
Abstract
The complete genome comprising three genomic RNAs of three Canadian and two Chinese isolates of Potato mop-top virus were sequenced and analysed. Two open reading frames (ORFs) were found in RNA1 of 6.1 kb, encoding a readthrough RNA-dependent RNA polymerase (RdRp). A coat protein (CP)-readthrough protein was encoded by RNA2 of 3.1 kb. Four ORFs that encoded the triple gene block proteins (TGBps) and a cysteine-rich protein were found in RNA3 (2.9 kb) of the Chinese isolate ‘Yunnan’; whereas in the remaining isolates (three Canadian isolates and the Chinese isolate ‘Guangdong’), only three ORFs encoding TGBps were observed in RNA3. A single nucleotide mutation of A2462 to G2462 abolished the start codon ‘AUG’ for the fourth putative ORF in RNA3 of these isolates. Based on phylogenetic and sequence similarity analysis of these isolates as well as those reported by others at the complete RNA sequence level, each of RNA1, RNA2 and RNA3 could be divided into at least two groups. In Canadian isolates ‘Ch9’, ‘Ch10’ and ‘Ch20’ and Chinese isolate ‘Guangdong’, all genomic RNAs belonged to group A; and in Chinese isolate ‘Yunnan’, all of its RNA belonged to group B. Interestingly, in Swedish isolate ‘Sw’, RNA1 and RNA2 belonged to group A while RNA3 belonged to group B. A duplex RT-PCR for differentiating groups A and B of RNA3 was developed and evaluated. All PMTV samples collected in Guangdong, China, and New Brunswick, Canada, possessed a RNA3 belonging to group A; whereas the samples collected in Yunnan, China, possessed a RNA3 belonging to group B.
Résumé
Trois ARN génomiques de trois isolats canadiens et de deux isolats chinois, issus du génome complet du virus du sommet touffu de la pomme de terre (VSTPT), ont été séquencés et analysés. Deux cadres de lecture ouverts (ORF) ont été trouvés dans l’ARN-1 de 6.1 kb, codant par translecture un ARN-polymérase sous la dépendance d’ARN (RdRp). L’ARN-2 de 3.1 kb codait pour une protéine capsidiaire (CP) et pour une protéine de translecture. Quatre ORF qui codaient les protéines du bloc de trois gènes (TGBp) ainsi qu’une protéine riche en cystéine ont été trouvés dans l’ARN-3 de l’isolat chinois « Yunnan », tandis que, chez les autres isolats (les trois isolats canadiens et l’isolat chinois « Guangdong »), nous n’avons observé que trois ORF codant les TGBp dans l’ARN-3. La mutation d’un seul nucléotide, de A2462 à G2462, a annulé le codon initiateur « AUG » du quatrième ORF putatif dans l’ARN-3 de ces isolats. En se basant sur les résultats des analyses phylogénétiques et de similarité de séquences, ainsi que sur ceux rapportés par d’autres quant à la séquence complète de l’ARN, chaque ARN-1, ARN-2 et ARN-3 pourrait être divisé en au moins deux groupes. Chez les isolats canadiens « Ch9 », « Ch10 » et « Ch20 » ainsi que chez l’isolat chinois « Guangdong », tous les ARN génomiques appartenaient au groupe A. Par ailleurs, chez l’isolat chinois « Yunnan », tous les ARN appartenaient au groupe B. Chose intéressante, chez l’isolat suédois « Sw », l’ARN-1 et l’ARN-2 appartenaient au groupe A, tandis que l’ARN-3 appartenait au groupe B. Une technique de RT-PCR duplex a été mise au point et évaluée pour différencier les groupes A et B de l’ARN-3. Dans tous les échantillons de VSTPT collectés au Guangdong, en Chine, et au Nouveau-Brunswick, au Canada, l’ARN-3 appartenait au groupe A, tandis que l’ARN-3 des échantillons collectés au Yunnan, en Chine, appartenait au groupe B.
Introduction
Potato mop-top virus (PMTV) is one of several viruses that are capable of inducing tuber necrosis, potentially leading to significant economic loss of the infected potato crops. The disease caused by PMTV is called spraing disease, a condition in which necrotic arcs and lines develop in infected tubers of sensitive potato cultivars (Sandgren et al. Citation2002). PMTV can also induce a range of foliar symptoms, such as stunting, mottling, chevrons and yellow blotches/rings, depending on potato cultivars and environmental conditions (Salazar Citation1996; Carnegie et al. Citation2010). PMTV is transmitted by Spongospora subterranea f. sp. subterranea (Sss), the causal agent of potato powdery scab disease. The resting spores in spore-balls of Sss remain infective in soil for years, and when released from the spore-balls, the zoospores can infect root hairs, stolons and developing tubers and thus introduce PMTV to the plant cells (Jones & Harrison Citation1969; Arif et al. Citation1994; Kirk Citation2008; Santala et al. Citation2010).
PMTV is the type species of the genus Pomovirus, family Virgaviridae, possessing three single-stranded positive-sense RNAs encapsidated in tubular particles (Torrance & Mayo Citation1997; Pringle Citation1999). All of the RNAs have a tRNA-like secondary structure at the 3ʹ-proximal end (Savenkov et al. Citation1999, Citation2003). The largest RNA, RNA1, encodes a viral RNA-dependent RNA polymerase (RdRp)-readthrough protein; the two smaller RNAs, which have been interchangeably termed RNA2 and RNA3 (Torrance et al. Citation1999; Sandgren et al. Citation2001; Latvala-Kilby et al. Citation2009), encode a coat protein (CP)-readthrough protein and the triple gene block proteins (TGBps), respectively (Savenkov et al. Citation1999).
Occurring commonly in northern Europe and the Andean regions in South America (Sandgren Citation1995; Salazar Citation1996; Nielsen & Nicolaisen Citation2003; Carnegie et al. Citation2010, Citation2012; Santala et al. Citation2010; Beuch et al. Citation2015), PMTV has recently been found in an increasing number of countries in South and North America (Lambert et al. Citation2003; Xu et al. Citation2004; Montero-Astúa et al. Citation2008; David et al. Citation2010; Crosslin Citation2011; Whitworth & Crosslin Citation2013; Ramesh et al. Citation2014; Beuch et al. Citation2015), Central Europe (Čeřovská et al. Citation2007; Budziszewska et al. Citation2010) and Asia (Nakayama et al. Citation2010; Arif et al. Citation2013; Hu et al. Citation2013). However, whether or not the occurrence of PMTV in these areas/countries is due to a recent introduction is unknown. Sequence analysis, especially analysis at the complete genome level, should provide new insights into the relationship among isolates from different countries/areas, and possibly shed light on the possible origins of the PMTV isolates.
In this study, the complete genome of two isolates of PMTV from China and three isolates from Canada were sequenced and analysed. The results demonstrate that genome reassortment might have occurred in PMTV; and together with single nucleotide mutation and sequence deletion, they contributed to PMTV evolution and diversification.
Materials and methods
Virus samples and symptom identification
Two sources of PMTV were identified in China, one in Guangdong province as described previously (Hu et al. Citation2013) and the other from Yunnan province. The potato tubers exhibited characteristic PMTV-like spraing symptoms and tested PMTV-positive by ELISA and RT-PCR. One tuber each from Guangdong (cultivar ‘Favorita’) and Yunnan (cultivar ‘Lishu 6’) provinces, designated isolates ‘Guangdong’ and ‘Yunnan’, respectively, were processed for sequencing of the complete genome at the Hunan Agricultural University, Changsha, Hunan province, China. In Canada, tubers of cultivar ‘Chieftain’ exhibiting external necrotic cracks and internal brown rings and arcs () were collected from a potato field in New Brunswick in 2012. ELISA and RT-PCR screening for common viruses as described in Hu et al. (Citation2013) and Crosslin and Hamlin (Citation2011) confirmed that PMTV was the causal agent for the spraing diseases in these tubers (data not shown). Three samples, designated isolates ‘Ch9’, ‘Ch10’ and ‘Ch20’ were processed for sequencing at Agriculture and Agri-Food Canada’s Fredericton Research and Development Centre.
Fig. 1 (Colour online) External and internal necrosis in potatoes ‘Chieftain’ collected in a field in New Brunswick, Canada. Photos were taken 3 months after harvesting.

Reverse transcription-polymerase chain reaction
Total RNA was isolated from PMTV-infected tubers as described in Nie & Singh (Citation2001). RNA concentration and quality was analysed using a Nanodrop 2000c Spectrophotometer (Thermo Fisher Scientific, Waltham, MA). Reverse transcription reactions were performed using 1 µg of total RNA. Complementary DNA was synthesized using SuperScript III™ (Life Technologies, Grand Island, NY) following the manufacturer’s protocol. Briefly, 1 µg of total RNA was mixed with 2 pmoles of specific reverse primer (Supplementary Table 1), and 10 mM dNTP mix and incubated for 5 min at 65°C. Samples were placed on ice, briefly centrifuged (8000 rpm) and a master mix of First Strand Buffer, 0.1 M DTT, 40 Units of RNaseOUT™, and 200 Units of SuperScript III were added. The samples were incubated for 50 min at 50°C and the reaction was terminated by incubating samples at 70°C for 15 min.
For the Canadian isolates, PCR was performed using Q5® High-Fidelity DNA polymerase (New England Biolabs, Canada) on a Bio-Rad C1000 thermal cycler (Bio-Rad Laboratories, Hercules, CA). In the 25-µL PCR mix, it contained 1 × Q5 reaction buffer, 10 mM dNTPs, 0.5 µM forward and reverse primers (Supplementary Table 1), 0.02 units of Q5® High-Fidelity DNA polymerase, and 1 µl cDNA. The PCR was performed using the following programme: 98°C for 30 s; 30 cycles of 98°C for 10 s, 53°C for 20 s and 72°C for 20 s/kb; an additional 2 min elongation step was used to ensure complete amplicon synthesis. Rapid amplification of cDNA ends (RACE) was used to clone the 5ʹ and 3ʹ ends for the RNAs of isolates ‘Ch9’, ‘Ch10’ and ‘Ch20’, following the protocol outlined in the RACE System for Rapid Amplification of cDNA Ends, version 2.0 (Life Technologies). For amplification of the 5ʹ end, reverse transcription for each of the three PMTV RNAs (i.e. RNA1, RNA2 and RNA3) was performed using gene specific primer 1 (GSP1) (Supplementary Table 1). Upon purification with a S.N.A.P.™ spin column (Invitrogen, Carlsbad, CA), the cDNA was tailed with dC using terminal deoxynucleotidyl transferase (Life Technologies). A PCR with GSP2 (Supplementary Table 1) and the anchored primer (provided with kit), followed by a nested PCR with 5ʹ RACE nested primer (Supplementary Table 1) and the abridged universal amplification primer (AUAP, provided with kit), was performed. For cloning the 3ʹ ends, the PMTV RNAs were first polyadenylated using E. coli poly(A) polymerase (Life Technologies) according to the manufacturer’s instructions. Complementary DNA was synthesized using an oligo-dT-AUAP primer (provided with kit). PCR with a GSP (Supplementary Table 1) and the AUAP was carried out, which was followed by nested PCR with 3ʹ RACE nested primer (Supplementary Table 1) and AUAP.
For the Chinese isolates, PCR was performed using TransTaq® DNA High Fidelity Polymerase (Transgen Biotech, Beijing, China) on a PTC-0200 DNA Engine thermal cycler (MJ Research, Waltham, MA). In the 25-µL PCR mix, it contained 1 × PCR buffer (10 mM Tris-HCl, pH 8.3, 50 mM KCl), 1.5 mM MgCl2, 0.2 mM of each dNTP, 50 ng of each forward and reverse primers, 0.625 U TransTaq® DNA High Fidelity Polymerase, and 2 µL cDNA. Samples were amplified for 30 cycles of 94°C for 30 s, 58°C for 30 s and 72°C for 60 s. A final extension of 10 min at 72°C was also included.
Cloning and sequencing
To obtain the complete sequences of RNA1, RNA2 and RNA3, primers spanning the entire genome of PMTV were designed using the Swedish isolate ‘Sw’ (RNA1, AG238607, RNA2, AJ243719 and RNA3, AG277556) as a reference. For the two Chinese isolates, 13 sets of primers (Supplementary Table 2), each resulting in an amplicon of ~0.5–1.3 kb, were used. The adjacent fragments contained an overlap of approximately 100 bp. For the three Canadian isolates, in addition to the above mentioned sequencing strategy but with different primers (Supplementary Table 1), 5ʹ and 3ʹ RACEs were used to obtain the 5ʹ- and 3ʹ-proximal ends of each of the three genomic RNAs.
PCR products were analysed by electrophoresis on a 1.5% low-melt agarose gel to ensure single fragment amplification. If multiple bands were detected, the amplicon of the expected size was extracted using QIAquick Gel Extraction Kit (Qiagen, Burlington, ON) as per manufacturer’s directions. For the Chinese isolates, purified fragments were cloned into a pMD™19-T cloning vector (Takara Biotechnology, Dalian, China) according to the manufacturer’s instructions. Two cDNA clones of each fragment were sent to Sangon Biological Engineering Technology & Services Co. Ltd (Shanghai, China) for forward and reverse sequencing using the T7 promoter and SP6 promoter primers, respectively. The contig was confirmed in an independent experiment by re-cloning and sequencing the overlapping fragments covering the complete genome. For the Canadian isolates, the PCR fragments were cloned into Zero Blunt® cloning vector (Life Technologies) using manufacturer’s protocol. Two clones of each fragment were sent to Robarts Research Institute (London, ON) for sequencing with the M13 forward and reverse primers. Sequence results were assembled and aligned using Vector NTI®Express Software (Life Technologies).
Sequence analysis
Sequence similarity was performed using National Center for Biotechnology Information’s BLAST (http://www.ncbi.nlm.nih.gov/BLAST). Multiple sequence alignments were performed with MUSCLE implemented in MEGA5 (Tamura et al. Citation2011) and conserved regions were determined using the Gblocks program (Talavera & Castresana Citation2007). Phylogenetic analyses were carried out with the Bayesian inference (BI) in MrBayes v3.22 (Ronquist et al. Citation2012). Before BI analyses, the best-fit substitution model for RNA1, RNA2 and RNA3 was HKY + I, HKY + G and HKY + I, respectively determined by MrModelTest (Nylander Citation2008) according to the Akaike’s information criterion (AIC). For BI analyses, four chains were processed with the generation set as 500 000 000 generations using the selected model for each dataset, Markov chains were sampled every 100 generations, and 25% of the converged runs were regarded as burn-in. Chain stationary and run parameter convergence were assessed by the average standard deviation of split frequencies (<0.01) and further checked using Tracer version 1.6 (Rambaut et al. Citation2014). For analysis of possible recombination events, complete nucleotide sequences of various PMTV isolates were aligned using ClustalW2 (Larkin et al. Citation2007). The aligned sequences served as inputs for similarity scanning using the program SimPlot (Lole et al. Citation1999) and RDP3 (Martin et al. Citation2010). The resulting similarities were plotted along the nucleotide sequences of the virus genome.
Results
General description of genome structure of PMTV isolates
Using primers derived from isolate ‘Sw’, the complete sequence of genomic RNA1, RNA2 and RNA3 sized at 6042 bp, 3134 bp and 2964 bp, respectively, were obtained in both Chinese isolates ‘Guangdong’ and ‘Yunnan’ (). The sizes of the RNA2 and RNA3 were identical to those of ‘Sw’; but the RNA1 in both ‘Guangdong’ and ‘Yunnan’ was one nucleotide (nt) shorter than that of ‘Sw’ due to a possible nt deletion (nt 90 of RNA1-Sw) in the 5ʹ untranslated region (5ʹ-UTR). Similar to that reported by Savenkov et al. (Citation1999), a tRNA-like secondary structure was predicted at the 3ʹ-proxymal ends for all of the three RNAs. Using 5ʹ- and 3ʹ-RACE in combination with primers derived from ‘Sw’, the genomic RNA sequences sized at 6005, 6042 and 6005 bp (RNA1); 3102, 3134 and 3134 (RNA2); and 2937, 2933 and 2934 (RNA3) for Canadian isolates ‘Ch9’, ‘Ch10’ and ‘Ch20’, respectively, were obtained (). Alignment of each of the genomic RNAs from the newly sequenced isolates revealed a shortage of 38 nt at the 5ʹ proximal ends of RNA1 for isolates ‘Ch9’ and ‘Ch20’ and one possible nt deletion (nt 90 of RNA1-‘Sw’) in the 5ʹ-UTR of RNA1 in all three Canadian isolates; shortages of 12 nt at 5ʹ end and 20 nt at 3ʹ end of RNA2 for isolate ‘Ch9’; and shortages of 27, 31 and 30 nt at 3ʹ end of RNA3 for ‘Ch9’, ‘Ch10’ and ‘Ch20’, respectively. The apparent missing of nucleotide occurring at the proximal ends of the RNAs did not affect the analysis of the RNAs, especially the open reading frames embedded in the RNAs. It is noteworthy that, like isolate ‘Sw’ as well as ‘Guangdong’ and ‘Yunnan’, the RNAs in each of ‘Ch9’, ‘Ch10’ and ‘Ch20’ shared a high sequence identity at the 3ʹUTR and a tRNA-like secondary structure (data not shown).
Table 1. Summary of genomic RNA sequences of Potato mop-top virus (PMTV) isolates in Canada and China as well as other PMTV isolates available in databases.
Analysis of RNA1
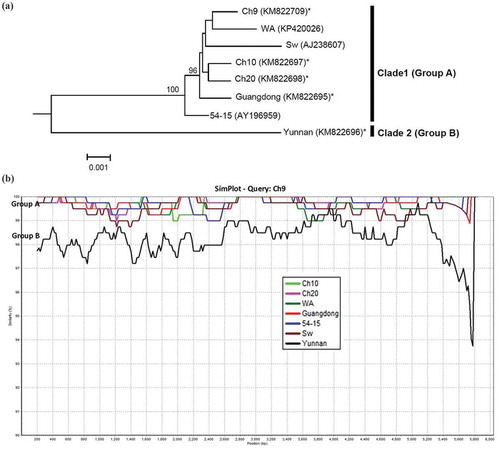
Two ORFs embedded in RNA1 were found in all isolates from China and Canada, similar to those retrieved from databases. The ORFs in the RNA led to a putative readthrough-RdRp (Savenkov et al. Citation1999) sized at 1812 aa (). Analysis of sequence similarities between different isolates in Canada and China revealed a high sequence identity at 98.9–99.7% at the amino acid level and 98.4–99.9% at the nucleotide level for the RdRp (), suggesting that the gene and gene product are highly conserved. Despite the limited sequence variation of RdRp among the isolates from Canada and China, ‘Yunnan’ appeared to be the most distant isolate, scoring the lowest sequence identities with others at both amino acid and nucleotide levels (). Phylogenetic analysis of the complete sequences of RNA1 of different isolates, including those available from GenBank (i.e. isolates ‘Sw’, ‘WA’ and ‘54–15’), placed ‘Yunnan’ as Clade 2 (Group B) whereas all other isolates were clustered to Clade 1 (Group A) (). Scanning the complete sequences of RNA1 for sequence similarities against isolate ‘Guangdong’ using software SimPlot (Lole et al. Citation1999) indicated that ‘Yunnan’ scored the lowest along the sequence (), consistent with the above observation.
Table 2. Nucleotide (shaded) and amino acid (non-shaded) sequence identities between different isolates of Potato mop-top virus in China and Canada.
Fig. 2 (Colour online) (a) Bayesian phylogenetic tree of the genomic RNA1 of Potato mop-top virus (PMTV) isolates. Phylogenetic analyses were carried out with the Bayesian inference (BI) in MrBayes v3.22. For each node, the Bayesian posterior probabilities are given above branches (only shown >50%) and isolate grouping based on genetic similarity and nodes with posterior probabilities greater than 70% are indicated. The nucleotide sequence accession number of each isolate is shown in parentheses. The newly sequenced isolates are indicated by ‘*’. The distance unit is substitutions/site. (b) Analysis of the sequence identities between ‘Guangdong’-RNA 1 (query sequence) and RNA1 of other isolates (reference sequences) using SimPlot. The similarities (y-axis, 0.9–1) were plotted along the nucleotide sequence (x-axis, nt 1–6000). The window covered 500 nucleotides and moved along the alignment with 20 nucleotides with every step. Other parameter settings were: distance model, Kimura (2-parameter); tree model, neighbour-joining; bootstrap replicates, 100; parental threshold, 70. Nucleotide sequence accession numbers for isolates are the same as shown in (a).
Analysis of RNA2
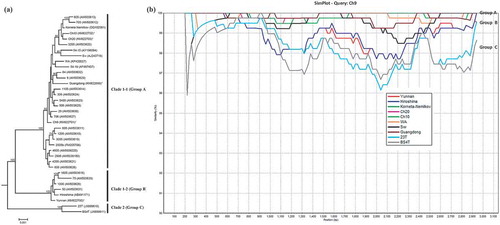
Two ORFs were found in RNA2 of the newly sequenced isolates. Similar to that in RNA1, the ORFs in RNA2 led to a putative readthrough protein termed coat protein (CP)-RT (Kashiwazaki et al. Citation1995; Sandgren et al. Citation2001) and sized at 825 aa (). Like RdRp, CP-RT of the newly sequenced isolates appeared to be rather conserved, with sequence identities at 99.0–100% and 98.7–99.9% at the amino acid and the nucleotide levels, respectively (). Phylogenetic analysis of the full and near-full length RNA2 sequences, including the newly sequenced isolates and those retrieved from GenBank, placed two Colombia isolates (‘23T’ and ‘BS4T’, accession numbers JX889610 and JX889611) as a branch (Clade 2) (Group C) whereas the rest as another branch (). The latter branch was further divided into two clades: Clade 1-1 (Group A) included all Canadian isolates, the Chinese isolate ‘Guangdong’ and many more; and Clade 1-2 (Group B) included the Chinese isolate ‘Yunnan’, Japanese isolate ‘Hiroshima’ as well as four Finnish isolates (e.g. ‘50’, ‘75’, ‘1006’ and ‘1605’). This grouping is consistent with the subgroups identified by Nielsen and Nicolaisen (Citation2003), Ramesh et al. (Citation2014) and Beuch et al. (Citation2015). Scanning the nucleotide sequences of representative isolates from the above identified branches for sequence identities against ‘Guangdong’ was in agreement with the phylogenetic tree: ‘23T’ and ‘BS4T’ being the most ‘distant’ group (Group C), followed by ‘Yunnan’ and ‘Hiroshima’ group (Group B), then by the rest (Group A) (). Previously, two partial length-CP sequences (HQ171916, HQ171917) of two Columbia isolates have been shown to be considerably ‘distant’ from other isolates around the world, with a sequence identity at ~77% (Gil et al. Citation2011; Hu et al. Citation2013). It is likely that a more distinct RNA2 of PMTV will be found in Colombia and countries in the Andean region where the potato originated.
Fig. 3 (Colour online) (a) Bayesian phylogenetic tree of the genomic RNA2 of Potato mop-top virus (PMTV) isolates. Phylogenetic analyses were carried out with the Bayesian inference (BI) in MrBayes v3.22. For each node, the Bayesian posterior probabilities are given above branches (only shown >50%) and isolate grouping based on genetic similarity and nodes with posterior probabilities greater than 70% are indicated. The nucleotide sequence accession number of each isolate is shown in parentheses. The newly sequenced isolates are indicated by ‘*’. The distance unit is substitutions/site. (b) Analysis of the sequence identities between ‘Guangdong’-RNA2 (query sequence) and RNA2 of other isolates (reference sequences) using SimPlot. The similarities (y-axis, 0.9–1) were plotted along the nucleotide sequence (x-axis, 0–3,100). The window covered 500 nucleotides and moved along the alignment with 20 nucleotides with every step. Other parameter settings were: distance model, Kimura (2-parameter); tree model, neighbour-joining; bootstrap replicates, 100; parental threshold, 70. Nucleotide sequence accession numbers are the same as shown in (a).
Analysis of RNA3
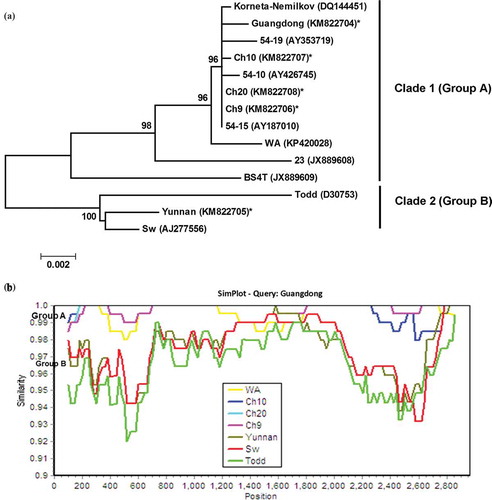
Three overlapping ORFs encoding the triple gene block proteins sized at 463, 119 and 190 aa, respectively, were found in RNA3 of the Canadian isolates as well as the Chinese isolate ‘Guangdong’; whereas four ORFs, encoding the TGBps 1–3 and a cysteine-rich protein (68 aa in length), respectively, were only found in the Chinese isolate ‘Yunnan’ (). Interestingly, RNA3 in isolates ‘Sw’ and ‘Todd’ has four ORFs (Scott et al. Citation1994; Savenkov et al. Citation2003) whereas in isolates ‘54–15’ and ‘Korneta-Nemilkov’, there are only 3 ORFs (Pečenková et al. Citation2004; Čeřovská et al. Citation2007). A switch from A2462 in ‘Yunnan’, ‘Sw’ and ‘Todd’ to G2462 in ‘Ch9’, ‘Ch10’, ‘Ch20’, ‘Guangdong’, ‘Korneta-Nemilkov’, ‘54–15’, ‘54–10’, ‘54–19’ and ‘WA’ abolished the start codon ‘AUG’ for the fourth ORF in RNA3.
Sequence similarity analysis among the isolates revealed slightly higher sequence variations for TGBps over RdRp and CP. For TGBp1, the sequence identities were at 98.3–100% at amino acid level and 98.1–100% at the nucleotide level (). Isolate ‘Yunnan’ showed the lowest identities for TGBp1 with other isolates. For TGBp2, the identities were at 96.6–100% and 98.0–100% at the amino acid and nucleotide levels, respectively. The lowest identities were found between isolate ‘Yunnan’ and the three Canadian isolates as well as the Chinese isolate ‘Guangdong’ (). For TGBp3, the identities were 95.8–100% and 94.7–100% at the amino acid and nucleotide levels, respectively. Again, the Chinese isolate ‘Yunnan’ exhibited the lowest identities for TGBp3 with other isolates ().
Phylogenetic analysis of available full-length (including the near-full length) sequences of RNA3 placed isolates ‘Ch9’, ‘Ch10’, ‘Ch20’, ‘Guangdong’, ‘54–10’, ‘54–15’, ‘54–19’, ‘Korneta-Nemilkov’, ‘WA’, ‘BS4T’ and ‘23T’ as Clade 1 (Group A), ‘Yunnan’, ‘Sw’ and ‘Todd’ as Clade 2 (Group B) (). ‘BS4T’ and ‘23T’ appeared to be the most ‘distant’ isolates in Clade I. Scanning the nucleotide sequences of representative isolates from the above identified branches for sequence identities against ‘Guangdong’ using software SimPlot (Lole et al. Citation1999) was in agreement with the phylogenetic tree.
Fig. 4 (Colour online) (a) Bayesian phylogenetic tree of the genomic RNA3 of Potato mop-top virus (PMTV) isolates. Phylogenetic analyses were carried out with the Bayesian inference (BI) in MrBayes v3.22. For each node, the Bayesian posterior probabilities are given above branches (only shown >50%) and isolate grouping based on genetic similarity and nodes with posterior probabilities greater than 70% are indicated. The nucleotide sequence accession number of each isolate is shown in parentheses. The newly sequenced isolates are indicated by ‘*’. The distance unit is substitutions/site. (b) Analysis of the sequence identities between RNA3 of ‘Guangdong’ (query sequence) and RNA3 of other isolates (reference sequences) using SimPlot. The similarities (y-axis, 0.9–1) were plotted along the nucleotide sequence (x-axis, 0–2900). The window covered 500 nucleotides and moved along the alignment with 20 nucleotides with every step. Other parameter settings were: distance model, Kimura (2-parameter); tree model, neighbour-joining; bootstrap replicates, 100; parental threshold, 70. Nucleotide sequence accession numbers for isolates are the same as shown in (a).
Development of RT-PCR for differentiation of RNA3 belonging to different sequence groups
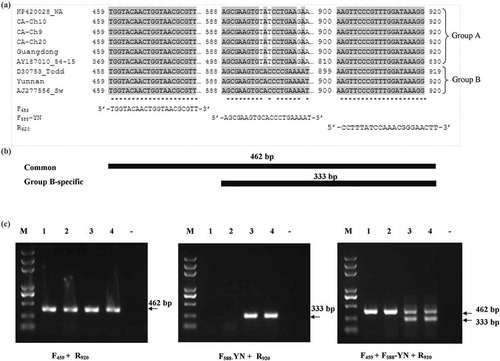
To effectively differentiate isolates possessing RNA3 belonging to groups A and B, a common reverse primer (R920, 5ʹ-CCTTTATCCAAACGGGAACTT-3ʹ), a common sense primer (F459, 5ʹ-TGGTACAACTGGTAACGCGTT-3ʹ) and a group B-specific primer (F588-YN, 5ʹ-AGCGAAGTGCACCCTGAAAAT-3ʹ) were designed after aligning RNA3 sequences (). As anticipated, simplex RT-PCR with F459 and R920 produced a 462-bp fragment in both isolates ‘Guangdong’ and ‘Yunnan’ (, left panel, lanes 1 and 4) whereas the simplex RT-PCR with F588-YN and R920 led to an amplicon of 333 bp only in isolate ‘Yunnan’ (, central panel, lanes 3 and 4). In duplex RT-PCR containing all the above primers, the common and the group B-specific fragments were amplified as projected: 462-bp fragment in both isolates ‘Guangdong’ and ‘Yunnan’ (, right panel, lanes 1–4) and 333-bp fragment only in isolate ‘Yunnan’ (, right panel, lanes 3–4). Application of the duplex RT-PCR to 14 field samples collected in Guangdong, four samples in Yunnan and 17 samples in New Brunswick unveiled that all samples in Guangdong and New Brunswick possessed a group A-RNA3 whereas all the samples from Yunnan contained a group B-RNA3.
Fig. 5 RT-PCR differentiation of RNA3 belonging to different phylogenetic groups in Potato mop-top virus (PMTV). (a) Alignment of sequences of RNA3 of PMTV for designing primers for detection and differentiation of RNA3 belonging to phylogenetic Group A (KP420028_Wa, CA-Ch10, CA-Ch9, CA-Ch20, Guangdong, AY187010_54–15) and Group B (D30753_Todd, Yunnan and AJ277556_Sw). Primers F459 and R920 are group-non-specific whereas primer F588-YN is group-B specific. (b) Schematic diagram of PCR amplification products for differentiation of PMTV-RNA3 belonging to phylogenetic Groups A and B. Target segments and resulting PCR products are illustrated. (c) RT-PCR assay results for differentiating RNA3 phylogenic groups. Left panel, simplex RT-PCR with primers F459 and R920; central panel, simplex RT-PCR with primers F588-YN and R920; right panel, duplex RT-PCR with F459, F588-YN and R920. Lanes 1–2, PMTV isolate ‘Guangdong’; lanes 3–4: PMTV isolate ‘Yunnan’; lane ‘-’, negative control; lane M = DNA ladder.
The GenBank accession numbers for the three genomic RNAs of five PMTV isolates are KM822695–KM822709.
Discussion
The RNA that encodes the CP-readthrough protein was once called ‘RNA3’ as it was the smallest among the three genomic RNAs in PMTV-S and PMTV-T (Kashiwazaki et al. Citation1995; Reavy et al. Citation1998), the very first isolates characterized at the molecular level. Subsequent studies on other isolates such as ‘Sw’ (Savenkov et al. Citation2003) revealed that the CP-readthrough protein encoding RNA is factually the second largest among the genomic RNAs, sized at ~3.1 kb. Consistent with ‘Sw’ as well as most other isolates reported to date, the CP-encoding RNA in the newly sequenced isolates from two continents were the second largest, thus supporting the nomenclature proposed by Sandgren et al. (Citation2001) to term the CP-encoding RNA as RNA2 and the TGBp-encoding RNA as RNA3. This nomenclature is consistent with that in other pomoviruses such as Beet soil-borne virus (BSBV) (Koenig et al. Citation1996) and Broad bean necrosis virus (Lu et al. Citation1998).
The size difference of RNA2 between isolates ‘PMTV-S’/‘PMTV-T’ and other isolates is attributed to deletions in the central part of the reading frame encoding CP-RT (Sandgren et al. Citation2001). Both ‘PMTV-S’ and ‘PMTV-T’ were maintained in the greenhouse in Nicotiana benthamiana or N. debneyi plants through mechanical inoculation (Scott et al. Citation1994; Kashiwazaki et al. Citation1995; Reavy et al. Citation1998; Sandgren et al. Citation2001). Whether this means of transmission might have caused the deletion in RNA2, as shown in RNA2 of soil-borne Wheat mosaic virus under similar circumstances (Chen et al. Citation1994, Citation1995), remains unanswered; nor is it known whether similar deletions occur in field isolates that are transmitted naturally by its fungal vectors. Regardless, the deletion clearly offers an effective means for virus adaptation and diversification under selection pressure.
Our research also reveals other means of diversification/evolution in PMTV, which include single nucleotide mutation and genome reassortment. Due to the lack of proofreading function in viral RdRp, single nucleotide mutations occur commonly in RNA viruses, leading to a cloud of mutant sequences (Roossinck & Schneider Citation2006), a typical feature of a viral quasispecies (Elena et al. Citation2008). Nevertheless, some of the mutants may not be viable and would be eliminated from the infected tissue (Haenni Citation2008). The alteration of A2462 in ‘Yunnan’ and ‘Sw’ to G2462 in ‘Ch9’, ‘Ch10’, ‘Ch20’, ‘Guangdong’, ‘Korneta-Nemilkov’, ‘54–15’, ‘54–10’, ‘54–19’ and ‘WA’, which abolished the start codon ‘AUG’ needed for the fourth ORF in RNA3 in the latter group, not only provided evidence of single nucleotide mutations in PMTV but also suggested that the 68-aa-long cysteine-rich protein is not essential for PMTV survival and potato spraing disease induction. It is necessary to point out that the fourth ORF is absent in most, if not all, Beet soil-borne virus RNA2 (accession numbers Z66493, EF545143, EF545142 and FJ971719) (Koenig et al. Citation1997; Wang et al. Citation2008), but is present in Broad bean necrosis virus (accession number D86638) (Lu et al. Citation1998).
Genome reassortment and RNA recombination have been reported in different plant RNA viruses (Fraile et al. Citation1997; Qiu et al. Citation1998; Sztuba-Solinska et al. Citation2011). Previously, Nielsen and Nicolaisen (Citation2003) reported two RNA2 subgroups within PMTV in Denmark; and Beuch et al. (Citation2015) reported two subgroups in RNA2 and RNA3, respectively, from PMTV isolates collected in Sweden, Denmark and USA, consistent with the phylogeny-based grouping of RNA2 and RNA3 ( and ) and the RT-PCR differentiation of RNA3 sequence variants (). However, no grouping on RNA1 has been reported to date. The evidence of genome reassortment in PMTV comes from the phylogenetic analysis and sequence similarity scanning of the complete RNA sequences among different isolates. As shown in –, for the isolates whose whole genome sequences are available (i.e. ‘Sw’, '54–15’, ‘Guangdong’, ‘Yunnan’, ‘Ch9’, ‘Ch10’ and ‘Ch20’), each of the three genomic RNAs of a particular isolate may have different phylogenetic relationship from that of other isolates. For both RNA1 and RNA2, isolates ‘Guangdong’, ‘Sw’, ‘54–15’, ‘Ch9’, ‘Ch10’ and ‘Ch20’ clustered together, respectively, whereas isolate ‘Yunnan’ stood alone; for RNA3, isolates ‘54–15’, ‘WA’, ‘Ch9’, ‘Ch10’, ‘Ch20’ and ‘Guangdong’ clustered together, ‘Yunnan’ and ‘Sw’ grouped together. This phylogenetic relationship suggests that, in a given isolate, the source of origin for each of the three genomic RNAs might be different (): in isolates ‘Ch9’, ‘Ch10’, ‘Ch20’, ‘Guangdong’, ‘WA’ and ’54–15’, all the RNAs appeared to come from one source (i.e. A); in isolate ‘Yunnan’, the RNAs probably came from another source (i.e. B); whereas in isolates ‘Sw’, RNA1 and RNA2 were from source A, and RNA3 from source B. Taken together, this research demonstrates that genomic RNA reassortment had taken place during the PMTV evolution and diversification process. Although the isolates analysed in this research were from different geographic locations which are far apart, it is likely that the isolates had emerged from a common place, and were then introduced to different locations. Given the fact that a greater molecular diversity was found in PMTV isolates in Colombia (Gil et al. Citation2011; Osorio-Giraldo et al. Citation2013), countries in the Andean region where the potato originated could be the probable source of origin for various PMTV isolates.
Table 3. Genomic RNA type of Potato mop-top virus based on the phylogenetic grouping and sequence similarity scanning.
Supplemental Material
Download MS Word (21.6 KB)Acknowledgements
We thank Jennifer O’Donnell and Angela Gallagher for PCR and ELISA analysis of the potato samples in Canada; Dr Fangluan Gao for phylogenetic analysis. The research in Canada was supported by Agriculture and Agri-Food Canada under the project #1110 (X. Nie) and in China it was supported by the Non-profit Industry Financial Programme of Ministry of Agriculture of China under the project #2012-03096 (X. Hu).
Supplemental data
Supplemental data for this article can be accessed online here: http://dx.doi.org/10.1080/07060661.2016.1189968.
Additional information
Funding
Related Research Data
References
- Arif M, Ali M, Rehman A, Fahim M. 2013. Occurrence of potato mop-top virus in northwest of Pakistan. Eur J Plant Pathol. 137:787–796.
- Arif M, Torrance L, Reavy B. 1994. Improved efficiency of detection of potato mop-top furovirus in potato tubers and in the roots and leaves of soil-bait plants. Potato Res. 37:373–381.
- Beuch U, Berlin S, Åkerblom J, Nicolaisen M, Nielsen SL, Crosslin JM, Hamm PB, Santala J, Valkonen JPT, Kvarnheden A. 2015. Diversity and evolution of potato mop-top virus. Arch Virol. 160:1345–1351.
- Budziszewska M, Wieczorek P, Nowaczyk K, Borodynko N, Pospieszny H, Obrêpalska-Stêplowska A. 2010. First report of Potato mop-top virus on potato in Poland. Plant Dis. 94:920.
- Carnegie SF, Cameron AM, Mccreath M. 2010. Foliar symptoms caused by Potato mop-top virus on potato plants during vegetative propagation in Scotland and their association with tuber yield, spraing and tuber infection. Potato Res. 53:83–93.
- Carnegie SF, Davey T, Saddler GS. 2012. Prevalence and distribution of Potato mop-top virus in Scotland. Plant Pathol. 61:623–631.
- Čeřovská N, Pečenková T, Filigarová M, Dědič P. 2007. Sequence analysis of the Czech Potato mop-top virus (PMTV) isolate Korneta-Nemilkov. Folia Microbiol. 52:61–64.
- Chen J, Macfarlane SA, Wilson TMA. 1994. Detection and sequence analysis of a spontaneous deletion mutant of soil-borne wheat mosaic virus RNA2 associated with increased symptom severity. Virology. 202:921–929.
- Chen J, Macfarlane SA, Wilson TMA. 1995. An analysis of spontaneous deletion sites in soil-borne wheat mosaic virus RNA2. Virology. 209:213–217.
- Crosslin JM. 2011. First report of Potato mop-top virus on potatoes in Washington State. Plant Dis. 95:1483.
- Crosslin JM, Hamlin LL. 2011. Standardized RT-PCR conditions for detection and identification of eleven viruses of potato and Potato spindle tuber viroid. Am J Potato Res. 88:333–338.
- David N, Mallik I, Crosslin JM. 2010. First report of Potato mop-top virus in North Dakota. Plant Dis. 94:1506.
- Elena SF, Agudelo-Romero P, Carrasco P, Codoner FM, Martin S, Torres-Barcel OC, Sanjuán R. 2008. Experimental evolution of plant RNA viruses. Heredity. 100:478–483.
- Fraile A, Aloso-Prados JL, Aranda MA, Bernal JJ, Malpica JM, Garcia-Arenal F. 1997. Genetic exchange by recombination or reassortment is infrequent in natural populations of a tripartite RNA plant virus. J Virol. 71:934–940.
- Gil JF, Gutiérrez PA, Cotes JM, González EP, Marín M. 2011. Genotypic characterization of Colombian isolates of Potato mop-top virus (PMTV, Pomovirus). Actualidades Biológicas. 33:69–84.
- Haenni AL. 2008. Virus evolution and taxonomy. In: Roossinck MJ, editor. Plant virus evolution. New York (NY): Springer Berlin Heidelberg; p. 205–217.
- Hu X, Lei Y, Xiong X, He C, Liu M, Nie X. 2013. Identification of Potato mop-top virus (PMTV) in potatoes in China. Can J Plant Pathol. 35:402–406.
- Jones RAC, Harrison BD. 1969. The behaviour of potato mop-top virus in soil, and evidence for its transmission by Spongospora subterranea (Wallr.) Lagerh. Ann Appl Biol. 63:1–17.
- Kashiwazaki S, Scott KP, Reavy B, Harrison BD. 1995. Sequence analysis and gene content of potato mop-top virus RNA 3: Further evidence of heterogeneity in the genome organization of furoviruses. Virology. 206:701–706.
- Kirk HG. 2008. Mop-top virus, relationship to its vector. Am J Potato Res. 85:261–265.
- Koenig R, Beier C, Commandeur U, Lüth U, Kaufmann A, Lüddeche P. 1996. Beet soil-borne virus RNA 3 - a further example of the heterogeneity of the gene content of furovirus genomes and of triple gene block-carrying RNAs. Virology. 216:202–207.
- Koenig R, Commandeur U, Loss S, Beier C, Kaufmann A, Lesemann DE. 1997. Beet soil-borne virus RNA 2: Similarities and dissimilarities to the coat protein gene-carrying RNAs of other furoviruses. J Gen Virol. 78:469–477.
- Lambert DH, Levy L, Mavrodieva VA, Johnson SB, Babcock MJ, Vayda ME. 2003. First report of Potato mop-top virus on potato from the United States. Plant Dis. 87:872–872.
- Larkin MA, Blachshields G, Brown NP, Chenna R, Mcgetigan PA, Mcwilliam H, Valentin F, Wallace IM, Wilm A, Lopez R, et al. 2007. ClustalW and ClustalX version 2.0. Bioinformatics 23:2947–2948.
- Latvala-Kilby S, Aura JM, Pupola N, Hannukkala A, Valkonen JPT. 2009. Detection of Potato mop-top virus in potato tubers and sprouts: Combinations of RNA2 and RNA3 variants and incidence of symptomless infections. Phytopathology. 99:519–531.
- Lole KS, Bollinger RC, Paranjape RS, Gadkari D, Kulkarni SS, Novak NG, Ingersoll R, Sheppard HW, Ray SC. 1999. Full-length human immunodeficiency virus type 1 genomes from subtype C-infected seroconverters in India, with evidence of intersubtype recombination. J Virol. 73:152–160.
- Lu X, Yamamoto S, Tanaka M, Hibi T, Namba S. 1998. The genome organization of the broad bean necrosis virus (BBNV). Arch Virol. 143:1335–1348.
- Martin DP, Lemey P, Lott M, Moulton V, Posada D, Lefeuvre P. 2010. RDP3: A flexible and fast computer program for analyzing recombination. Bioinformatics. 26:2462–2463.
- Montero-Astúa M, Vasquéz V, Turechek WW, Merz U, Rivera C. 2008. Incidence, distribution, and association of Spongospora subterranea and Potato mop-top virus in Costa Rica. Plant Dis. 92:1171–1176.
- Nakayama T, Maoka T, Hataya T, Shimizu M, Fuwa H, Tsuda S, Mori M. 2010. Diagnosis of Potato mop-top virus in soil using bait plant bioassay and RT-PCR-microplate hybridization. Am J Potato Res. 87:218–225.
- Nie X, Singh RP. 2001. Differential accumulation of Potato virus A and expression of pathogenesis-related genes in resistant potato cv Shepody upon graft inoculation. Phytopathology. 91:197–203.
- Nielsen SL, Nicolaisen M. 2003. Identification of two nucleotide sequence sub-groups within Potato mop-top virus. Arch Virol. 148:381–388.
- Nylander JAA. 2008. MrModeltest v2.3. Program distributed by the author. Evolutionary Biology Centre, Uppsala University, Uppsala, Sweden.
- Osorio-Giraldo I, Gutiérrez-Sánchez P, Marín-Montoya M. 2013. Genetic variability of colombian isolates of Potato mop-top virus (PMTV). Agronomía Mesoamericana. 24:1–15.
- Pečenková T, Moravec T, Filigarová M, Rosecká P, Čeřovská N. 2004. Extended sequence analysis of three Danish Potato mop-top virus (PMTV) isolates. Vir Genes. 29:249–255.
- Pringle CR. 1999. Virus taxonomy – 1999. The Universal System of Virus Taxonomy, updated to include the new proposals ratified by the International Committee on Taxonomy of Viruses during 1998. Arch Virol. 144:421–429.
- Qiu WP, Geske SM, Hickey CM, Moyer JW. 1998. Tomato spotted wilt tospovirus genome reassortment and genome segment-specific adaptation. Virology. 244:186–194.
- Rambaut A, Suchard MA, Xie D, Drummond AJ. 2014. Tracer v1.6, Available from: HTTP://BEAST.BIO.ED.AC.UK/TRACER.
- Ramesh SV, Raikhy G, Brown CR, Whitworth JL, Pappu HR. 2014. Complete genomic characterization of a potato mop-top virus isolate from the United States. Arch Virol. 159:3427–3433.
- Reavy B, Arif M, Cowan GH, Torrance L. 1998. Association of sequences in the coat protein/readthrough domain of potato mop-top virus with transmission by Spongospora subterranea. J Gen Virol. 79:2343–2347.
- Ronquist F, Teslenko M, van der Mark P, Ayres DL, Darling A, Hohna S, Larget B, Liu L, Suchard MA, Huelsenbeck JP. 2012. MrBayes 3.2: Efficient Bayesian phylogenetic inference and model choice across a large model space. Syst Biol. 61:539–542.
- Roossinck MJ, Schneider WL. 2006. Mutant clouds and occupation of sequence space in plant RNA viruses. In: Domingo E, editor. Quasispecies: Concept and implications for virology. New York (NY): Springer Berlin Heidelberg; p. 337–348.
- Salazar LF. 1996. Potato viruses and their control. Lima: International Potato Center; p. 23–46 and 83–166.
- Sandgren M. 1995. Potato mop-top virus (PMTV): Distribution in Sweden, development of symptoms during storage and cultivar trials in field and glasshouse. Potato Res. 38:379–389.
- Sandgren M, Plaisted RL, Watanabe KN, Olsson S, Valkonen JPT. 2002. Evaluation of some North and South American potato breeding lines for resistance to Potato mop-top virus in Sweden. Am J Potato Res. 79:205–210.
- Sandgren M, Savenkov EI, Valkonen JPT. 2001. The readthrough region of Potato mop-top virus (PMTV) coat protein encoding RNA, the second largest RNA of PMTV genome, undergoes structural changes in naturally infected and experimentally inoculated plants. Arch Virol. 146:467–477.
- Santala J, Samuilova O, Hannukkala A, Latvala S, Kortemaa H, Beuch U, Kvarnheden A, Persson P, Topp K, Ørstad K, et al. 2010. Detection, distribution and control of Potato mop-top virus, a soil-borne virus, in northern Europe. Ann Appl Biol. 157:163–178.
- Savenkov EI, Germundsson A, Zamyatnin Jr AA, Sandgren M, Valkonen JPT. 2003. Potato mop-top virus: The coat protein-encoding RNA and the gene for cysteine-rich protein are dispensable for systemic virus movement in Nicotiana benthamiana. J Gen Virol. 84:1001–1005.
- Savenkov EI, Sandgren M, Valkonen JPT. 1999. Complete sequence of RNA 1 and the presence of tRNA-like structures in all RNAs of Potato mop-top virus, genus Pomovirus. J Gen Virol. 80:2779–2784.
- Scott KP, Kashiwazaki S, Reavy B, Harrison BD. 1994. The nucleotide sequence of potato mop-top virus RNA 2: A novel type of genome organization for a furovirus. J Gen Virol. 75:3561–3568.
- Sztuba-Solinska J, Urbanowicz A, Figlerowicz M, Bujarski JJ. 2011. RNA-RNA recombination in plant virus replication and evolution. Annu Rev Phytopathol. 49:415–443.
- Talavera G, Castresana J. 2007. Improvement of phylogenies after removing divergent and ambiguously aligned blocks from protein sequence alignments. Syst Biol. 56:564–577.
- Tamura K, Peterson D, Peterson N, Stecher G, Nei M, Kumar S. 2011. MEGA5: Molecular evolutionary genetics analysis using maximum likelihood, evolutionary distance, and maximum parsimony methods. Mol Biol Evol. 28:2731–2739.
- Torrance L, Cowan GH, Sokmen MA, Reavy B. 1999. A naturally occurring deleted form of RNA 2 of Potato mop-top virus. J Gen Virol. 80:221–2215.
- Torrance L, Mayo MA. 1997. Proposed re-classification of furoviruses. Arch Virol. 142:435–439.
- Wang B, Li M, Han C, Li D, Yu J. 2008. Complete genome sequences of two Chinese Beet soil-borne virus isolates provide evidence that the genome is highly conserved. J Phytopathol. 156:487–488.
- Whitworth JL, Crosslin JM. 2013. Detection of Potato mop-top virus (Furovirus) on potato in southeast Idaho. Plant Dis. 97:149.
- Xu H, De Haan T-L, De Boer SH. 2004. Detection and confirmation of Potato mop-top virus in potatoes produced in the United States and Canada. Plant Dis. 88:363–367.