
Abstract
A simple and inexpensive method for extraction of genomic DNA from mycelia of filamentous fungi was developed and the standardization of the protocol is described. The protocol includes both mycelium culturing and DNA extraction. By incorporating a selected commercial sand product into the culture media, the collection and the grinding of mycelia was simple and quick. The protocol of DNA extraction was derived from the alkaline lysis method for plasmid DNA extraction and modified to facilitate efficient extraction of fungal genomic DNA. It has increased efficiency and fidelity compared to the other simple DNA extraction protocols. Relative to a commonly used commercial DNA extraction kit, this extraction protocol is faster, easier and can generate more DNA without compromising quality. From a single sample of mycelium, genomic DNA can be prepared within 20 min, including concentration measurement. The DNA can be used in restriction enzyme digestion and as a template in PCR to amplify DNA fragments larger than 3000 base pairs. This method is recommended for DNA extraction in fungal phylogenetic studies when hundreds or thousands of isolates from one or a few closely-related species are subjected to PCR-based genotyping.
Résumé
Une méthode simple et bon marché pour extraire l’ADN génomique du mycélium des champignons filamenteux a été développée et nous décrivons la standardisation du protocole. Le protocole inclut la culture du mycélium et l’extraction de l’ADN. En incorporant un certain sable commercial dans le milieu de culture, la collecte et la pulvérisation du mycélium est simple et rapide. Le protocole d’extraction de l’ADN est dérivé de la lyse alcaline utilisée pour l’extraction de l’ADN plasmidique et est modifié pour faciliter l’extraction efficace de l’ADN génomique fongique. Par comparaison aux autres protocoles simples d’extraction de l’ADN, ce dernier en a amélioré l’efficacité et la fidélité. Semblable au protocole utilisé avec une trousse commerciale d’extraction d’ADN, ce protocole d’extraction est plus rapide, plus simple et peut produire plus d’ADN sans en compromettre la qualité. À partir d’un échantillon unique de mycélium, il est possible d’obtenir l’ADN génomique en moins de 20 minutes, y compris la mesure de la concentration. L’ADN peut être utilisé dans la digestion par enzyme de restriction et comme gabarit dans le cadre d’une PCR pour amplifier des fragments d’ADN contenant plus de 3 000 paires de bases. Cette méthode est recommandée pour l’extraction d’ADN dans les études phylogénétiques portant sur les champignons, lorsque des centaines, voire des milliers d’isolats provenant d’une ou des quelques espèces voisines sont soumis au génotypage par PCR.
Introduction
Extraction of genomic DNA from plant pathogenic fungi is a common practice in plant pathology laboratories. Many protocols for the extraction of fungal DNA are available (Cenis Citation1992; Min et al. Citation1995; Tapia-Tussell et al. Citation2006), generally derived from the method originally developed for plant DNA extraction (Saghai-Maroof et al. Citation1984). These methods are simple and inexpensive but time consuming, especially when multiple samples are subject to DNA extraction. Commercial kits are available from many companies. The advantage of the kit is the high quality of the extracted DNA and the consistency from different starting materials. However, most kits being used for fungal DNA extraction were designed for plant DNA, and thus include reagents and steps that are necessary for plant material but not for fungi. For example, kits may require either grinding the samples in liquid nitrogen or a prolonged incubation time in the lysis buffer. The kits, however, are preferable to other methods to extract DNA from a smaller number of samples, especially when the quality of the DNA is extremely important.
In plant pathology or plant diagnostics laboratories, genomic DNA often needs to be extracted from many fungal isolates for phylogenetic studies or DNA barcoding, in which the DNA is mainly used for PCR or qPCR (e.g. Feng et al. Citation2010a; Rong et al. Citation2015; Tamburic-Ilincic et al. Citation2015; Liban et al. Citation2016). In such a situation, a simple, inexpensive and timesaving method is preferred. This study describes the standardization of a new protocol, including the culturing of mycelia and the DNA extraction from the resultant mycelia. This protocol has been extensively used in the Alberta Plant Health Lab (APHL) for extraction of genomic DNA from many fungal species, such as those isolated from diseased samples of various plant species, and the province-wide collections of canola blackleg pathogens (Leptosphaeria maculans and L. biglobosa) and cereal head blight/corn ear blight pathogens (Fusarium species). The resultant DNA was used as a template in PCR and qPCR for species or sub-species identification. It was expected that it will be a useful tool for other research groups conducting similar studies. For simplicity, this protocol is referred to as the APHL (Alberta Plant Health Lab) protocol in this paper.
Materials and methods
Chemicals and standard techniques
All chemicals were purchased from Fisher Scientific Canada (Ottawa, ON) unless otherwise specified. Primers () were synthesized by Integrated DNA Technologies (Coralville, IA). All PCR reactions were conducted in Promega PCR master mix (Promega, Madison, WI) with a T100 thermal cycler (Bio-Rad Canada, Mississauga, ON). Electrophoresis was performed in 1% (w/v) agarose gel stained with SYBR Green. Other molecular techniques, if not specified, were performed according to the protocols described by Sambrook and Russell (Citation2001).
Table 1. Oligonucleotide primers used in this study.
Fungal materials
Three fungal strains – Fusarium graminearum strain PH1, Stagonospora nodorum strain Sn15 and L. maculans strain C19 – were used in this study. The first two are the original strains for whole genome sequencing of the corresponding species (Cuomo et al. Citation2007; Hane et al. Citation2007). The C19 was derived from the S4 strain (Feng et al. Citation2014) tagged with a red fluorescent protein (RFP). The three fungal strains were stocked in −80°C as conidia suspension at 1 × 106 spores/mL in 16% (v/v) glycerol.
Preparation of fungal mycelia for DNA extraction
Two culture media were used to increase fresh mycelia: YPG (0.5% yeast extract, 1% peptone and 1.5% glucose; w/v) and YPG supplemented with sand (Cat: S5631; Sigma-Aldrich Canada, Oakville, ON) at a concentration of 0.5% (w/v). The sand was chosen above others because of the ability of the particles to remain suspended in water when being shaken at 150 rpm in 1.5-mL tubes. The sand was pre-treated by soaking in 50× volumes of 100 mM Tris buffer (pH 7.5) for 4 h and then rinsing with water. The YPG and YPG/sand were autoclaved and 400-uL aliquots of each medium were prepared in 1.5-mL microcentrifuge tubes.
Both YPG and the YPG/sand media were inoculated with the three fungal strains by two methods: with spores and with mycelia. The spore inoculum was 10 uL of stock solution of conidia (1 × 106 conidia/mL in 16% glycerol). To prepare the mycelial inoculum, a drop of the stock solutions was inoculated on the centre of a 6-cm-diameter Petri dish containing 4 mL of 4% (w/v) PDA. The inoculation of the three species was conducted on different dates to ensure the colonies of the three species could grow to cover two-thirds of the Petri dishes on the same day. PDA culture plugs at 1 mm-diameter were prepared by punching the edge of the colonies with the tip of a glass Pasteur pipet. Three plugs were used to inoculate the 400 uL media in the microcentrifuge tube. For each species-medium-inoculum combination, six tubes were prepared. The tubes were shaken at 150 rpm at room temperate (22°C). After 48 h, the mycelia in the YPG/sand culture were examined using an AX10 microscope (Carl Zeiss Canada, Toronto, ON). The mycelia were pelleted by centrifugation at 14 000 rpm (16 873 ×g) for 2 min. The supernatant was removed by a vacuum pump (for the YPG/sand medium) or pipetting (for YPG). The collected mycelia were kept in a −20°C freezer for less than one week until DNA extraction.
DNA extraction
The tubes with the collected mycelia were divided into two sets. Each set consisted of 36 tubes that included three replicates for each species-medium-inoculum combination. The two sets of mycelia were subjected to DNA extraction with either a DNeasy Plant Mini Kit (Qiagen Canada, Toronto, ON) or the APHL protocol. In the DNA extraction process using both methods, the experiment was treated as randomized complete blocks with three blocks (repeats), in which the tubes in one repeat were performed in one extraction process with a random order. Unless specified by the instruction manual of the Qiagen kit, all centrifugations were performed in an Eppendorf 5418 microcentrifuge at the maximum speed of 14 000 rpm (16 873 ×g).
In the APHL protocol, after adding 0.4 mL lysis buffer (100 mM Tris base, 50 mM EDTA, 1% SDS (w/v), 1% N-lauroyl sarcosine sodium salt (w/v) and 10 µg mL−1 RNase A; pH 8.2), the mycelium pellet in the tubes were ground with a pellet pestle (Fisher cat: 12-141-364) driven by an electric drill at 1000 rpm for either 5 s (for mycelia derived from the YPG/sand medium) or 5–20 s until the mycelium lumps were ground (for mycelia derived from YPG). The tubes were inverted several times, and then 100 µL of potassium acetate solution (3.0 M, pH 6.5) was added. The tube was inverted several times and centrifuged for 2 min. A 0.4 mL aliquot of supernatant was transferred into a new tube containing 0.5 mL of isopropanol, which was then inverted several times and centrifuged for 2 min to precipitate DNA. The supernatant was removed and the DNA pellet was washed with 0.75 mL 70% (v/v) ethanol. After centrifugation for 1 min, the ethanol was removed and the DNA pellet was air dried for 2 min. The DNA was dissolved in 50 µL sterile distilled water.
DNA extraction by the Qiagen kit was performed according to the manufacturer’s instructions except the following two modifications: (1) after adding the lysis buffer, the mycelia were ground with the pestle as described above and (2) the extracted DNA was eluted in 50 µL of elution buffer.
Quantity and quality assessment of the extracted DNA
The concentrations and the 260/280 ratio of the extracted DNA were measured using a NanoDrop Lite Spectrophotometer. The concentration data from each inoculum (spores or mycelia) for each species were analysed separately, for each of which, a two-way ANOVA was performed using Microsoft Excel with extraction method and medium as the two factors.
DNA samples extracted with the APHL protocol were subjected to further analyses (those from kit extraction were excluded hereafter). PCR was conducted against all DNA, using two pairs of primers, ITS1/ITS4 (White et al. Citation1990) amplifying the internal transcribed spacers in the rDNA region and H729/H730 (Hill et al. Citation2004) amplifying a fragment of the cpn60 gene. In addition, another primer pair, 13059/13062, specific to a gene encoding a hypothetical protein (GenBank accession number: XP_003844770) was used in PCR against all the L. maculans DNA samples. The sequences of primers and the important PCR conditions are listed in . The other PCR conditions are: an initial denaturation at 95°C for 3 min, followed by 35 cycles of denaturation at 95°C for 30 s, annealing at 52–58°C () for 45 s, elongation at 72°C for 45 s–3 min (). The final elongation is at 72°C for 5 min. One DNA sample was randomly chosen from the three repeats in each species-medium-inoculum combination and digested with the restriction enzyme Hind III (NEB Canada, Whitby, ON) at 37°C for 2.5 h. The reaction volume was 50 µL containing 3 µg of DNA and 20 units of the enzyme.
Results
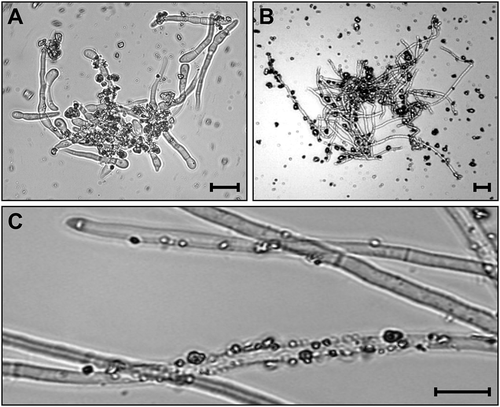
It was expected that the sand in YPG would increase the efficiency of mycelium collection and grinding. After 48 h of culturing, sand particles were found adhering to the mycelia generated from spores () or mycelium plugs (). These mycelia could be easily centrifuged down, resulting in neat pellets and clear supernatant. In contrast, centrifugation for 2 min could not efficiently spin down the mycelia in the YPG medium without sand. When the pellet consisting of sand-attached mycelia were vortexed in 40 mL water for 10 s, many sand particles still clung to the hyphae (). When grinding the mycelia without sand, extensive care was needed because the mycelium lumps were floating in the lysis buffer and circulating with the whirlpool created by the pellet pestle. There was no such problem when the mycelia with sand were being ground, in which the mycelia are pelleted at the bottom of the tube and situated between the pestle and the tube wall during the grinding process. Furthermore, it was believed that the sand helped to break the fungal cell wall during grinding.
Fig. 1 Adhesion of sand particles on hyphae and mycelia of Fusarium graminearum in a liquid medium supplemented with sand. a, b, The medium was incubated at 150 rpm in 30°C for 48 h after inoculated with F. graminearum conidia (a) and mycelia (b). c, The mycelia in (b) after washing with 40 mL of water. Bars = 10 μm.
Genomic DNA was extracted from all the species-medium-inoculum-extraction method combinations. The time period between removing the liquid cultures from the shaker incubator to finishing the concentration measurement of the extracted DNA was evaluated. For one sample, the APHL protocol needed 20 min, whereas the Qiagen kit needed 50 min; for 18 samples, the APHL protocol needed one hour and the Qiagen kit needed 3.5 h. The DNA dissolved in 50 µL water or elution buffer, produced by the APHL protocol had a concentration of 22.17–121.23 ng/µL, with a 260/280 ratio of 1.61–1.85, where those from the Qiagen kit were between 1.93 and 4.33 ng/µL, with a 260/280 ratio of 1.63–2.00 (). From each of the three fungal species in either medium with either inoculum, there was a significant difference in DNA concentrations between the two extraction methods, in which the APHL protocol produced more DNA than the Qiagen kit (). No difference was observed between the media (YPG vs. YPG/sand) and the interaction of extraction method-medium.
Table 2. Quantity and range of the 260/280 ratio for genomic DNA extracted from mycelia.
Table 3. Calculated possibilities (p-values) of a two-way ANOVA to investigate the variations in DNA quantity.
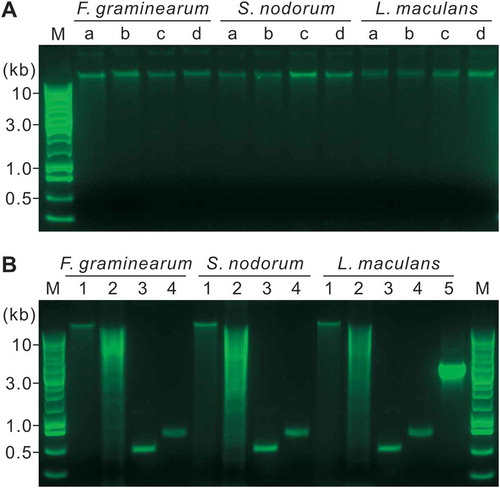
All the DNA samples extracted by the APHL protocol showed a sharp band on 1% (w/v) agarose gel (). After incubation at 37°C for 2.5 h, the sharp bands could still be produced without DNA degradation (). After digestion with Hind III, the DNA showed clear smears on the gel (). The two primer pairs, ITS1/ITS4 and H729/H730, could amplify a single, sharp band from all the tested DNA samples (). The size of the bands was visually equal to the expected size () of the amplicons. Furthermore, a gene fragment at more than 3 kb was amplified from all the tested L. maculans DNA using the primer pair 13059/13062 ().
Fig. 2 (Colour online) Quality assessment of the DNA extracted by the APHL protocol on 1% agarose gels. M, Promega 1-kb DNA ladder. a, Five-hundred ng of DNA in a volume of 10 μL was analysed. The DNA was extracted from mycelia derived from YPG media inoculated with conidia (a) or mycelia (b), or YPG/sand inoculated with conidia (c) or mycelia (d). b, The DNA in (c) were further analysed. 1, Three-hundred ng of DNA in a volume of 10 μL were incubated at 37°C for 2.5 h; 2, Three μg DNA in a volume of 50 μL were digested with Hind III at 37°C for 2.5 h; 3–5, PCR products from 50 ng DNA templates using the primer pair ITS1/ITS4 (3), H729/H730 (4) or 13059/13062 (5).
Discussion
The YPG medium was modified from the Standard Methods Agar (SMA) medium (Atlas Citation2004) with increased glucose content to promote the growth of fungal mycelia. The other conditions for fungal culturing were defined to ensure that the fungus used up most of the nutrients when the mycelia were being collected. It was expected that such a design would generate a similar amount of mycelia from different isolates of a certain fungal species, albeit there were variations in the initial inoculum in respect of age and quantity. The use of sand in the media facilitated the collection and the grinding of the mycelia, which would decrease the variation of the final materials subjected to DNA extraction among isolates.
Although simple and quick, the APHL protocol includes all of the important steps of standard DNA extraction (Sambrook & Russell Citation2001). Grinding with a pestle with the presence of sand efficiently breaks the fungal cell wall. At the same time, SDS denatures proteins and disrupts the cell membrane (Sambrook & Russell Citation2001), resulting in the release of nucleic acids into the cell lysis mixture. EDTA and N-lauroyl sarcosine sodium salt inhibit the activity of DNA-degrading enzymes by chelating the cations functioning as cofactors of the enzymes and providing a reductive environment to disrupt the disulphide bonds in the enzymes, respectively. The RNase A degrades RNA that would otherwise precipitate together with the DNA. Addition of potassium acetate into the lysis mixture results in the replacement of sodium ions by potassium ions, which efficiently precipitate proteins and SDS (Ish-Horowicz & Burke Citation1981). Potassium ions also neutralize the charge on the sugar-phosphate backbone of the DNA, which is essential for DNA precipitation by isopropanol (Sambrook & Russell Citation2001). Furthermore, potassium acetate changes the pH from 8.2 to 7.0 and protonates Tris to strengthen its buffering capacity. In the final step, the volume of water or buffer used to dissolve the DNA pellet can be adjusted to provide the appropriate concentration of DNA needed for specific applications.
Using the APHL protocol, genomic DNA was extracted from three fungal species. The quantity of the DNA was higher than that of the DNA extracted by the Qiagen kit; the quality, as measured by the 260/280 ratio, was similar between the DNA extracted by the two methods. DNA extracted by the APHL protocol was stable after incubated at 37°C for 2.5 h. Furthermore, the DNA could be used in restriction enzyme digestion and as template in PCR using both regular primers (ITS1/ITS4) targeting a multiple copied gene and degenerated primers (H729/H730) targeting a single-copied gene (Hill et al. Citation2004). The DNA could also be used in PCR to amplify DNA fragments larger than 3 kb. In many plant pathology studies, the purpose for DNA extraction was to prepare a template for PCR or qPCR, for which the APHL protocol proved to be very useful.
Many simple protocols for DNA extraction from fungal mycelia are available (Al-Samarrai & Schmid Citation2000; Saitoh et al. Citation2006; Feng et al. Citation2010b). Among others, the APHL protocol is not the simplest one, but it is designed to produce more DNA by adding a step of grinding. The grinding procedure was expedited by the supplement of sand in the liquid culture. In the Alberta Plant Health Lab, this protocol has been extensively used. Based on our experience, it is highly recommended in phylogenetic studies where hundreds or even thousands of isolates from one species or closely-related species were subjected to DNA extraction.
Acknowledgements
The authors acknowledge funding support from the Alberta Crop Industry Development Fund (ACIDF).
Additional information
Funding
References
- Al-Samarrai TH, Schmid J. 2000. A simple method for extraction of fungal genomic DNA. Lett Appl Microbiol. 30:53–56.
- Atlas RM. 2004. Handbook of Microbiological Media. London: CRC Press.
- Cenis JL. 1992. Rapid extraction of fungal DNA for PCR amplification. Nucleic Acids Res. 20:2380.
- Cuomo CA, Guldener U, Xu J-R, Trail F, Turgeon BG, Di Pietro A, Walton JD, Ma L-J, Baker SE, Rep M, et al. 2007. The Fusarium graminearum genome reveals a link between localized polymorphism and pathogen specialization. Science. 317:1400–1402.
- Feng J, Hwang R, Chang KF, Hwang SF, Strelkov SE, Gossen BD, Conner RL, Turnbull GD. 2010a. Genetic variation in Fusarium avenaceum causing root rot on field pea. Plant Pathol. 59:845–852.
- Feng J, Hwang R, Chang KF, Hwang SF, Strelkov SE, Gossen BD, Zhou Q. 2010b. An inexpensive method for extraction of genomic DNA from fungal mycelia. Can J Plant Pathol. 32:396–401.
- Feng J, Zhang H, Strelkov SE, Hwang S-F. 2014. The LmSNF1 gene is required for pathogenicity in the canola blackleg pathogen Leptosphaeria maculans. PloS One. 9:e92503.
- Hane JK, Lowe RG, Solomon PS, Tan K-C, Schoch CL, Spatafora JW, Crous PW, Kodira C, Birren BW, Galagan JE, et al. 2007. Dothideomycete-plant interactions illuminated by genome sequencing and EST analysis of the wheat pathogen Stagonospora nodorum. Plant Cell. 19:3347–3368.
- Hill JE, Penny SL, Crowell KG. 2004. cpnDB: a chaperonin sequence database. Genome Res. 14:1669–1675.
- Ish-Horowicz D, Burke JF. 1981. Rapid and efficient cosmid cloning. Nucleic Acids Res. 9:2989–2998.
- Liban SH, Cross DJ, Kutcher HR, Peng G, Fernando WGD. 2016. Race structure and frequency of avirulence genes in the western Canadian Leptosphaeria maculans pathogen population, the causal agent of blackleg in brassica species. Plant Pathol. 65:1161–1169.
- Min J, Aroanoza T, Ohrnberger J, Xu C, Akins RA. 1995. Alternative methods of preparing whole-cell DNA from fungi for dotblot, restriction analysis, and colony filter hybridization. Anal Biochem. 225:94–100.
- Rong S, Feng J, Li Q, Fei W, Ahmed HU, Liang Y, Hwang S-F, Strelkov SE. 2015. Pathogenic variability and prevalence of Avr genes in Leptosphaeria maculans populations from Alberta, Canada. J Plant Dis Protect. 122:161–168.
- Saghai-Maroof MA, Soliman KM, Jorgensen RA, Allard RW. 1984. Ribosomal DNA spacer-length polymorphisms in barley: Mendelian inheritance, chromosomal location, and population dynamics. Proc Natl Acad Sci USA. 81:8014–8018.
- Saitoh K-I, Togashi K, Arie T, Teraoka T. 2006. A simple method for a mini-preparation of fungal DNA. J Gen Plant Pathol. 72:348–350.
- Sambrook J, Russell DW. 2001. Molecular cloning: a laboratory manual. 3rd ed. Cold Spring Harbor (NY): Cold Spring Harbor Laboratory Press.
- Tamburic-Ilincic L, Wragg A, Schaafsma A. 2015. Mycotoxin accumulation and Fusarium graminearum chemotype diversity in winter wheat grown in southwestern Ontario. Can J Plant Sci. 95:931–938.
- Tapia-Tussell R, Lappe P, Ulloa M, Quijano-Ramayo A, Cáceres-Farfán M, Larqué-Saavedra A, Perez-Brito D. 2006. A rapid and simple method for DNA extraction from yeasts and fungi isolated from Agave fourcroydes. Mol Biotechnol. 33:67–70.
- White TJ, Bruns T, Lee S, Taylor J. 1990. Amplification and direct sequencing of fungal ribosomal RNA genes for phylogenetics. In: Innes MA, Gelfand DH, Sninsky JJ, White TJ, editors. PCR protocols: a guide to methods and applications. San Diego: Academic Press; p. 315–322.