobacter calcoaceticus-a Baumannii complex isolated from blood cultures and antimicrobial susceptibility testing of the isolates. J Clin Microbiol. 46:2052–2059.
- Lamka GL, Hill JH, McGee DC, Braun EJ. 1991. Development of an immunosorbent assay for seedborne Erwinia stewartii in corn seeds. Phytopathology. 81:839–846.
- Lievens B, Brouwer M, Vanachter AC, Levesque CA, Cammue BP, Thomma BP. 2005. Quantitative assessment of phytopathogenic fungi in various substrates using a DNA macroarray. Environ Microbiol. 7:1698–1710.
- Lin YT, Vaneechoutte M, Huang AH, Teng LJ, Chen HM, Su SL, Chang TC. 2010. Identification of clinically important anaerobic bacteria by an oligonucleotide array. J Clin Microbiol. 48:1283–1290.
- Mergaert J, Verdonck L, Kersters K. 1993. Transfer of Erwinia ananas (synonym Erwinia uredovora) and Erwinia stewartii to the genus Pantoea emend. as Pantoea ananas (Serrano 1928) comb. nov. and Pantoea stewartii (Smith 1898) comb. nov., respectively, and description of Pantoea stewartii subsp. indologenes subsp. nov. Intern J Syst Bacteriol. 43:162–173.
- Michener PM, Pataky JK, White DG. 2002. Transmission of Erwinia stewartii from plants to kernels and reactions of corn hybrids to Stewart’s wilt. Plant Dis. 86:167–172.
- Panicker G, Bej AK. 2005. Real-time PCR detection of Vibrio vulnificus in oysters: comparison of oligonucleotide primers and probes targeting vvha. Appl Environ Microbiol. 71:5702–5709.
- Schaad NW, Berthier SY, Sechler A, Knorr D. 1999. Detection of Clavibacter michiganensis subsp. sepedonicus in potato tubers by BIO-PCR and an automated real-time fluorescence detection system. Plant Dis. 83:1095–1100.
- Schneider CA, Rasband WS, Eliceiri KW. 2012. NIH image to ImageJ: 25 years of image analysis. Nat Methods. 9:671–675.
- Smid EJ, Hausen AHJ, Gorris LGM. 1995. Detection of Erwinia carotovora subsp. atroseptica and Erwinia chrysanthemi in potato tubers using polymerase chain reaction. Plant Pathol. 44:1058–1069.
- Stewart FC. 1897. A bacterial disease of sweet corn. New York Agric Exp Stn Bullettin. 130:422–439.
- Tambong JT, de Cock AW, Tinker NA, Levesque CA. 2006. Oligonucleotide array for identification and detection of Pythium species. Appl Environ Microbiol. 72:2691–2706.
- Tambong JT, Mwange KN, Bergeron M, Ding T, Mandy F, Reid LM, Zhu X. 2008. Rapid detection and identification of the bacterium Pantoea stewartii in corn by Taqman real-time PCR assay targeting the cpsD gene. J Appl Microbiol. 104:1525–1537.
- Tambong JT, Xu R, Kaneza C-A, Nshogozabahizi J-C. 2014. An in-depth analysis of a multilocus phylogeny identifies leuS as a reliable phylogenetic marker for the genus Pantoea. Evol Bioinformatics. 10:115–125.
- Tamura K, Peterson D, Peterson N, Stecher G, Nei M, Kumar S. 2011. MEGA5: molecular evolutionary genetics analysis using maximum likelihood, evolutionary distance, and maximum parsimony methods. Mol Biol Evol. 28:2731–2739.
- Tung SK, Teng LJ, Vaneechoutte M, Chen HM, Chang TC. 2007. Identification of species of Abiotrophia, Enterococcus, Granulicatella and Streptococcus by sequence analysis of the ribosomal 16S-23S intergenic spacer region. J Med Microbiol. 56:504–513.
- Wensing A, Zimmermann S, Geider K. 2010. Identification of the corn pathogen Pantoea stewartii by mass spectrometry of whole-cell extracts and its detection with novel pcr primers. Appl Environ Microbiol. 76:6248–6256.
- Wernersson R, Pedersen AG. 2003. RevTrans: multiple alignment of coding DNA from aligned amino acid sequences. Nucl Acids Res. 31:3537–3539.
- Wilson WJ, Wiedmann M, Dillard HR, Batt CA. 1994. Identification of Erwinia stewartii by a ligase chain reaction assay. Appl Environ Microbiol. 60:278–284.
- Xu R, Chen Q, Djama ZR, Tambong JT. 2010. Miniprimer PCR assay targeting multiple genes: a new rapid and reliable tool for genotyping Pantoea stewartii subsp. stewartii. Lett Appl Microbiol. 50:216–222.