
Abstract
High throughput sequencing (HTS) revealed the existence of complexes of fabaviruses infecting two ‘Staccato(R) ’ sweet cherry plants (Prunus avium). Species classified as members of the genus Fabavirus are positive sense RNA viruses that possess bipartite genomes. The cherry plants Stac-3B (symptomatic) and Stac-4B (asymptomatic) were each determined to be infected with multiple RNA1 and RNA2 segments of Prunus virus F (PrVF) and the putative cherry virus F (CVF). The RNA1 and RNA2 segments Stac-3B_C3 and Stac-3B_c11, respectively, are proposed to represent an isolate identified as PrVF-CP1; while the RNA1 and RNA2 segments Stac-3B_C4 and Stac-3B_c7 are proposed to represent an isolate CVF-CC1. The termini of these four RNA segments were confirmed by RACE revealing that the first 28 nucleotides of the 5ʹuntranslated region (UTR) of all four RNA segments is conserved and is predicted to form a hairpin structure. The 3ʹUTRs possess various repeat sequences. Very interestingly, a group of PrVF sequences, exemplified by SwC 74_2c, were identified as the products of an interspecies recombination event, with PrVF 8816_s1 as a possible major parent and an isolate of CVF being the minor parent. This was supported by all methods in the RDP4 program. No association was made of any of the fabavirus complexes with any disease symptoms. This is the first description of the detection of CVF in Canada.
Résumé
Le séquençage à haut débit a révélé l’existence de complexes de fabavirus infectant deux cerisiers sauvages (Prunus avium) ‘Staccato(R)’. Les espèces classifiées en tant que membres du genre Fabavirus sont des virus à ARN positif qui possèdent un génome biparti. Il a été établi que les cerisiers Stac-3B (symptomatique) et Stac-4B (asymptomatique) étaient infectés par des segments multiples d’ARN1 et d’ARN2 de Prunus virus F (PrVF) et par le virus putatif F du cerisier (CVF). Les segments d’ARN1 et d’ARN2 Stac3B_3C et Stac-3B_c11, respectivement, sont prévus représenter un isolat identifié en tant que PrVF-CP1, tandis que les segments d’ARN1 et d’ARN2 Stac-3B_C4 et Stac-3B_c7 sont prévus en représenter un autre identifié en tant que CVF-CC1. Les extrémités de ces quatre segments d’ARN ont été confirmées par RACE, révélant que les 28 premiers nucléotides de la région 5ʹ non traduite (UTR) des quatre segments d’ARN sont conservés et prévus former une structure en épingle à cheveux. L’UTR3ʹ possède diverses séquences répétées. D’ailleurs, un groupe de séquences de PrVF, illustré par SwC 74_2c, était identifié comme produit d’une recombinaison interspécifique, avec PrVF 8816_s1 comme parent primaire possible et un isolat de CVF comme parent secondaire. Cela était soutenu par toutes les méthodes du programme RDP4. Aucune association n’a été faite entre les complexes de fabavirus et quelque symptôme que ce soit. Il s’agit de la première description de la détection de CVF au Canada.
Introduction
Virus species in the genus Fabavirus, family Secoviridae, have a wide host range, can cause a range of symptoms in economically important crops and are transmitted by aphids in a non-persistent manner (Thompson et al., Citation2017). It seems also that there are cases of seed or pollen transmission among some members of the family Secoviridae (Sanfaçon et al., Citation2011).
Broad bean wilt virus 1 (BBWV1) is the type species of the genus Fabavirus and members of the genus possess positive sense, 3ʹ polyadenylated single-stranded RNA, bipartite genomes (Ferriol et al., Citation2014; Thompson et al., Citation2017). RNA1 is mono-cistronic with a single open reading frame (ORF) that encodes a polyprotein that is cleaved into a putative protease co-factor (Co-Pro), a helicase protein (HEL), a genome-linked viral protein (VPg), a protease (PRO) and the RNA-dependent RNA polymerase (RdRp). RNA2 possesses also a single ORF that encodes a polyprotein that is cleaved into the putative movement protein (MP) and two coat protein subunits (CPL and CPS). Putative protease cleavage sites associated with both polyproteins of fabaviruses include the dipeptides Q/S, Q/A and Q/G (Sanfaçon et al., Citation2011). Other dipeptides have been associated with putative cleavage sites, for example Q/D at the Co-Pro/HEL junction (Dong et al., Citation2012).
Recently, the known natural host range of fabaviruses has been expanded to include stone fruit trees (Prunus spp.) with the detection of the new fabavirus Prunus virus F (PrVF) in sweet cherry (Prunus avium) (Villamor et al., Citation2017; James et al., Citation2018; Koloniuk et al., Citation2018) and in sour cherry (P. cerasus) (Šafářová et al., Citation2017; Koloniuk et al., Citation2018); detection of the putative fabavirus cherry virus F (CVF) in sweet and sour cherry (Koloniuk et al., Citation2018); and the detection of the putative fabavirus peach leaf pitting-associated virus (PLPaV) in peach (P. persica) (He et al., Citation2017). Some new dipeptides associated with putative protease cleavage sites and shared by stone fruit-infecting fabaviruses were identified (He et al., Citation2017; Villamor et al., Citation2017). These include Q/F at the Co-Pro/HEL and PRO/RdRp junctions and with Q/L at the CP(L)/CP(S) junction of PrVF (Villamor et al., Citation2017), while PLPaV has R/A at the CP(L)/CP(S) junction (He et al., Citation2017).
Stone fruits are economically important crops and are cultivated globally (Byrne, Citation2005), with many virus-related diseases that present a serious challenge to producers (James et al., Citation2017a). However, as yet no symptoms have been associated definitively with the presence of PrVF or CVF in sweet or sour cherry (Šafářová et al., Citation2017; Villamor et al., Citation2017; James et al., Citation2018; Koloniuk et al., Citation2018). PLPaV was detected in peach trees displaying symptoms that included small and cracked fruit; however, several other co-infecting pathogens were also detected (He et al., Citation2017). These included the viruses Apple chlorotic leaf spot virus (ACLSV) and Plum bark necrosis stem pitting-associated virus (PBNSPaV), as well as the viroids Peach latent mosaic viroid (PLMVd) and Hop stunt viroid (HSVd) (He et al., Citation2017). Additionally some symptomatic samples did not appear to be infected with PLPaV (He et al., Citation2017). Bioassay analysis of PLPaV using the peach indicator host GF305 did result in the development of pitting symptoms on leaves, suggesting a definite potential for this virus to cause symptoms in some peach genotypes (He et al., Citation2017).
In this study, thorough analysis of high-throughput sequencing (HTS) data obtained from two ‘Staccato(R)’ sweet cherry plants revealed the presence of complexes of fabaviruses that included RNA1 and RNA2 segments of PrVF and CVF. We present the results of these analyses and possible clues to establishing RNA1/RNA2 relationships. Since fabaviruses are aphid transmitted (Thompson et al., Citation2017) and perhaps in some cases pollen and/or seed transmitted (Sanfaçon et al., Citation2011), their spread might be difficult to control and mixed infections of isolates/species are quite probable. Even though no definitive evidence of disease relationships has as yet been established, clearly stone fruits have the potential to be reservoirs for fabaviruses that might affect susceptible crops in close proximity. A better understanding of the genetic diversity and distribution of fabaviruses in stone fruits may help in determining their biological significance in these hosts.
Materials and methods
Virus source
‘Staccato(R)’ sweet cherry trees (P. avium) growing in the Okanagan Valley, BC, Canada, were observed showing a range of symptoms that included leaf deformity and dieback. Selected plants were sampled and were analysed using HTS. James et al. (Citation2018) identified two plants that were infected with PrVF; one symptomatic cherry plant (OKStac-3B) and a non-symptomatic control plant (OKStac-4B). Thorough analysis of HTS data from both plants, identified in this study as Stac-3B and Stac-4B, respectively () was carried out, revealing infections of complexes of fabaviruses.
Table 1. Table of illumina HiSeq 2500 paired end reads obtained and contigs assembled from the Stac-3B and from Stac-4B samples of sweet cherry (Prunus avium) ‘Staccato’.
Total RNA extraction
Total RNA extractions were carried out using QIAGEN RNeasy Plant Total RNA Kit (QIAGEN), following a modified protocol as described by Kalinowska et al. (Citation2012) and including an on-column DNase digestion step. Total RNA was eluted with 50 µL warm (37°C) nuclease-free H2O after a 5 min incubation on the column.
Complementary (c)DNA production and high-throughput sequencing (HTS)
Complementary (c)DNA and HTS were carried out as described by Messmer et al. (Citation2017), with sequencing performed at The Centre for Applied Genomics at Toronto’s Hospital for Sick Children (Toronto, ON).
Genome assembly
HTS data assembly was carried out using CLC Genomics Server 8.5 (CLCBio, www.clcbio.com). Initially reads were paired and trimmed using the default parameters for quality, ambiguous nucleotides, and adapter sequence. Trimmed reads were subjected to a custom workflow. Briefly, reads were mapped to host sequence which allowed for in silico removal of host reads. Contigs were generated by de novo assembly of the remaining non-host reads. Contigs > 500bp were analysed for potential open reading frames (ORFs) and then translated into protein sequence where possible. Viral contigs were identified by nucleotide and protein BLAST alignment (BLASTN and BLASTP algorithm, respectively) to viral National Center of Biotechnology Information (NCBI) derived local databases. Contigs of interest were extracted and reads were mapped to confirm and extend the sequences as necessary.
5ʹ RACE, cloning and sanger sequencing
The 5ʹ termini of selected RNA1 and RNA2 fabavirus segments were determined using Invitrogen 5ʹ RACE System version 2.0 (cat no. 18 374–058) following the manufacturer’s protocol. Due to their genetic diversity, potential relationships and the availability of plant tissue, two RNA1-associated contigs (Stac-3B_C3 and Stac-3B_C4) and two RNA2-associated contigs (Stac-3B_c11 and Stac-3B_c7) from Stac3B were selected for confirmation of their 5ʹ and 3ʹ termini by RACE and/or targeted RT-PCR. Primer design for detection and resolution of the 5ʹ termini was aided by Clone Manager 9 (Professional Edition (c) 1994–2010, Scientific and Educational Software, Cary, NC, USA). Primers used in RACE analyses are shown in . Amplified cDNA fragments were separated on 1% agarose gels, with fragments of interest excised and gel extracted using QIAGEN’s MinElute gel extraction kit (Qiagen, Ontario, cat no. 28 606). Gel extracts were then ligated into pCR 2.1-TOPO(R) vector and cloned using the TOPO TA cloning kit as described by the manufacturer (Thermo-Fisher, Waltham, MA, cat no. K450001). Three independently obtained plasmids containing inserts of the expected sizes were prepared using the Quantum Prep Plasmid Miniprep kit (BioRad, cat no. 7 326 100), then subjected to Sanger sequencing with an Applied Biosystems 3730 DNA Analyzer at the Nucleic Acid Protein Service Unit, UBC (Vancouver, BC).
Table 2. Oligonucleotide primers based on the sequences of fabaviruses infecting sweet cherry (Prunus avium) ‘Staccato’.
RT-PCR analysis
RT-PCR was carried out as follows. For cDNA synthesis, a 13 μL volume of pre-mix, containing 1 μL total RNA, 2 pmol of the primer PrVF-3ʹambiR (), and 2.5 nanomoles of each dNTP, was incubated at 65°C for 5 min. To this pre-mix was added 7 μL of a cDNA synthesis mix containing First Strand buffer, DTT, RNase OUT and SuperScript III Reverse Transcriptase in amounts sufficient to match the manufacturer’s recommended concentrations in the final 20 μL reaction volume. This mixture was incubated at 55°C for 60 min, followed by a 70°C termination step for 15 min. Next, 2 μL of this mixture was used in a 25 μL (final volume) PCR reaction containing 0.2 μM each of the target-specific sense primer and the 3ʹambiR antisense primer, plus MgSO4, Hi-Fi PCR buffer and Hi-Fi Taq Polymerase at the manufacturer’s recommended concentrations. After an initial denaturation step for 2 min at 94°C, cycling conditions were as follows: 35 cycles of 94°C for 30 s, 55°C for 30 s, and 72°C for 45 s, with a 5 min final elongation step at 72°C. Thermal cycling was performed in an Eppendorf® Mastercycler® Pro S. Amplicons of the expected sizes were excised and gel purified using a Qiagen MinElute gel extraction kit, fragments were cloned using the TOPO-TA technique and Sanger sequenced as described above.
Sequence analysis
From the HTS data, several fabavirus sequences were identified. To determine associations between RNA1 and RNA2 segments, reads were mapped to contigs at a length and similarity fraction of 0.95 and relative average coverage quantified (CLCBio, www.clcbio.com). RNA1 and RNA2 sequences of CVF and PrVF were aligned in MEGA6 using CLUSTAL W (Larkin et al., Citation2007; Tamura et al., Citation2013). In an attempt at sorting the RNA1 and RNA2 PrVF sequences from mixed samples into biologically relevant pairs, additional alignments were performed using Clone Manager 9 (Professional Edition (c) 1994–2010, Scientific and Educational Software, Cary, NC, USA) to search for any homologous regions shared between the two genomic components. The molecular weights of the deduced proteins were determined using Compute pI/Mw tool, http://web.expasy.org/compute_pi (Gasteiger et al., Citation2005). The ORF Finder program at https://www.ncbi.nlm.nih.gov/orffinder/(Rombel et al., Citation2002) was used to check for additional open reading frames of potential biological significance.
Investigations of fabavirus-associated 5ʹ ‘signature sequences’ as outlined by Ferriol et al. (Citation2015) were performed using MEGA6, which was also used for the comprehensive multiple sequence alignments used in phylogenetic analysis, as well as pairwise comparisons between isolates, using the ‘pairwise deletion’ option to handle missing data. Separate alignments focused on specific subsets of sequences or specific portions of the genome (ex. 5ʹ and 3ʹUTRs) were created for additional analyses. Accessions of related viral sequences were obtained from GenBank (www.ncbi.nlm.nih.gov). Genedoc 2.7 (Nicholas et al., Citation1997) was used to display identity shading using the default settings, to enable visual comparisons between RNA1 and RNA2 segments. Most sequences of CVF/PrVF deposited in GenBank have not been described as having RACE confirmed termini, making it difficult to distinguish between sequences that were truly ‘truncated’ in vivo as opposed to simply incomplete. As such, sequences were only included in alignments if the extent of their 5ʹ and 3ʹ termini were similar to that of the RACE confirmed sequences in GenBank and this study. Microsoft WordPad 6.0 and Apache OpenOffice 4.1.2 were used for formatting and file conversion, with Inkscape 0.91 used for figure construction.
The 3ʹUTR of some plant viruses are known to possess interesting repeat sequences (van der Vlugt et al., Citation2015), hence the 3ʹUTR of selected PrVF and CVF isolates were analysed using the Tandem Repeats Finder program (Benson, Citation1999). Selected sequences showing any evidence of repeats were analysed further by RT-PCR (see details above) to exclude the possibility of HTS data assembly artifacts. Oligonucleotide primers PrVF-A-3064F and PrVF-B-3107F () were designed for this purpose to further analyse the 3ʹUTR of PrVF RNA2 Stac-3B_c11 and the 3ʹUTR of CVF RNA2 Stac-3B_c7, respectively. The primers were designed to be specific for their respective target when combined with the universal reverse primer PrVF-3ʹambiR. The sequences of the primers used for 3ʹUTR confirmation and their locations are given in .
Phylogenetic analysis
Nucleotide substitution model testing was performed using the PhyML Smart Model Selection webserver (Lefort et al., Citation2017) available at http://www.atgc-montpellier.fr/sms/. The Bayesian Information Criterion option was used. A parallel set of model tests was also performed with the options available in MEGA6 (Tamura et al., Citation2013). Analyses were performed using MrBayes 3.2.5 (Ronquist et al. Citation2012) using 1 M generations for each 10 taxa present in the inputted multiple sequence alignment. Except where otherwise indicated, analyses of inferred protein sequences used the ‘mixed’ model option. Rambaut’s FigTree v.1.4.0 (unpublished) was used to visualize phylogram figures, which were further processed in Inkscape 0.91.
RNA folding
To identify secondary structures of possible functional significance, the Quikfold server at http://unafold.rna.albany.edu/?q=DINAMelt/Quickfold was employed, using the 2.3 version with default settings (Zuker, Citation2003). Folding tests were performed on the 5ʹ and 3ʹUTRs of the four Canadian ‘RACE-confirmed’ sequences featured in this study (Stac-3B_C3, _C4, _c11 and _c7), using the first 28 nt of the 5ʹ end, and a well-conserved 106 nt stretch in the 3ʹUTR (spanning, for reference, nt 5941 to 6046 in Stac-3B_C3). Structures of potential interest were selected on the basis of how consistently they appeared in the four individual result sets, and the original alignments were referred to in creating figures displaying nucleotide diversity. As a follow-up test, multiple sequence alignments encompassing the full PrVF/CVF sequence diversity for both RNA1 and RNA2 were analysed on the RNAalifold webserver (Bernhart et al., Citation2008) available at http://rna.tbi.univie.ac.at/cgi-bin/RNAWebSuite/RNAalifold.cgi.
Recombination analysis
Recombination analysis was performed using the Recombination Detection Program, RDP version 4 (Martin et al., Citation2015). Analyses were carried out to detect any evidence of recombination among isolates of PrVF and CVF using the default parameters, with the exception that the ‘disentangle overlapping signals’ option was selected in the general settings, and the ‘use internal and external references’ option was selected in the RDP settings. Separate analyses were performed for RNA1 segments and for RNA2 segments. The composition of the MEGA6 alignment was biased in favour of including sequences that were considered to be as complete as possible, and especially those with 5ʹ and 3ʹ termini that have been confirmed using RACE analysis as reported in the literature. Putative recombination events were investigated further using a focused subset of sequences. Recombination events identified were deemed notable if they scored a P value of 1 × 10−5 or lower in at least three of the programs used in the RDP4 suite (RDP (Martin & Rybicki, Citation2000), GENECONV (Padidam et al., Citation1999), Chimaera (Posada & Crandall, Citation2001), MaxChi (Maynard Smith, Citation1992), BOOTSCAN (Martin et al., Citation2005), SISCAN (Gibbs et al., Citation2000) and 3Seq (Lam et al., Citation2018).
Results
HTS data and coverage
After trimming, a total of 96 172 065 reads were obtained from the Stac-3B cherry sample, with 2 956 823 reads (3.07%) mapping to species in the genus Fabavirus (). The average coverage ranged from approximately 1.9 K–13.9 K reads across the RNA1 segments and 8.6 K–30 K reads for the RNA2 segments (). In the case of cherry sample Stac-4B, a total of 83 270 390 reads were obtained after trimming, with 1 997 505 reads (2.4%) mapping to species in the genus Fabavirus, with the average coverage ranging from approximately 0.7 K–16.3 K reads for the RNA1s and 1.2 K–30.4 K for the RNA2 segments ().
General genome assembly and analysis
Assembly and analysis of virus-related reads derived from the Stac-3B sample resulted in the assembly of six distinct fabavirus-associated contigs, three identified as RNA1 segments and three as RNA2 segments. In the case of the Stac-4B sample, contigs were assembled and determined to represent four fabavirus-associated RNA1 segments and three RNA2 segments (). The genome sequences are deposited in GenBank and their accession numbers are listed in .
Based on BLAST searches and pairwise comparisons of the nucleotide and the deduced aa sequences, fabavirus complexes were identified infecting both Stac-3B and Stac-4B, which included co-infections of variants/isolates of PrVF and CVF ( and ; Supplementary Table 1 and Supplementary Table 2). Analysis of the Stac-3B contigs revealed one PrVF-associated RNA1 segment and one PrVF RNA2 segment; also two CVF-associated RNA1 segments and two CVF RNA2 segments ( and ; Supplementary Table 1 and Supplementary Table 2). Analysis of Stac-4B contigs revealed two PrVF RNA1 segments but notably only one PrVF-associated RNA2 segment; also two CVF RNA1 segments and two CVF RNA2 segments ( and ; Supplementary Table 1 and Supplementary Table 2). Interestingly, higher identities were observed for the nt and deduced aa sequences of the RNA1 segments (Supplementary Table 1 and , respectively), compared with the nt and deduced aa sequences of the RNA2 segments (Supplementary Table 2 and , respectively).
Table 3. Pairwise comparisons of the deduced amino acid sequences of the RNA1 segments of the fabaviruses infecting ‘Staccato’ cherry, including also fabavirus sequences in GenBank with Prunus spp. as hosts. The fabavirus-associated sequences detected in ‘Staccato’ cherry are bolded. Sequences determined to be cherry virus F are marked with an asterisk. Numbers below the diagonal show the identities (%) for each comparison and the number of residues that differed for each comparison are shown above the diagonal. The data were obtained using MEGA6 (Tamura et al. Citation2013).
Table 4. Pairwise comparisons of the deduced amino acid sequences of the RNA2 segments of the fabavirus-associated sequences detected in ‘Staccato’ cherry, including also fabavirus sequences in GenBank with Prunus spp. as hosts. The fabavirus-associated sequences detected in ‘Staccato’ cherry are bolded. Sequences determined to be cherry virus F are marked with an asterisk. Numbers below the diagonal show the identities (%) for each comparison and the number of residues that differed for each comparison are shown above the diagonal. The data was obtained using MEGA6 (Tamura et al. Citation2013).
Analysis of RACE verified RNA segments
Based on their relative HTS read counts (), their phylogenetic relationships () and by their identity at the 3ʹ termini, the PrVF-associated RNA1 and RNA2 segments Stac-3B_C3 and Stac-3B_c11 are proposed to represent a single virus isolate identified as PrVF-CP1. Similarly, the CVF-associated RNA1 and RNA2 segments Stac-3B_C4 and Stac-3B_c7, are proposed to represent the virus isolate CVF-CC1. Stac-3B_C3 and Stac-3B_c11 are 98.2% identical at a shared 227 nt section of their 3ʹ termini, while Stac-3B_C4 and Stac-3B_c7 are 92.7% identical at their 3ʹ termini (a 234 nt section). Stac-3B_C4 is only 53.4% identical to the corresponding region of Stac-3B_c8 (234 nt section), the only other CVF-associated RNA-2 fragment detected in Stac-3B. Stac-3B_c8 was not confirmed by RACE; however the complete 3ʹ terminus was assembled using HTS reads.
Fig. 2 (Colour online) Schematic 3diagram of the genome organization of the proposed and RACE-confirmed Prunus virus F (PrVF) isolate CP1 (Stac-3B_C3 plus Stac-3B_c11) and cherry virus F (CVF) isolate CC1 (Stac-3B_C4 plus Stac-3B_c7). RNA1 and RNA2 segments are shown in blue and green, respectively, with CVF in darker shades than PrVF. Untranslated regions (UTRs) are shown in yellow. Vertical lines demarcating gene products in the coding sequences of each segment are placed at the protease cleavage sites. Abbreviations are as follows: NTP-BPP = nucleoside triphosphate binding protein peptide; RdRp = RNA dependent RNA polymerase; MP = movement protein; CP(L) = large capsid protein; CP(S) = small capsid protein. The putative genome-linked protein (VPg) is located between NTP-BPP and 2protease and is marked with an asterisk. Figure layout is adapted from Koloniuk et al. (Citation2018).
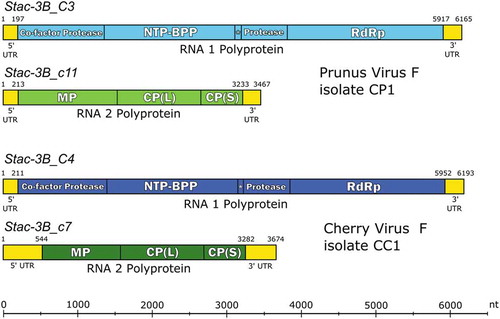
Stac-3B_C3 RNA1 is 6165 nt in size, monocistronic with 5ʹUTR and 3ʹUTR of 196 nt and 251 nt, respectively (). The ORF encodes a putative polyprotein (1906 aa residues, Mr 215.6 kDa) that is cleaved into mature functional proteins; Co-Pro (43.6 kDa), HEL (65.8 kDa), VPg (3.4 kDa), PRO (23 kDa) and the RdRp (79.9 kDa) (, ), with the signature RdRp motif, the tripeptide GDD, at aa position 1596–1598 of the polyprotein (data not shown). Stac-3B_c11 RNA2 is 3467 nt in size, monocistronic with 5ʹ and 3ʹUTRs of 212 nt and 234 nt, respectively. The ORF encodes a polyprotein (1006 aa residues, Mr 110.7 kDa) and is cleaved into mature proteins that include the movement protein (MP, 49.2 kDa), the large coat protein subunit (CP(L), 40.3 kDa) and the small coat protein subunit (CP(S), 21.3 kDa) (, ). The putative start codons (capitalized) were identified as ccuaAUGg (RNA1) and cccuAUGc (RNA2), with pyrimidines in both the −3 and +4 positions, except in the case of the RNA1 start codon which has the purine guanine (g) at the +4 position. Purines in the −3 and +4 positions relative to the start codons are optimal for initiating translation (Kozak, Citation1987; Dinesh-Kumar & Miller, Citation1993), however no other AUG triplet was identified in a suitable frame.
Table 5. Deduced proteins, predicted sizes and associated protease cleavage sites of Prunus virus F (PrVF) putative isolate CP1 (Stac-3B_C3/_c11). The corresponding values for cherry virus F (CVF) putative isolate CC1 (Stac-3B_C4/_c7) are indicated in parentheses, except in the case of the cleavage sites that are shown in separate columns. The unique CP(L)/CP(S) dipeptide cleavage site for CVF-CC1 is bolded.
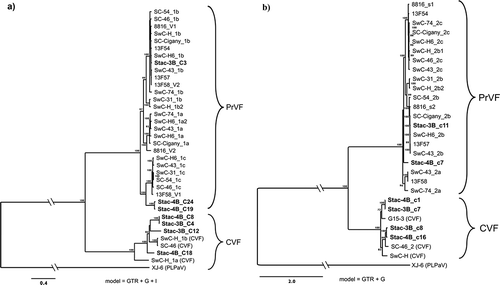
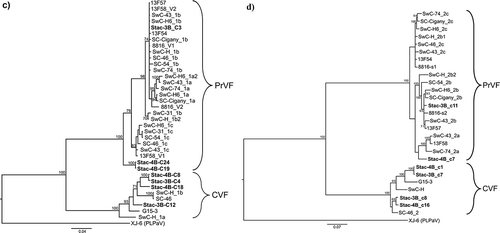
Fig. 1 Bayesian phylogenetic trees illustrating the relationships between the isolates of fabaviruses detected in sweet cherry cv. Stacatto in Canada and published sequences of fabaviruses infecting Prunus spp. Trees based on available RNA1 genome sequences (a) RNA2 genome sequences (b), deduced amino acid sequences (aa) of the Pro-Pol region (c) and the deduced aa sequences of the CP (S + L) region (d) are shown. Posterior probabilities of 70% or higher are shown at group nodes. Scale bars indicate the number of substitutions per residue.
Stac-3B_C4 RNA1 is 6193 nt in size, monocistronic with 5ʹUTR and 3ʹUTR of 210 nt and 241 nt, respectively (). The ORF encodes a polyprotein (1913 aa residues, Mr 215.3 kDa) that is cleaved into mature functional proteins that include Co-Pro (44.4 kDa), HEL (65.5 kDa), VPg (3.4 kDa), PRO (22.8 kDa) and the RdRP (79.3 kDa) (, ). Stac-3B_c7 RNA2 is 3674 nt in size, monocistronic with 5ʹUTR and 3ʹUTR of 543 nt and 395 nt, respectively. The ORF encodes a polyprotein (912 aa residues, Mr 100.2 kDa) and is cleaved into mature proteins that include the MP (37.7 kDa), CP(L) (40.4 kDa) and the CP(S) (22.1 kDa) (, ).
The putative start codons associated with CVF-CC1 were identified as uacaAUGg (RNA1) and uaucAUGu (RNA2), in good context to initiate translation with purines in the −3 and +4 positions (Kozak, Citation1987; Dinesh-Kumar & Miller, Citation1993), except in the case of the RNA2 start codon which has the pyrimidine uracil (u) in the +4 position. The putative dipeptide proteolytic cleavage sites are similar to those described by Koloniuk et al. (Citation2018), except the putative cleavage site at the CP(L)-CP(S) junction was determined to be Q/F. This is different from other PrVF isolates, isolates of CVF (Villamor et al., Citation2017; Koloniuk et al., Citation2018) and PLPaV (He et al., Citation2017).
Untranslated regions (UTRs)
The 5ʹUTRs of the RNA1 and RNA2 of PrVF-CP1 (196 nt and 212 nt, respectively, ) are 61% identical; however in both cases they share a conserved 28 nt region at the 5ʹ-terminus of both sequences. The 5ʹUTR of the RNA1 and RNA2 of CVF-CC1 (210 nt and 543 nt, respectively) are 32% identical, with the first 42 nt conserved among both sequences (98% identity). Interestingly, the first 28 nt at the 5ʹ terminus of all four RNA segments are conserved (93–100% identical) and are predicted to form similar stem-loop or hairpin structures (), similar also to the initial 5ʹ hairpin of the predicted secondary structure of the 5ʹUTR of both the RNA1 and RNA2 of PLPaV (He et al., Citation2017). Mutations on one side of the stem were accompanied by complementary ‘co-variations’ on the other side that maintained the predicted structure (). As with the case of PrVF, the 5ʹUTRs of CVF RNA1 and RNA2 show copies of the fabavirus ‘signature sequence’ (Ferriol et al., Citation2015). One copy that is well-conserved between isolates, with sequence AACCGCUUUC, can be found at nucleotides 18–27 (using sequence Stac-3B_C4 as reference). At least two possible additional copies are present (at nt 37–46 and 52–61 of the same sequence), but are highly divergent between CVF isolates, especially in the first four positions of the signature sequence.
Fig. 3 (Colour online) Predicted secondary structures for: a highly conserved 28 nucleotide (nt) region at the proximal end of the 5ʹUTR (a) and a 106 nt region that is conserved among the 3ʹUTR (b) of the RNA1 and RNA2 of the putative Canadian virus isolates PrVF-CP1 and CVF-CC1 that were RACE-confirmed. Variable nucleotide positions are identified where a single bar above and a double bar below indicate that the mutation occurs in CVF RNA1 and CVF RNA2, respectively. An asterisk indicates a wobble G-U pairing that may not occur in all isolates considered and the nt in parentheses indicates a mutation identified in PrVF-CP1 RNA2.
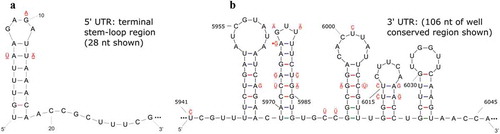
The 3ʹUTRs of the RNA1 and RNA2 of PrVF-CP1 (248 nt and 234 nt, respectively) are 87% identical, but the final 226 nt of both segments are conserved and are approximately 99% identical. The 3ʹUTRs of the RNA1 and RNA2 of CVF-CC1 (241 nt and 392 nt, respectively) are 86% identical, with the last 55 nt of both segments conserved (approximately 96% identity). A highly conserved region within the 3ʹUTRs of the RNA1 and RNA2 of both PrVF-CP1and CVF-CC1 and consisting of 106 nt was selected for folding analysis (). The predicted secondary structure was similar for corresponding regions of the RNA1 and RNA2 segments of the proposed PrVF-CP1 and CVF-CC1 (). Mutations observed were accompanied by complementary ‘co-variations’ that served to maintain the predicted hairpin structures, and in a few cases putative G-U wobble base pairings were identified ().
Using the RACE confirmed 5ʹ and 3ʹUTRs of Stac-3B_C3 (RNA1) and Stac-3B_c11 (RNA2) sequences as references (proposed PrVF-CP1) it is clear that universally among both PrVF and CVF isolates the 3ʹUTRs have greater stretches of sequences that are highly conserved (100%), compared with the 5ʹUTR (S3). This may reflect the shared and important functional significance of the 3ʹUTR.
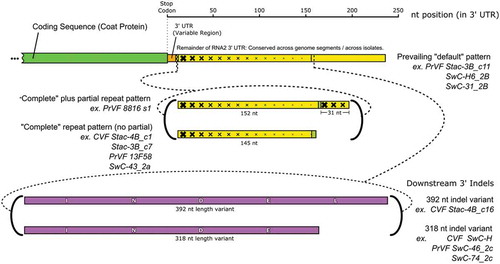
In general, the 3ʹUTRs of the RNA2 segments of both PrVF and CVF are characterized by repeats, ‘partial’ repeats and putative indel events (Supplementary and ). There appears to be a more common or prevailing pattern among the 3ʹUTRs of the Prunus-infecting fabaviruses. For the purposes of this study, we have identified this pattern as the ‘default’ pattern, with three PrVF isolates given as examples of this pattern (). Full repeats, additionally sometimes with partial repeats, were identified and a suitable example, PrVF 8816_s1, is given in . Analysis using the Tandem Repeats Finder program (Benson, Citation1999) revealed a stretch of approximately 150 nt that appear to be present as two adjacent copies, 95% identical. In the case of the RNA2 3ʹUTR region of Stac-3B_c7 (a CVF segment), the two copies occupy a region stretching from nt 3299 to 3598 (). Several isolates appear to have this ‘full repeat’ pattern and include isolates of both CVF and PrVF. Interestingly, a co-infecting PrVF isolate Stac-3B_c11 lacks this repeat pattern (). Additionally, downstream of the region characterized by repeats, several RNA2 3ʹUTR possess what appears to be large indel events that contribute to 3ʹUTRs of large and variable sizes (). Indel events of 392 nt and 318 nt are shown in (in magenta colour). In some cases, the repeat patterns were accompanied by short sequences that have no counterpart in the default sequences, therefore of unknown origin (, brownish-green colour).
Fig. 4 (Colour online) Schematic diagram showing the variability that was identified in the 3ʹUTR of RNA2 for cherry virus F (CVF) and Prunus virus F (PrVF). The location of partial CP coding sequence and the conserved region of the 3ʹ UTR are shown in green and yellow, respectively. Graduated crosshatching is included to illustrate the nature of repeats found in the sequences of various PrVF isolates, focussing on PrVF isolate 8816_s2. The upstream variable region of the 3ʹ UTR is shown in orange. The large downstream indels are shown in purple, labelled INDEL (392 nt) and INDE (truncated, 318 nt). Short olive-coloured sections are associated with the repeat sequence and do not seem to have a counterpart in the prevailing default pattern. The two locations in the 3ʹUTR at which the known ‘non-default’ features occur are indicated with dashed lines. At both of these locations, multiple variants have been seen. These are indicated with bold curved brackets, and representative isolates of both CVF and PrVF bearing these features are listed as examples.
Phylogenetic analysis
Phylogenetic analysis was carried out based on available and corresponding RNA1 and RNA2 genome sequences (nt) of fabaviruses infecting Prunus spp., the deduced aa sequences of RNA1 and RNA2 polyproteins, the deduced aa sequences of the Pro-Pol region (the region between the CG motif of the protease and the GDD motif of the polymerase) and the deduced CP (L) + (S) aa sequences. The relationships observed for the various analyses were consistent. show analyses based on available RNA1 genome sequences (nt), RNA2 nt sequences, the deduced aa sequences of the Pro-Pol region and the deduced aa sequences of CP (L) + (S), respectively. PLPaV is used as an outgroup as it is phylogenetically distinct from PrVP and CVF (He et al., Citation2017; Koloniuk et al., Citation2018).
RNA1 segments (nt and Pro-Pol deduced aa) of isolates of PrVF form two distinct clades (, respectively), with Canadian, American and European isolates represented in both clades and showing no obvious geographic grouping. These relationships are supported by high posterior probabilities. Four Canadian RNA1 segments (2 Stac-3B and 2 Stac-4B) group as CVF isolates that are phylogenetically distinct from isolates of PrVF (). The RNA2 segments (nt) and the deduced aa sequences of CP (L) + (S) appear to form two major clades (), with one major clade separating into two subclades (more distinct in analysis based on the deduced aa sequences of CP (L) + (S)). The Canadian RNA2 segments (Stac-3B_c11 and Stac-4B) group cluster one each into the two subclades (). The CVF-associated RNA2 sequences are phylogenetically distinct from the corresponding PrVF sequences. Two Stac-3B and two Stac-4B RNA2 segments were identified as CVF-associated segments (). It is interesting to note that among the PrVF and CVF isolates in these analyses phylogenetic distribution does not appear to be influenced by host or region of origin.
Recombination analysis
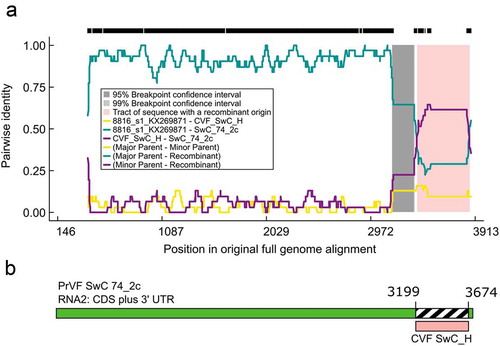
Putative recombination events were detected in both RNA1 and RNA2 segments and in some cases were inconclusive and/or not well supported. However, we were able to confirm PrVF 13F28_v1 (RNA1) as a recombinant as described by Koloniuk et al. (Citation2018). A phylogenetically related group of six PrVF RNA2 segments were identified as recombinants. These include SwC-74_2c, SC-Cigany_2c, SwC-H6_2c, SwC-H_2b1, SwC-46_2c and SwC-43_2c (). Using SwC_74_2c as a representative of this group, a clear and well supported recombination event was identified, extending from the 3ʹ section of the CP-S coding region (nt 3199) to the 3ʹUTR (nt 3674), with PrVF 8816_s1RNA2 identified as the major parent and CVF SwC_H RNA2 as the minor parent (, ). This recombination event was supported by all methods within the RDP4 program (). Of the six putative recombinant PrVF RNA2 segments, five were detected in sweet cherry and one in sour cherry. The sequences included for analysis in our study that resulted in the identification of the SwC_74_2c ‘group’ of recombinants included the five sequences considered by Koloniuk et al. (Citation2018), with the addition of CVF SwC_H.
Table 6. RDP4 results following the analysis of Prunus virus F (PrVF) isolate SwC 74_2c indicating that it is a recombinant with PrVF 8816_s1 as the major parent and cherry virus F isolate SwC_H as the minor parent. This was supported by all seven methods used for screening. The P values obtained and the references for the individual methods are provided.
Fig. 5 (Colour online) Schematic representation of a putative recombination event detected in some Prunus virus F (PrVF) isolates with SwC 74_2c as a representative and showing PrVF 8816_s1 as the major parent and cherry virus F (CVF) isolate SwC_H as the minor parent, illustrated using an RDP Plot (a) and a schematic of the SwC 74_2c genome showing the recombination event at the 3ʹ end of the virus (b). The 5ʹ termini were excluded from the multiple sequence alignment for ease of RDP4 analysis. The numbers on the RDP Plot and the genome map indicate nucleotide positions (nt) and the putative nt positions of recombination breakpoints, respectively.
Discussion
The symptomatic cherry tree Stac-3B and the asymptomatic cherry tree Stac-4B were found to be infected with fabavirus complexes that included phylogenetically distinct RNA1 and RNA2 segments of PrVF and CVF. Stac-3B was infected with one PrVF RNA1 segment and one PrVF RNA2 segment, assumed to represent a single PrVF isolate, tentatively named PrVF-CP1. Stac-3B was found to be infected also with two phylogenetically distinct CVF RNA1 segments and two CVF RNA2 segments, with 3B_C4 and 3B_c7 assumed to represent a single virus isolate tentatively identified as CVF-CC1. It was determined that the asymptomatic cherry Stac-4B was infected with two closely related PrVF RNA1 segments and one PrVF RNA2 segment; and also with two distinct CVF RNA1 and RNA2 segments. It is indeed possible that the two PrVF RNA1 segments detected in Stac-4B are variants of a single isolate, with the single 4B RNA2 segment (4B_c7) as its counterpart. The proposed RNA1/RNA2 combinations representing putative virus isolates are supported by relative HTS read counts, phylogenetic relationships and a region of over 200 nt at the 3ʹ terminus of the associated RNA1/RNA2 segments that is conserved (over 92% identity).
Complexes of fabaviruses infecting sweet and sour cherry (P. avium and P. cerasus, respectively) were described previously. Villamor et al. (Citation2017) described complexes that consisted only of isolates/variants of PrVF. Later Koloniuk et al. (Citation2018) described fabavirus complexes that consisted only of isolates/variants of PrVF in some samples and of complexes of both PrVF and CVF in other samples. Koloniuk et al. (Citation2018) were the first to detect and describe CVF. In the case of the only other Prunus sp. described as a fabavirus host, He et al. (Citation2017) described co-infection of peach (P. persica) with pathogens that included two viruses (ACLSV and PBNSPaV), two viroids (HSVd and PLMVd) and the novel fabavirus PLPaV. It seems that no complex of fabaviruses was detected in peach. Koloniuk et al. (Citation2018) speculated that these complexes or mixtures of variants might be linked to unknown epidemiological features. Fabaviruses are aphid transmissible (Thompson et al., Citation2017), but it seems that some viruses in the family Secoviridae (fabaviruses are members of this family) may be transmissible by seed and/or pollen (Sanfaçon et al., Citation2011). Therefore, the existence of these fabavirus complexes in cherry is not surprising. Commercially cultivated cherry trees are usually combinations of rootstock and scion. The use of either as undetected infected germplasm material can lead to co-infections or virus complexes (James et al., Citation2017b). Another factor that will influence the occurrence of mixed infections is tree location, relative to other plants or hosts that may or may not be infected with fabaviruses or other pollen/seed/insect transmissible pathogens.
Despite the seemingly common occurrence of infections involving complexes of fabaviruses in cherry (Villamor et al., Citation2017; Koloniuk et al., Citation2018; this study), only two well supported recombination events are described. Koloniuk et al. (Citation2018) identified PrVF 13F28_v1 (RNA1) as a recombinant segment with the recombination event in the protease coding region; and also a recombination event in the 3ʹUTR of 8816_s1 (RNA2), but in this case the minor parent was not clearly determined. Only one parent and the recombinant need to be in an alignment for the detection of a recombination event (Martin et al., Citation2015). Interestingly, it seems that Koloniuk et al. (Citation2018) did not include any isolates of CVF in their recombination analyses. We were able to confirm that 8816_s1 (RNA2) or an ancestor with similar sequence was indeed likely involved in a recombination event although it seems more likely to be the major parent rather than the actual recombinant, with some ancestral CVF isolate as the minor parent, contributing a portion of sequence that includes a variant of the ‘downstream indel’ identified in the 3ʹUTR. This recombination event was supported by all seven methods in the RDP4 recombination detection program. Six PrVF RNA2 sequences possess what is likely a single event. Five were detected in sweet cherry and one in sour cherry, suggesting that virus transmissibility is not affected by this event. Since co-infections of PrVF and CVF do occur (Koloniuk et al., Citation2018; this study) the possibility of interspecies recombination cannot be excluded. Recombination is likely the main mechanism contributing to the variation in size seen among the RNA2 3ʹUTRs. The presence/absence pattern observed in some isolates/variants can only be explained by at least two or possibly three events that may include both recombination and deletion events. Given that the outgroup PLPaV seems to contain partially homologous sequences approximately the same size as the indel and in the same location, it seems that the original source for this sequence block may be an ancestor of some as yet undiscovered fabavirus.
Bipartite RNA viruses often have 3ʹUTRs that are characterized by sequence homology between RNA segments and the occurrence of repeat sequences (van der Vlugt et al., Citation2015). Both phenomena were observed in fabaviruses analysed in this study. Additionally, putative ‘partial’ repeat sequences and also novel sequences were identified that did not appear to originate from the proposed default sequence and may be the result of recombination events or perhaps error-prone replication events. When datasets contain several recombinant sequences it is difficult for RDP4 to accurately identify recombination events (Martin et al., Citation2015), which may be a factor contributing to the low number of events identified and the differences in size/location of the recombination event described (Koloniuk et al., Citation2018; this study). Reliable detection of recombination events is influenced also by genetic diversity (James et al., Citation2016).
Use of HTS/next-generation sequencing as a diagnostic tool for virus detection (Boonham et al., Citation2014; Massart et al., Citation2014) may reveal a greater prevalence of co-infections than was described previously. These virus/viroid complexes may include multiple virus/viroid species and/or multiple variants/isolates of a single species. This may have implications for disease aetiology and symptomatology (James et al., Citation2017b), such as increased severity or alternatively the modulation of symptoms. RNA viruses usually exist as genetically diverse populations (Roossinck, Citation2003) due to frequent mis-incorporation of nucleotides by RdRp (Korboukh et al., Citation2014). This may result in viral quasispecies defined as closely related viral genomes undergoing a process of variation, competition and selection (Domingo et al., Citation2012). Insect- and pollen transmissible RNA viruses add another level of complexity to understanding the variability observed and the genesis of the composition of virus populations. There is no easily identifiable difference between mutations representing quasispecies-level variation of a single viral population and those that separate two or more overlapping but operationally distinct populations, also complexes that may result from transmission events. Better understanding is required of the virus replication process in the case of bipartite viruses, for example, where seemingly uneven numbers of RNA1 and RNA2 segments exist in a plant (Koloniuk et al., Citation2018; this study), differentiating mixed infections from re-assortment, also determining cellular and systemic distribution of genotypes. It is possible that different combinations of RNA segments/fabaviruses might have different biological properties. Different virus combinations are known to influence virus replication and disease symptoms (Gil-Salas et al., Citation2011). Also, spatiotemporal separation of RNA virus populations in plant cells and/or tissues is known to occur (Dietrich & Maiss, Citation2003; Tromas et al., Citation2014). This has important implications for virus evolution and for reliable diagnostics as HTS analysis at different times or using different samples may give different virus profiles/results. The available data seem to suggest that fabavirus complexes are relatively common. Competition is known to occur within some mixed virus populations (Carrasco et al., Citation2007; Predajna et al., Citation2012). It would be interesting to understand the mechanisms of replication and how these mixed fabavirus virus populations, either of the same species or multiple species, are maintained and if over time any change in the composition of the virus populations occurs. The fact that we have identified an interspecies recombination event suggests that simultaneous infection of individual cells by PrVF and CVF is possible.
In summary, this is the first description of the detection of CVF in Canada and a well-supported interspecies recombination event between PrVF and CVF was identified. As yet there has been no definitive association of fabaviruses detected in cherry with any disease symptoms (Šafářová et al., Citation2017; Villamor et al., Citation2017; James et al., Citation2018; Koloniuk et al., Citation2018). Their presence may influence symptom severity of known harmful viruses (James et al., Citation2017a), but as yet this has not been demonstrated. Also, the fact that these viruses may be insect/pollen/seed transmitted (Sanfaçon et al., Citation2011; Thompson et al., Citation2017) means that cherry trees can serve as a reservoir for these viruses that may or may not affect other plants in the vicinity. Based, for example, on the HTS detection by Al Rwahnih et al. (Citation2016) of a novel putative fabavirus infecting grapevine, it is clear that fabaviruses have a greater host range than was suspected previously.
Supplemental Material
Download MS Word (193.2 KB)Acknowledgements
We wish to express our gratitude to Gayle Jesperson, Ministry of Agriculture, Kelowna, British Columbia, Canada for kindly providing samples of sweet cherry (cv. Staccato).
Supplementary material
Supplemental data for this article can be accessed online here: https://doi.org/10.1080/07060661.2019.1566179.
Additional information
Funding
Related Research Data
References
- Al Rwahnih M, Alabi OJ, Westrick NM, Golino D, Rowhani A. 2016. Near-complete genome sequence of grapevine fabavirus, a novel putative member of the genus Fabavirus. Genome Announc. 4:e00703–16.
- Benson G. 1999. Tandem repeats finder: a program to analyze DNA sequences. Nuc Acids Res. 27:573–580.
- Bernhart SH, Hofacker IL, Will S, Gruber AR, Stadler PF. 2008. RNAalifold: improved consensus structure prediction for RNA alignments. BMC Bioinformatics. 9:474.
- Boonham N, Kreuze J, Winter S, van der Vlugt R, Bergervoet J, Tomlinson J, Mumford R. 2014. Methods in virus diagnostics: from ELISA to next generation sequencing. Virus Res. 186:20–31.
- Byrne DH. 2005. Trends in stone fruit cultivar development. HortTechnology. 15:494–500.
- Carrasco P, Daros JA, Agudelo-Romero P, Elena SF. 2007. A real-time RT-PCR assay for quantifying the fitness of tobacco etch virus in competition experiments. J Virol Methods. 139:181–188.
- Dietrich C, Maiss E. 2003. Fluorescent labelling reveals spatial separation of potyvirus populations in mixed infected Nicotiana benthamiana plants. J Gen Virol. 84:2871–2876.
- Dinesh-Kumar SP, Miller WA. 1993. Control of start codon choice on a plant viral RNA encoding overlapping genes. Plant Cell. 5:679–692.
- Domingo E, Sheldon J, Perales J. 2012. Viral quasispecies evolution. Microbiol Mol Biol Rev. 76:159–216.
- Dong S-W, Xiang H-Y, Shang Q-X, Li W-D, Yu J-L, Han C-G. 2012. Complete genomic sequence analysis reveals a novel fabavirus infecting curcubits in China. Arch Virol. 157:597–600.
- Ferriol I, Ferrer RM, Luis-Arteaga M, Guerri J, Moreno P, Rubio L. 2014. Genetic variability and evolution of broad bean wilt virus 1: role of recombination, selection and gene flow. Arch Virol. 159:779–784.
- Ferriol I, Rangel EA, Panno S, Davino S, Han C, Olmos A, Rubio L. 2015. Rapid detection and discrimination of fabaviruses by flow‐through hybridisation with genus‐ andspecies‐specific riboprobes. Ann Appl Biol. 167:26–35.
- Gasteiger E, Hoogland C, Gattiker A, Duvaud S, Wilkins MR, Appel RD, Bairoch A. 2005. Protein identification and analysis tools on the ExPASy server. In: Walker JM, editor. The proteomics protocol handbook. Totowa (New Jersey): Humana Press; p. 571–607.
- Gibbs MJ, Armstrong JS, Gibbs AJ. 2000. Sister-scanning: A Monte Carlo procedure for assessing signals in recombinant sequences. Bioinformatics. 16:573–582.
- Gil-Salas FM, Peters J, Boonham N, Cuadrado IM, Janssen D. 2011. Yellowing disease in zucchini squash produced by mixed infections of Cucurbit yellow stunting disorder virus and Cucumber yellow veining virus. Phytopathology. 101:1365–1372.
- He Y, Cai L, Zhou L, Yang Z, Hong N, Wang G, Li S, Xu W. 2017. Deep sequencing reveals the first fabavirus infecting peach. Sci Rep. 7:11329.
- James D, Cieslinska M, Pallas V, Flores R, Candresse T, Jelkmann W. 2017a. Viruses, viroids, phytoplasmas and genetic disorders of cherry. In: Quero-Garcia JQ, Iezzoni A, Pulawska J, Lang G, editors. Cherries: botany, production and uses. Wallingford (UK): CABI Publishing; p. 386–419.
- James D, Phelan J, Jesperson G 2017b. The Geneva complex – an interesting disease perspective based on the results of NGS analysis. Proceedings of the 24th International Conference on Virus and Other Graft Transmissible Diseases of Fruit Crops (ICVF) Thessaloniki, Greece; 43.
- James D, Phelan J, Jesperson G. 2018. First report of Prunus virus F infecting sweet cherry (Prunus avium cv. ‘StaccatoTM’) in Canada. Plant Dis. 102:1468.
- James D, Sanderson D, Varga A, Sheveleva A, Chirkov S. 2016. Genome sequence analysis of new isolates of the Winona strain of Plum pox virus and the first definitive evidence of intrastrain recombination events. Phytopathology. 106:407–416.
- Kalinowska E, Chodorska M, Paduch-Cichal E, Mroczkowska K. 2012. An improved method for RNA isolation from plants using commercial extraction kits. Acta Biochim Polon. 59:391–393.
- Koloniuk I, Sarkisova T, Petrzik K, Lenz O, Přibylová J, Fránová J, Špak J, Lotos L, Beta C, Katsiani A, et al. 2018. Variability studies of two Prunus-infecting fabaviruses with the aid of high-throughput sequencing. Viruses. 10:204.
- Korboukh VK, Lee CA, Acevedo A, Vignizzi M, Xiao Y, Arnold JJ, Hemperly S, Graci JD, August A, Andino R, et al. 2014. RNA virus population diversity, an optimum for maximal fitness and virulence. J Biol Chem. 289:29531–29544.
- Kozak M. 1987. Possible role of flanking nucleotides in recognition of the AUG initiation codon by eukaryotic ribosomes. Nucleic Acids Res. 15:8125–8148.
- Lam HM, Ratmann O, Boni MF. 2018. Improved algorithmic complexity for the 3SEQ recombination detection algorithm. Mol Biol Evol. 35:247–251.
- Larkin MA, Blackshields G, Brown NP, Chenna R, McGettigan PA, McWilliam H, Valentin F, Wallace IM, Wilm A, Lopez R, et al. 2007. ClustalW and ClustalX version 2. Bioinformatics. 23:2947–2948.
- Lefort V, Longueville J-E, Gascuel O. 2017. SMS: smart model selection in PhyML. Mol Biol Evol. 34:2422–2424.
- Martin DP, Murrell B, Golden M, Khoosal A, Muhire B. 2015. RDP4: detection and analysis of recombination patterns in virus genomes. Virus Evol. 1:1–5.
- Martin DP, Posada D, Crandall KA, Williamson C. 2005. A modified bootscan algorithm for automated identification of recombinant sequences and recombination breakpoints. AIDS Res Hum Retroviruses. 21:98–102.
- Martin DP, Rybicki E. 2000. RDP: detection of recombination amongst aligned sequences. Bioinformatics. 16:562–563.
- Massart S, Olmos A, Jijakli H, Candresse T. 2014. Current impact and future directions of high throughput sequencing in plant virus diagnostics. Virus Res. 188:90–96.
- Messmer A, Sanderson D, Braun G, Serra P, Flores R, James D. 2017. Molecular and phylogenetic identification of unique isolates of hammerhead viroid-like RNA from ‘Pacific Gala’ apple (Malus domestica) in Canada. Can J Plant Pathol. 39:342–353.
- Nicholas KB, Nicholas HB, Deerfield DW. 1997. GeneDoc: analysis and visualization of genetic variation. Embnew News. 4:14.
- Padidam M, Sawyer S, Fauquet CM. 1999. Possible emergence of new geminiviruses by frequent recombination. Virology. 265:218–225.
- Posada D, Crandall KA. 2001. Evaluation of methods for detecting recombination from DNA sequences: computer simulations. Proc Natl Acad Sci USA. 98:13757–13762.
- Predajna L, Subr Z, Candresse T, Glasa M. 2012. Evaluation of the genetic diversity of Plum pox virus in a single plum tree. Virus Res. 167:112–117.
- Rombel IT, Sykes KF, Rayner S, Johnston SA. 2002. ORF-FINDER: a vector for high- throughput gene identification. Gene. 282:33–41.
- Ronquist F, Teslenko M, van der Mark P, Ayres DL, Darling A, Höhna S, Larget B, Liu L, Suchard MA, Huelsenbeck JP. 2012. MrBayes 3.2: efficient Bayesian phylogenetic inference and model choice across a large model space. Syst Biol. 61:539–542.
- Roossinck M. 2003. Plant RNA virus evolution. Curr Opin Microbiol. 6:406–409.
- Šafářová D, Faure C, Marais A, Suchá J, Paprštein F, Navrátil M, Candresse T. 2017. First report of Prunus virus F infecting sour cherry in the Czech Republic. Plant Dis. 101:1828.
- Sanfaçon H, Iwanami T, Karasev AV, van der Vlugt R, Wellink J, Wetzel T, Yoshikawa N. 2011. Secoviridae. In: King AMQ, Adams MJ, Carstens EB, Lefkowitz EJ, editors. Virus Taxonomy, ninth report of the international committee on taxonomy of viruses. London: Elsevier Academic Press; p. 881–899.
- Smith JM. 1992. Analyzing the mosaic structure of genes. J Mol Evol. 34:126–129.
- Tamura K, Stecher G, Peterson D, Filipski A, Kumar S. 2013. MEGA6: molecular evolutionary genetics analysis version 6.0. Mol Biol Evol. 30:2725–2729.
- Thompson JR, Dasgupta I, Fuchs M, Iwanami T, Karasev AV, Petrzik K, Sanfaçon H, Tzanetakis I, van der Vlugt R, Wetzel T, et al. 2017. ICTV Virus taxonomy profile: secoviridae. J Gen Virol. 98:529–531.
- Tromas N, Zwart MP, Lafforgue G, Elena SF. 2014. Within-host spatiotemporal dynamics of plant virus infection at the cellular level. PLoS Genet. 10:e1004186.
- van der Vlugt RAA, Verbeek M, Dullemans AM, Wintermantel WM, Cuellar WJ, Fox A, Thompson JR. 2015. Torradoviruses. Annu Rev Phytopathol. 53:485–512.
- Villamor DEV, Pillai SS, Eastwell KC. 2017. High throughput sequencing reveals a novel fabavirus infecting sweet cherry. Arch Virol. 162:811–816.
- Zuker M. 2003. Mfold web server for nucleic acid folding and hybridization prediction. Nucl Acids Res. 31:3406–3415.