
Abstract
Cercospora beticola causes cercospora leaf spot (CLS) on sugar beet and table beet. Accurate identification of this pathogen is critical to disease diagnosis and effective research outcomes for improved management. Several PCR assays have been described for identifying C. beticola; however, the specificity has either not been adequately tested or cross-reactions with related Cercospora species have occurred. Comparison of the three published assays for specificity to C. beticola indicated that each also amplified DNA from closely related Cercospora species. This study describes the development and subsequent assessment of conventional and quantitative PCR assays specific for detection of C. beticola. Assay specificity was confirmed across a broad range of Cercospora and other common fungal species using public DNA sequence databases and PCR. A conventional PCR assay was designed with fast PCR conditions and completed in under 40 min. The quantitative PCR assay detected 0.001–10 ng of C. beticola DNA. The effectiveness of the quantitative PCR assay to detect C. beticola DNA in diseased leaf tissue and diseased leaf tissue mixed with soil was also demonstrated. These assays provide an improved method for specific identification and quantification of C. beticola, and a valuable tool for enhancing studies into the biology of C. beticola and epidemiology of CLS.
Résumé
Cercospora beticola cause la cercosporose chez la betterave et la betterave sucrière. Afin d’améliorer la gestion de cette maladie, l’identification précise de l’agent pathogène est essentielle au diagnostic et à l’obtention de résultats de recherche concrets. Plusieurs analyses visant à identifier C. beticola par PCR ont été décrites. Toutefois, ou bien la spécificité n’a pas été adéquatement testée ou des réactions croisées avec des espèces parentes de Cercospora se sont produites. Une comparaison des trois analyses publiées relatives à la spécificité de C. beticola a indiqué que chacune de ces analyses amplifiait également l’ADN d’espèces de Cercospora étroitement apparentées. Cette étude décrit la conception et l’évaluation subséquente des analyses par PCR, traditionnelle et quantitative, spécifique de la détection de C. beticola. La spécificité des analyses a été confirmée quant à un large éventail de Cercospora et d’autres espèces fongiques courantes à l’aide de bases de données publiques de séquences d’ADN et de la PCR. Une analyse traditionnelle par PCR a été conçue en fonction de la rapidité d’exécution et s’est déroulée en moins de 40 minutes. L’analyse par PCR quantitative a détecté de 0,001 à 10 ng d’ADN de C. beticola. L’efficacité de la PCR quantitative quant à la détection de l’ADN de C. beticola dans les tissus infectés de feuilles et dans les tissus infectés de feuilles mélangés à de la terre a également été démontrée. Ces analyses fournissent une méthode améliorée pour identifier et quantifier spécifiquement C. beticola, et constituent un précieux outil pour parfaire les études sur la biologie de l’agent ainsi que sur l’épidémiologie de la cercosporose.
Introduction
Accurate identification and detection of plant pathogens is essential for the development of robust disease management strategies and control tactics. In the genus Cercospora, in vitro and in vivo morphological characteristics have traditionally been used to describe and identify species. However, these methods have limitations due to the similarity between species (Groenewald et al. Citation2005; Cai et al. Citation2011; Bakhshi et al. Citation2015). Host range has also been used to distinguish between certain plant pathogenic species within Cercospora (Chupp Citation1954). A taxonomic revolution based upon DNA sequencing of multiple, phylogenetically informative genes has demonstrated the limitations of relying solely on morphology and host range for species characterization (Crous et al. Citation2015). This has led to detailed phylogenetic studies which have identified gene sequences that are informative for differentiating species within Cercospora (Bakhshi et al. Citation2018; Crous et al. Citation2000; Goodwin et al. Citation2001; Groenewald et al. Citation2006; Vaghefi et al. Citation2018).
Fig. 3 Lesion diameter and Cercospora beticola biomass over 21 days on inoculated ‘Ruby Queen’ table beet leaves in two replicated experiments (a and b). Means (n = 10) for adjacent time points were compared using a Wilcoxon signed rank test for lesion diameters and a paired t-test for C. beticola biomass (log [x + 1] transformed). Bars represent the standard error, an asterisk indicates a significant difference in lesion diameters between time points and a star indicates a significant difference in C. beticola biomass between time points (P < 0.05).
![Fig. 3 Lesion diameter and Cercospora beticola biomass over 21 days on inoculated ‘Ruby Queen’ table beet leaves in two replicated experiments (a and b). Means (n = 10) for adjacent time points were compared using a Wilcoxon signed rank test for lesion diameters and a paired t-test for C. beticola biomass (log [x + 1] transformed). Bars represent the standard error, an asterisk indicates a significant difference in lesion diameters between time points and a star indicates a significant difference in C. beticola biomass between time points (P < 0.05).](/cms/asset/c30b6f18-625b-4c94-97ca-3133830e321e/tcjp_a_1621380_f0003_b.gif)
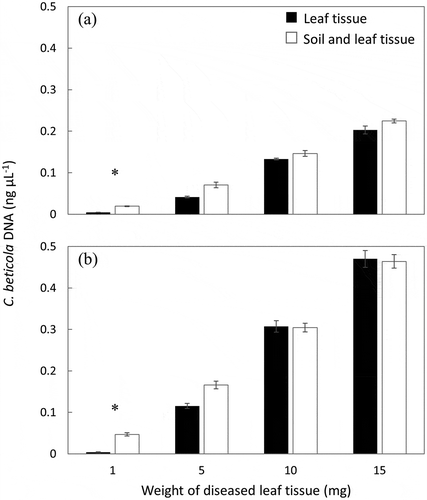
Fig. 4 Comparison of Cercospora beticola DNA concentrations in four weights of ‘Ruby Queen’ table beet leaf tissue inoculated with C. beticola isolates (a) Tb14-081 (ICMP 21691) and (b) Tb14-085 (ICMP 21692). Each weight was assessed in triplicate as either leaf tissue or leaf tissue mixed with pasteurized soil to a total of 100 mg. Bars represent the standard error, and an asterisk indicates a significant difference between treatments (P < 0.01).
Cercospora beticola, the cause of the destructive disease cercospora leaf spot (CLS) on sugar beet and table beet (Beta vulgaris ssp. vulgaris) (Weiland and Koch Citation2004), has undergone extensive DNA sequence-based phylogenetic analysis (Groenewald et al. Citation2005, Citation2006; Vaghefi et al. Citation2018). These studies have estimated the relationship of C. beticola to other closely related Cercospora species using a set of five partial gene sequences, including the internal transcribed spacers and 5.8S rRNA, actin, translation elongation factor 1-α, calmodulin and histone H3, and provided consistent evidence for its distinction as a species. Groenewald et al. (Citation2005) identified sequences of the calmodulin gene as phylogenetically informative for distinguishing between C. beticola, C. apii and 14 other Cercospora species, leading to the design of species-specific primers based on this gene.
Species-specific identification of C. beticola using PCR of actin and calmodulin gene sequences has been described in several studies (Lartey et al. Citation2003; Groenewald et al. Citation2005; De Coninck et al. Citation2012). However, while the interpretation of these studies indicated species-specificity of the primers, this has been questioned in subsequent studies (Bakhshi et al. Citation2013; Vaghefi et al. Citation2017b). Bakhshi et al. (Citation2013) does not provide information on the Cercospora species detected by the CBACTIN959L and CBACTIN949R primers in addition to C. beticola. Moreover, Vaghefi et al. (Citation2017b) demonstrated that PCR of C. chenopodii DNA using calmodulin sequence based CercoCal-beta and CercoCal-R primers (Groenewald et al. Citation2005) amplified fragments similar in size to those reported for C. beticola.
Quantitative PCR (qPCR) is an important research tool for detection of genomic DNA of plant pathogens (Schaad and Frederick Citation2002), and has allowed the quantification of pathogen growth in plants, along with providing supporting evidence of host resistance (De Coninck et al. Citation2012; Knight et al. Citation2012). qPCR has also been used to quantify inoculum density in soil and inform management strategies for soil-borne diseases (Ophel-Keller et al. Citation2008). Experimental methods may utilize broad-range or species-specific assays, depending on the research focus. For example, artificial inoculations may not encounter external pathogens, thus specificity may not be required. Field studies have aimed to simultaneously detect a range of similar species in a disease complex based on shared toxin related sequences (Strausbaugh et al. Citation2005) or attempted to quantify one or more specific pathogen species (Knight and Sutherland Citation2017; Abdullah et al. Citation2018). The qPCR assay reported by De Coninck et al. (Citation2012) compared quantities of C. beticola in leaf tissues of sugar beet cultivars in artificially inoculated trials; however, species-specificity testing in assay design was not described and would require further assessment prior to field-based application. Accuracy of species-specific assays is essential to achieve research outcomes to facilitate effective disease management decisions.
The ability to specifically target polymorphic sequences between species is reliant on the inclusion of relevant species and isolates in DNA databases, the robustness of the primer design method and the availability of relevant culture collections. For studies using these assays, it is imperative to validate published species-specific primers in respect to the rapid flux in species concepts, available sequence data and local environments. The aim of this study was to design an improved assay for detection and quantification of C. beticola. To demonstrate potential applications of the improved assay, detection and quantification of C. beticola were assessed in inoculated plant and soil samples. A complementary objective was to compare specificity with that of the published C. beticola-specific PCR assays.
Materials and methods
Design of C. beticola specific primers and probe
Primers specific for C. beticola were designed using sequences of the calmodulin gene, which encompasses a DNA region demonstrated to contain polymorphisms informative for the discrimination of closely related Cercospora species (Groenewald et al. Citation2005; Vaghefi et al. Citation2018). A multiple sequence alignment was performed using the National Center for Biotechnology Information (NCBI) Basic Local Alignment Search Tool (BLAST) 2.8.0+ (Zhang et al. Citation2000) using the partial calmodulin sequence of C. beticola strain CBS 116 456 (GenBank Acc. No. AY840425.1) as the query against calmodulin sequences of 86 C. beticola isolates and 71 isolates of 10 closely related Cercospora species (Supplementary Table 1). Sequence polymorphisms consistently discriminating between C. beticola and other Cercospora species were identified for primer and probe design. Primers were designed to include polymorphisms at the 3´ terminus (Petruska et al. Citation1988). The probe was aligned to the adjacent sequence to capture multiple polymorphisms discriminating between C. beticola and other Cercospora species. Primer3 v. 0.4.0 (Koressaar and Remm Citation2007; Untergasser et al. Citation2012) was used to assess potential primers and probes for suitable lengths and associated melting temperatures. Primer and probe sequences were assessed for specificity by a BLAST search of the GenBank nucleic acid database using the Others (nr etc.) database: Nucleotide Collection (nr/nt). Alternative primer sequences were designed with one or two deliberate mismatches adjacent to the 3´ terminus to evaluate PCR efficiency and specificity (Stadhouders et al. Citation2010).
Beta vulgaris primers
To reduce the risk of false negatives, multiplex PCR assays may include an internal positive control. Primers reported to amplify sugar beet DNA (De Coninck et al. Citation2012) were assessed for this purpose. Limited information regarding these primers was available, thus the sugar beet specific primer sequences SbEc1-F and SbEc1-R (De Coninck et al. Citation2012) were evaluated using Primer-BLAST (Ye et al. Citation2012) in the Refseq mRNA database, with the Organism restricted to Beta (taxid:3554), to determine expected fragment size and predicted gene region. Amplification of target DNA across 15 table beet cultivars (‘Avalanche’, ‘Boldor’, ‘Chioggia Guardsmark’, ‘Detroit Dark Red’, ‘Detroit Supreme’, ‘Merlin’, ‘Red Ace’, ‘Rhonda’, ‘Robin’, ‘Rodina’, ‘Ruby Queen’, ‘Scarlet Supreme’, ‘Soldier OP’, ‘Touchstone Gold’ and ‘Vulture’) using the qPCR conditions described below was evaluated in duplicate reactions. Using the method described below, DNA was extracted from two 16 mm leaf discs collected from a fully expanded leaf of two individual four-week-old plants.
DNA extraction
DNA was extracted from fungal and table beet leaf tissue using the Wizard Genomic DNA Extraction kit (Promega), following the manufacturer’s plant DNA extraction protocol. Fungal mycelia was harvested from 14-day-old cultures grown in 10% (v/v) clarified V8 broth (Miller Citation1955; Secor and Rivera Citation2012) at 25°C, and dried overnight in a laminar flow cabinet. Leaf tissues were transferred to 2 mL microfuge tubes and placed in a gravimetric drying oven at 60°C for 12 h. Prior to addition of the Nuclei Lysis solution, mycelia was ground in a microfuge tube using a polypropylene pestle, and separated into 10 mg sub-samples, while leaf tissues were weighed and ground with two 4.5 mm ball bearings (Daisy Outdoor Products) using a TissueLyser (Qiagen) for 16 s at 30 Hz. DNA concentrations were determined using a Qubit 2.0 Fluorometer (Thermo Fisher Scientific) with the Qubit dsDNA HS or BR Assay kit (Thermo Fisher Scientific) as appropriate. DNA samples were stored at −20°C.
Fast conventional PCR conditions
The conventional PCR assay containing the CbCAL primers designed in this study was performed in 20 µL volumes using a T100 thermal cycler (Bio-Rad). Reactions contained autoclaved high purity water, 0.4 units Taq DNA polymerase (New England Biolabs), 1× Standard Taq Buffer (containing 1.5 mM MgCl2), 250 µM dNTPs, 0.25 µM CbCAL-F and CbCAL-R primers (Integrated DNA Technologies; ) and 5 µL of diluted DNA (10 ng µL−1) template. Thermal cycling was completed in 35 min and consisted of an initial denaturation for 5 min at 95°C and then 30 cycles of 92°C for 1 s, 64°C for 5 s and 72°C for 10 s, followed by a final extension of 72°C for 2 min. PCR products were visualized after electrophoresis on a 2% (w/v) agarose gel in Tris-acetate-EDTA containing 0.001% (v/v) GelRed (Biotium). Product size was determined against a 100-bp DNA Ladder (Axygen).
Table 1. Primer/probe sets multiplexed for quantification of Cercospora beticola and Beta vulgaris DNA.
Specificity of published conventional PCR assays
Two conventional PCR assays reported to amplify only C. beticola DNA (Lartey et al. Citation2003; Groenewald et al. Citation2005) were evaluated for species-specificity in 20 µL volumes as described above, with reagent concentrations modified from published conditions. Reactions based on Lartey et al. (Citation2003) contained 0.25 µM CBACTIN959L and CBACTIN959R primers (Integrated DNA Technologies). Thermal cycling consisted of an initial denaturation of 5 min at 95°C and then 35 cycles of 95°C for 60 s, 52°C for 30 s, followed by a final extension at 72°C for 60 s. Reactions based on Groenewald et al. (Citation2005) contained 0.25 µM CercoCal-R and CercoCal-beta primers and 0.125 µM CercoCal-F primer (Integrated DNA Technologies). Thermal cycling consisted of an initial denaturation for 5 min at 95°C and then 35 cycles of 95°C for 30 s, 58°C for 30 s and 72°C for 30 s, followed by a final extension at 72°C for 7 min. PCR products were visualized as previously described.
Quantitative PCR conditions
The CbCAL primer/probe assay designed in the current study and the CercoCal1 primer/probe assay reported by De Coninck et al. (Citation2012) were assessed in multiplex reactions with the SbEc1 (B. vulgaris) primer/probe assay designed by De Coninck et al. (Citation2012; ) to amplify B. vulgaris DNA as an internal positive control. Multiplex qPCR was performed using white 96 well Multiplate PCR plates (catalogue number MLL9651, Bio-Rad) and Microseal ‘B’ seals (catalogue number MSB1001, Bio-Rad) in a C1000 Touch thermal cycler, CFX96 real-time system (Bio-Rad).
Reactions for both assays were performed in 20 µL volumes containing 10 µL Immomix (Bioline) and 5 µL of DNA template. The CbCAL assay contained 150 nM CbCAL-P 5´ CAL Fluor Gold 540/3´ BHQ-1-labelled hydrolysis probe, 150 nM SbEc1 5´ FAM/3´ BHQ-1-labelled hydrolysis probe (LGC Biosearch Technologies), and 0.25 µM CbCAL-F, CbCAL-R, SbEc1-F, and SbEc1-R primers (Integrated DNA Technologies). Thermal cycling conditions consisted of an initial denaturation for 10 min at 95°C followed by 35 cycles of 95°C for 15 s and 64°C for 60 s. The CercoCal1 assay contained 420 nM CercoCal1 5´ CAL Fluor Gold 540/3´ BHQ-1-labelled hydrolysis probe, 210 nM SbEc1 5´ FAM/3´ BHQ-1-labelled hydrolysis probe (LGC Biosearch Technologies), 0.31 µM CercoCal1-F and CercoCal1-R primers, and 0.21 µM SbEc1-F and SbEc1-R primers (Integrated DNA Technologies), plus autoclaved high-purity water. Thermal cycling conditions consisted of 10 min at 95°C followed by 35 cycles of 95°C for 15 s and 60°C for 60 s. The Bio-Rad CFX Manager 3.1 software collected fluorescence data for both fluorophores during the 60/64°C step of each cycle.
Controls in every assay included no-template controls and genomic DNA standards (positive or negative) for C. beticola and B. vulgaris. For quantification, each PCR included a 10-fold serial dilution of pure genomic C. beticola DNA and B. vulgaris DNA in duplicate. DNA concentrations of standards were determined using a Qubit 2.0 Fluorometer (Thermo Fisher Scientific) with the Qubit dsDNA HS or BR Assay kit (Thermo Fisher Scientific) as appropriate. Four 10-fold serial dilutions for C. beticola DNA (approximate range of 10 ng to 1 pg) and three 10-fold serial dilutions for B. vulgaris DNA (approximate range of 50 ng to 50 pg) were included. Standard curves for C. beticola DNA and B. vulgaris DNA were generated using the Bio-Rad CFX Manager 3.1 software by plotting the log of standard DNA quantities against their quantification cycle. The coefficient of determination (R2), slope, y-intercept and reaction efficiency (E) were reported by the software for each standard curve. Quantities of target DNA in experimental samples were determined by plotting the quantification cycle on the standard curve.
Primer and probe specificity
The CbCAL primers designed in this study and the two conventional PCR assays reported by Lartey et al. (Citation2003) and Groenewald et al. (Citation2005) were assessed for C. beticola-specificity using DNA of C. beticola (Tb14-080), C. apii (Ch15-001), C. cf. flagellaris (Tb16-178), C. chenopodii (Lq15-012) and C. zebrina (Tb16-177).
The fast conventional and qPCR assays designed for the CbCAL primers were further tested for specificity using DNA of 22 C. beticola isolates, including representatives from the three phylogenetic lineages described by Vaghefi et al. (Citation2018), and 16 other fungal species (). Conventional PCR products were visualized as previously described. qPCR results were assessed for increases in fluorescence recognized as quantification cycles.
Table 2. Fungi used for specificity testing of the Cercospora beticola specific CbCAL primers in fast conventional PCR and quantitative PCR.
Selective detection in planta
Leaf inoculation experiments were performed using C. beticola isolate Tb14-085 (ICMP 21692). After 14 days of growth on potato dextrose agar, 6 mL of sterile distilled water was added to the plate and the mycelia was disrupted from the surface using a spatula. The mycelial suspension (600 µL) was spread onto five clarified V8 agar plates (10% (v/v) clarified V8 broth, 1.5% (w/v) agar) (Miller Citation1955; Secor and Rivera Citation2012) and incubated for six days with a 8 h photoperiod. Conidia were collected by flooding plates with 2 mL of sterile distilled water and disrupting the surface with a glass spreader, before filtering through several layers of cheesecloth, to a volume of 10 mL. The concentration of the conidial suspension was quantified using a haemocytometer (Hausser Scientific) and diluted to 105 conidia mL−1.
Four fully expanded leaves of five-week-old ‘Ruby Queen’ table beet plants were marked with a circle (20 mm diameter) in six positions, avoiding major veins. Plants (five per inoculated and control treatments) were placed in a growth chamber and 2 µL of inoculum was placed in the centre of each circle. Control plants were treated with 2 µL of sterile distilled water. After inoculation, plants were sealed in a transparent plastic box in the growth chamber for 24 h. Growth chamber conditions were 25°C with 16 h of light at 430 lux (110 W fluorescent mercury bulbs, Philips). Relative humidity was maintained at >90%.
Harvest of ten 16 mm diameter leaf discs was performed at 0, 3, 6, 9, 12, 15, 18 and 21 days after inoculation (dai). Each time-course was replicated in independent experiments. At harvest, the diameter of each lesion was measured and presence of conidia and pseudostromata evaluated under 63× magnification using a SZX10 stereo microscope (Olympus) with a KL 1600 LED light source (Schott). Leaf discs were placed separately into pre-weighed microfuge tubes and dried for 24 h at 60°C and weighed. DNA of the leaf discs was extracted using the Wizard Genomic DNA Extraction kit and C. beticola DNA was quantified using the CbCAL qPCR assay described above. Normalisation of C. beticola DNA quantities from leaf tissue was performed by dividing the C. beticola DNA quantity by the weight of extracted tissue (nanograms of C. beticola DNA per milligram dry weight of tissue) to produce values to quantify relative C. beticola biomass.
Selective detection in inoculated soil
Detection of C. beticola in soil was assessed by mixing dried, ground, diseased leaf tissue with a pasteurized (71°C for 1 h) top soil:sand mix (4:1) (Sensenig’s Landscape Supply), sieved to 0.38 mm. Diseased leaf tissue was produced by spray-inoculating leaves of five-week-old ‘Ruby Queen’ table beet plants with 12 mL of a conidial suspension, produced as described above, of C. beticola isolates Tb14-081 (ICMP 21691) and Tb14-085 (ICMP 21692). Control plants were sprayed with 12 mL of sterile distilled water. Leaves were collected four weeks after inoculation, dried for 24 h at 60°C and ground to a powder. DNA was extracted from triplicate samples containing 1, 5, 10 and 15 mg of leaf tissue from each of the three sources, as well as the same weights of tissue made up to 100 mg with soil (including a control of 100 mg soil only), using the DNeasy PowerLyzer PowerSoil Kit (Qiagen). DNA from diseased and control leaf and leaf-soil samples was assessed using the CbCAL qPCR assay described above.
Data analysis
Statistical tests were performed in the R package stats v. 3.4.1 (R Core Team Citation2017). Time-course C. beticola biomass values were transformed using log (x + 1), assessed for normality using the Shapiro-Wilk normality test (Shapiro and Wilk Citation1965; Royston Citation1995) in the shapiro.test function and compared between time points using a Student’s t-test (Snedecor and Cochran Citation1989) of paired data in the t.test function. Lesion diameters were compared between time points using the Wilcoxon rank sum and signed rank test (Wilcoxon et al. Citation1963) in the wilcox.test function. Homogeneity of variance between corresponding times in each time-course experiment were assessed using Levene’s Test (Levene Citation1960) in the leveneTest function in the R package car v. 3.0–2. Spearman’s rank correlation coefficient (ρ) (Best and Roberts Citation1975) was calculated in the cor.test function and used to assess the association between lesion diameters and transformed C. beticola biomass values. A linear regression model (Wilkinson and Rogers Citation1973; Chambers and Hastie Citation1992) was fitted to the relationship between lesion diameters and transformed C. beticola biomass values using the lm function. C. beticola DNA concentrations were compared between leaf and leaf-soil mixtures using a Student’s t-test of paired data in the t.test function.
Results
Improved assay design
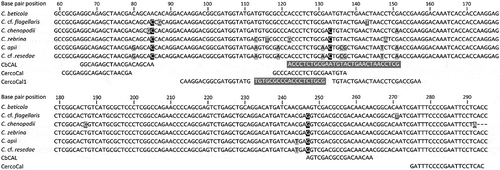
Multiple sequence alignment of the calmodulin gene region identified two single nucleotide polymorphisms as unique to C. beticola, and in appropriate positions for primer design (Fig. 1). Both primer designs incorporated a specific single nucleotide polymorphism at the 3´ terminus, while the probe sequence spanned a region containing multiple polymorphisms in the calmodulin gene region (; ).
Fig. 1 Alignment of CbCAL (current study), CercoCal (Groenewald et al. Citation2005) and CercoCal1 (De Coninck et al. Citation2012) primer (black text) and probe (white text on grey) sequences against calmodulin sequences (237 bp) of six related Cercospora species. Polymorphisms between Cercospora beticola and the remaining species sequences are indicated by shading (white text on black indicates >3, black text on grey indicates 2 to 3 and white text on grey indicates 1 polymorphism). GenBank accession numbers for calmodulin sequences were C. beticola (AY840425.1), C. cf. flagellaris (JX142876.1), C. chenopodii (KJ885793.1), C. zebrina (JX143020.1), C. apii (AY840417.1) and C. cf. resedae (DQ233395.1). C. beticola (AY840425.1) was used for base pair reference positions. A (-) indicates the absence of sequence information in the GenBank accession.
Primer-BLAST of the sugar beet primer sequences SbEc1-F and SbEc1-R reported a 90 bp product length from a predicted chloroplastic omega-amidase mRNA sequence (GenBank Acc. No.: XM_019248562). PCR fragments of the predicted length were amplified from DNA of each of the 15 table beet cultivars (data not shown).
Species specificity in published primer assays
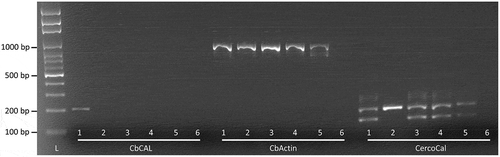
The conventional PCR assay reported by Lartey et al. (Citation2003) amplified approximately 959 bp sized fragments from DNA of C. beticola (Tb14-080), C. apii (Ch15-001), C. cf. flagellaris (Tb16-178), C. chenopodii (Lq15-012) and C. zebrina (Tb16-177) (Fig. 2). The two-fragment PCR assay described by Groenewald et al. (Citation2005) as specific for the Cercospora genus and C. beticola amplified two fragments (approximately 234 bp and 176 bp) from DNA of C. beticola (Tb14-080), C. cf. flagellaris (Tb16-178), C. chenopodii (Lq15-012) and C. zebrina (Tb16-177) (). Only the Cercospora genus-specific fragment (234 bp) was amplified using C. apii (Ch15-001) DNA. The qPCR assay described by De Coninck et al. (Citation2012) reported similar quantification cycles for C. beticola, C. cf. flagellaris and C. chenopodii (17.3–20.6), while C. apii reported a quantification cycle of 32 (). Alignment of the primers described by Groenewald et al. (Citation2005) and De Coninck et al. (Citation2012) with the calmodulin gene region of six Cercospora species () indicated the potential for primer binding to DNA of multiple Cercospora species.
Table 3. Mean quantification cycles for DNA of five Cercospora species using previously published and improved Cercospora beticola-specific quantitative PCR (qPCR) assays.
Fig. 2 Comparison of the specificity of three PCR assays reported to amplify DNA of Cercospora beticola. Each assay was performed with DNA of (1) C. beticola, (2) C. apii, (3) C. cf. flagellaris, (4) C. chenopodii, and (5) C. zebrina, including (6) a no template control. A DNA ladder (L) was included for fragment size comparison. Primer pair CbCAL (designed in the current study) is reported to amplify a 199 base pair (bp) fragment, primer pair CbActin a 959 bp fragment (Lartey et al. Citation2003), and the three primer CercoCal assay a 234 bp fragment specific for the Cercospora genus, and a 176 bp fragment specific for C. beticola (Groenewald et al. Citation2005).
Species specificity in improved assay
Fast conventional PCR using CbCAL primers and fungal DNA (1–5 ng µL−1), extracted from mycelia of 57 fungal isolates from 17 species (), amplified a single 199 bp sized fragment only from C. beticola DNA (). The optimal quantity of C. beticola DNA for detection ranged from 1 to 10 ng and the number of cycles was set to 30 to limit false positive results and provide a rapid assay. The CbCAL qPCR assay reported non-C. beticola species quantification cycles as absent or late (C. cf. flagellaris [32.6] and C. zebrina [34.6]), while all C. beticola isolates had quantification cycles of 18–22 (). A single ten-fold dilution of the DNA did not produce quantification cycles for non-C. beticola species and increased the quantification of C. beticola by approximately three cycles. Primers containing deliberate mismatches did not provide improved specificity for C. beticola DNA and reduced PCR efficiency (data not shown).
Detection thresholds for qPCR
A cut-off point, defined as the cycle number above which any sample response value (quantification cycle) was considered a false positive due to non-specific fluorescence, was set at 35 cycles. For detection and quantification, 35 cycles were sufficient for standard curve production and detection of small quantities of target DNA. C. beticola DNA was quantifiable from 0.001 to 10 ng and B. vulgaris DNA from 0.001 to 10 ng. The multiplexed CbCAL-SbEc1 primer/probe assay displayed high precision over a range of five orders of magnitude. The standard curve of the quantification cycle and log of standard DNA quantities was calculated for C. beticola (R2 > 0.99, y = – 3.27x + 24.7, E = 1.02) and B. vulgaris (R2 > 0.99, y = – 3.1x + 26.3, E = 1.09). The respective mean values (n = 10) and coefficient of variation (CV) of a single mixed DNA sample assessed for intra-assay variability were 5 pg C. beticola DNA (CV = 12.6%) and 60 pg B. vulgaris DNA (CV = 26.6%). The respective mean values (n = 10) and CV of the mixed DNA sample assessed for inter-assay variability were 8 pg C. beticola DNA (CV = 18.5%) and 58 pg B. vulgaris DNA (CV = 36.7%).
Quantitative PCR validation in biological matrices
C. beticola DNA was detected in inoculated leaf tissues as early as three days after inoculation, along with the initial appearance of CLS lesions. C. beticola biomass (ng C. beticola DNA/mg leaf tissue) and lesion diameter increased up to 21 dai (Fig. 3). Due to differences in variances between replications, each experiment was analyzed separately. C. beticola biomass was positively associated with lesion diameter in experiment 1 (ρ = 0.91, P < 0.001) and experiment 2 (ρ = 0.91, P < 0.001). A linear relationship between C. beticola biomass and lesion diameter was described in experiment 1 (y = 0.224x + 0.08, R2 = 0.84, residual standard deviation = 0.60, P < 0.001) and experiment 2 (y = 0.238x + 0.14, R2 = 0.84, residual standard deviation = 0.61, P < 0.001). Inhibition of PCR reactions was detected in the original DNA samples extracted from inoculated (14%) and control (22%) leaf disc tissues, based upon the absence of fluorescence from the B. vulgaris probe. A ten-fold dilution of the DNA solution restored the fluorescence from the B. vulgaris probe, but impeded detection of small quantities of C. beticola DNA. No C. beticola DNA was detected in control samples.
C. beticola DNA was detected in diseased leaf and leaf-soil mixtures with sensitivity to 1 mg of diseased leaf tissue in 99 mg of soil (Fig. 4). Samples with at least 5 mg of diseased leaf tissue added to soil reported similar quantities (P > 0.05) of C. beticola DNA as the pure diseased leaf tissue samples. However, 1 mg of diseased leaf material in 99 mg of soil resulted in significantly greater (P < 0.01) C. beticola DNA concentrations. No C. beticola DNA was detected in control samples.
Discussion
Specificity of primers for the target of interest is key to providing reliable detection and relevant results, especially for field-based research. However, with rapidly evolving taxonomy and availability of DNA sequences, specificity of assays may be compromised. Two assays reported to detect only C. beticola (Lartey et al. Citation2003; Groenewald et al. Citation2005) have both been found to amplify DNA from other Cercospora species (Bakhshi et al. Citation2013; Vaghefi et al. Citation2017b). This study confirmed the lack of specificity of these assays (Lartey et al. Citation2003; Groenewald et al. Citation2005), and disputes C. beticola specificity in another (De Coninck et al. Citation2012). Improved C. beticola-specific primers were designed and validated for detection with conventional PCR and quantification using multiplex-hydrolysis probe-based qPCR. The ability to specifically detect and identify C. beticola is fundamental for effective disease management and research outcomes. These assays will be a valuable tool for experiments evaluating the relative role of inoculum sources in CLS epidemics.
The description of the design used to determine specificity of an assay is critical for reporting assay robustness and may have major effects on subsequent research. This study used complementary methods to confirm and report species-specificity for C. beticola. The process consisted of identification of a polymorphic DNA region, alignment of sequences of multiple isolates of C. beticola and related species (based on current literature through in silico evaluation), identification of species-specific polymorphisms, appropriate primer design and BLAST assessments of primer sequences. Specificity of primers was confirmed using PCR across DNA of multiple Cercospora spp. and other fungi present in the local environment (Vaghefi et al. Citation2018). Five gene regions have predominantly been sequenced for phylogenetic assessment of the Cercospora genus; however, only the calmodulin gene region has allowed consistent discrimination between C. beticola and other closely related Cercospora species (Groenewald et al. Citation2005; Vaghefi et al. Citation2018). The identification of two single nucleotide polymorphisms unique to C. beticola within the calmodulin gene region limited primer design to two locations and necessitated stringent assessment of PCR specificity. A single nucleotide polymorphism at the 3´ terminus of a primer has a significant effect on PCR efficiency (Petruska et al. Citation1988), however extension of the primer will still occur. Therefore, while the primers designed in the current study result in inefficient amplification of non-target sequences, further assay refinement was required to limit detection of inefficient amplification and avoid false positives. This was achieved through determination of optimal DNA quantities and limiting PCR cycles. A trade-off is that assay sensitivity was limited to 0.001 ng of C. beticola DNA. Optimal identification or quantification of C. beticola will require DNA quantities to be within a relevant range to ensure detection, and inclusion of appropriate controls is required to support the accuracy of the results. Alternative technologies for quantification of PCR products, such as digital PCR (Sykes et al. Citation1992; Vogelstein and Kinzler Citation1999), may also provide a platform which enables improved detection sensitivity of pathogen DNA in complex samples. The assays described in this study provide improved detection of C. beticola; however, as with all species-specific assays, it is recommended to confirm the specificity in future projects across a range of isolates and species which are relevant to the location and time.
The development of an improved C. beticola-specific assay was necessitated by the detection of multiple Cercospora species using assays reported to detect only C. beticola (Bakhshi et al. Citation2013; Vaghefi et al. Citation2017b). The design process described for these assays varied in robustness. The assay described by Groenewald et al. (Citation2005) utilized a three primer PCR to amplify a fragment of the calmodulin gene considered specific for the Cercospora genus, along with a fragment designed to be specific for C. beticola. Specificity was assessed across DNA of 51 isolates from 16 Cercospora spp., dominated by C. apii (n = 11), C. beticola (n = 18) and a Cercospora sp. (n = 9) later described as C. apiicola (Groenewald et al. Citation2006). No in silico evaluation was described. Primers reported by Lartey et al. (Citation2003) were designed based on partial actin gene sequences of two isolates of C. beticola, and the qPCR assay described by De Coninck et al. (Citation2012) utilized a calmodulin gene sequence of a single C. beticola isolate, but assessment of homology with other Cercospora species was not reported in either study. Considering the limitations of these assays, it would be beneficial to establish a set of guidelines for reporting species-specific PCR assays, similar to those suggested for qPCR development (Bustin et al. Citation2009), including the requirement for alignment of sequences from related species, and indications of the polymorphic regions and associated primer positions.
Species-specific assays have a range of applications, including the quantification of target DNA in complex biological matrices. The quantification of C. beticola in planta demonstrated the utility of the qPCR assay for assessing DNA extracted from diseased table beet leaf tissue and reflected the quantification abilities reported for the assay designed by De Coninck et al. (Citation2012). The detection of C. beticola DNA from diseased leaf tissue mixed in soil samples indicates a potential for the assay as a tool for detecting C. beticola inoculum in field soil. This technique has been applied in other pathosystems and is a valuable tool to inform growers of risk levels for soil-borne diseases (Ophel-Keller et al. Citation2008). A major focus of CLS research, particularly in New York table beet fields, has been the investigation of potential sources of inoculum (Pethybridge et al. Citation2018). Population genetics studies have provided evidence for local and external sources of inoculum, including alternative hosts, infested plant debris and seed, which may be spread by wind, rain or human mediated dispersal (McKay and Pool Citation1918; Vaghefi et al. Citation2017a, Citation2017b; Knight et al. Citation2018). The worldwide importance and distribution of CLS on sugar beet and table beet (McGrath et al. Citation2007; Franc Citation2010; Lartey et al. Citation2010) offers potential applications for a C. beticola-specific PCR assay, including epidemiological studies, evaluation of cultivar susceptibility, and the ability to inform disease management and risk assessment strategies.
Supplementary Table 1
Download MS Word (48.5 KB)Acknowledgements
This research was supported by the United States Department of Agriculture National Institute of Food and Agriculture Hatch project NYG-625424, and the Federal Capacity Funds Initiative (2015-16-118) managed by Cornell AgriTech, Geneva, New York, USA. We thank Carol Bowden, Charli Morgan, Sean Murphy and Alex Silva for excellent technical support. We also thank Dr Frank Hay (Cornell AgriTech) for providing fungal isolates for specificity testing. The time-course disease assessment was performed by Charli Morgan (Hobart and William Smith Colleges, Geneva, New York, USA) at Cornell AgriTech in partial fulfilment of an undergraduate Summer Research Scholars Program project partly funded by the Brenda and David Rickey Foundation.
Related Research Data
References
- Abdullah AS, Turo C, Moffat CS, Lopez-Ruiz FJ, Gibberd MR, Hamblin J, Zerihun A. 2018. Real-time PCR for diagnosing and quantifying co-infection by two globally distributed fungal pathogens of wheat. Front Plant Sci. 9:1086.
- Bakhshi M, Arzanlou M, Babai-Ahari A. 2013. Identification of Cercospora beticola isolates the causal agent of Cercospora leaf spot disease of sugar beet, using species-specific primers. Iran J Plant Pathol. 49:31–32.
- Bakhshi M, Arzanlou M, Babai-Ahari A, Groenewald JZ, Crous PW. 2015. Is morphology in Cercospora a reliable reflection of generic affinity? Phytotaxa. 213:22–34.
- Bakhshi M, Arzanlou M, Babai-Ahari A, Groenewald JZ, Crous PW. 2018. Novel primers improve species delimitation in Cercospora. IMA Fungus. 9:299–332.
- Best D, Roberts D. 1975. Algorithm AS 89: the upper tail probabilities of Spearman’s rho. J Roy Stat Soc Ser C (Appl Stat). 24:377–379.
- Bustin SA, Benes V, Garson JA, Hellemans J, Huggett J, Kubista M, Mueller R, Nolan T, Pfaffl MW, Shipley GL, et al. 2009. The MIQE Guidelines: minimum information for publication of quantitative real-time PCR experiments. Clin Chem. 55:611–622.
- Cai L, Giraud T, Zhang N, Begerow D, Cai G, Shivas RG. 2011. The evolution of species concepts and species recognition criteria in plant pathogenic fungi. Fungal Divers. 50:121.
- Chambers JM, Hastie TJ. 1992. Statistical models in S, Vol. 251. Pacific Grove (CA): Wadsworth & Brooks/Cole.
- Chupp C. 1954. A monograph of the fungus genus Cercospora. Ithaca (NY): Published by the author.
- Crous PW, Aptroot A, Kang J-C, Braun U, Wingfield MJ. 2000. The genus Mycospharella and its anamorphs. Stud Mycol. 45:107–122.
- Crous PW, Hawksworth DL, Wingfield MJ. 2015. Identifying and naming plant-pathogenic fungi: past, present, and future. Annu Rev Phytopathol. 53:247–267.
- De Coninck BMA, Amand O, Delauré SL, Lucas S, Hias N, Weyens G, Mathys J, De Bruyne E, Cammue BPA. 2012. The use of digital image analysis and real-time PCR fine-tunes bioassays for quantification of Cercospora leaf spot disease in sugar beet breeding. Plant Pathol. 61:76–84.
- Franc GD. 2010. Ecology and epidemiology of Cercospora beticola. In: Lartey RT, Weiland JJ, Panella L, Crous PW, Windels CE, editors. Cercospora leaf spot of sugar beet and related species. St. Paul (MN): APS Press; p. 7–19.
- Goodwin SB, Dunkle LD, Zismann VL. 2001. Phylogenetic analysis of Cercospora and Mycosphaerella based on the internal transcribed spacer region of ribosomal DNA. Phytopathology. 91:648–658.
- Groenewald M, Groenewald JZ, Braun U, Crous PW. 2006. Host range of Cercospora apii and C. beticola and description of C. apiicola, a novel species from celery. Mycologia. 98:275–285.
- Groenewald M, Groenewald JZ, Crous PW. 2005. Distinct species exist within the Cercospora apii morphotype. Phytopathology. 95:951–959.
- Knight NL, Martin A, Sutherland MW, Herde DJ. 2012. Assessment of infection by Fusarium pseudograminearum in wheat seedling tissues using quantitative PCR and a visual discoloration scale. Plant Dis. 96:1661–1669.
- Knight NL, Sutherland MW. 2017. Assessment of Fusarium pseudograminearum and F. culmorum biomass in seedlings of potential host cereal species. Plant Dis. 101:2116–2122.
- Knight NL, Vaghefi N, Hansen ZR, Kikkert JR, Pethybridge SJ. 2018. Temporal genetic differentiation of Cercospora beticola populations in New York table beet fields. Plant Dis. 102:2074–2082.
- Koressaar T, Remm M. 2007. Enhancements and modifications of primer design program Primer3. Bioinformatics. 23:1289–1291.
- Lartey RT, Weiland JJ, Caesar-TonThat T, Bucklin-Comiskey S. 2003. PCR protocol for rapid detection of Cercospora beticola in sugarbeet tissues. J Sugar Beet Res. 40:1.
- Lartey RT, Weiland JJ, Panella L, Crous PW, Windels CE. 2010. Cercospora leaf spot of sugar beet and related species. St Paul (MN): APS Press.
- Levene H. 1960. Robust tests for equality of variances. In: Olkin I, editor. Contribution to probability and statistics: essays in honor of Harold Hotelling. Stanford (CA): Stanford University Press; p. 278–292.
- McGrath JM, Saccomani M, Stevanato P, Biancardi E. 2007. Beet. In: Kole C, editor. Genome mapping and molecular breeding in plants, vol 5: vegetables. Heidelberg (Germany): Springer-Verlag; p. 191–207.
- McKay MB, Pool VW. 1918. Field studies of Cercospora beticola. Phytopathology. 8:119–136.
- Miller PM. 1955. V-8 juice agar as a general purpose medium for fungi and bacteria. Phytopathology. 45:461–462.
- Ophel-Keller K, McKay A, Hartley D, Herdina CJ. 2008. Development of a routine DNA-based testing service for soilborne diseases in Australia. Australas Plant Pathol. 37:243–253.
- Pethybridge S, Kikkert J, Hanson L, Nelson S. 2018. Challenges and prospects for building resilient disease management strategies and tactics for the New York table beet industry. Agronomy. 8:112.
- Petruska J, Goodman MF, Boosalis MS, Sowers LC, Cheong C, Tinoco I. 1988. Comparison between DNA melting thermodynamics and DNA polymerase fidelity. Proc Natl Acad Sci USA. 85:6252–6256.
- R Core Team. 2017. R: a language and environment for statistical computing. Vienna (Austria): R Foundation for Statistical Computing.
- Royston P. 1995. Remark AS R94: a remark on algorithm AS 181: the W-test for normality. J Roy Stat Soc Ser C (Appl Stat). 44:547–551.
- Schaad NW, Frederick RD. 2002. Real-time PCR and its application for rapid plant disease diagnostics. Can J Plant Pathol. 24:250–258.
- Secor GA, Rivera VV. 2012. Fungicide resistance assays for fungal plant pathogens. In: Bolton MD, Thomma BPHJ, editors. Plant fungal pathogens: methods and protocols. Totowa (NJ): Humana Press; p. 385–392.
- Shapiro SS, Wilk MB. 1965. An analysis of variance test for normality (complete samples). Biometrika. 52:591–611.
- Snedecor GW, Cochran WG. 1989. Statistical methods 8th edition. Ames (IA): State University Press.
- Stadhouders R, Pas SD, Anber J, Voermans J, Mes THM, Schutten M. 2010. The effect of primer-template mismatches on the detection and quantification of nucleic acids using the 5′ nuclease assay. J Mol Diagn. 12:109–117.
- Strausbaugh CA, Overturf K, Koehn AC. 2005. Pathogenicity and real-time PCR detection of Fusarium spp. in wheat and barley roots. Can J Plant Pathol. 27:430–438.
- Sykes P, Neoh S, Brisco M, Hughes E, Condon J, Morley A. 1992. Quantitation of targets for PCR by use of limiting dilution. BioTechniques. 13:444–449.
- Untergasser A, Cutcutache I, Koressaar T, Ye J, Faircloth BC, Remm M, Rozen SG. 2012. Primer3—new capabilities and interfaces. Nucleic Acids Res. 40:e115–e115.
- Vaghefi N, Kikkert JR, Bolton MD, Hanson LE, Secor GA, Nelson SC, Pethybridge SJ. 2017a. Global genotype flow in Cercospora beticola populations confirmed through genotyping-by-sequencing. PLoS One. 12:e0186488.
- Vaghefi N, Kikkert JR, Hay FS, Carver GD, Koenick LB, Bolton MD, Hanson LE, Secor GA, Pethybridge SJ. 2018. Cryptic diversity, pathogenicity, and evolutionary species boundaries in Cercospora populations associated with Cercospora leaf spot of Beta vulgaris. Fungal Biol. 122:264–282.
- Vaghefi N, Nelson SC, Kikkert JR, Pethybridge SJ. 2017b. Genetic structure of Cercospora beticola populations on Beta vulgaris in New York and Hawaii. Sci Rep. 7:1726.
- Vogelstein B, Kinzler KW. 1999. Digital PCR. Proc Natl Acad Sci USA. 96:9236–9241.
- Weiland J, Koch G. 2004. Sugarbeet leaf spot disease (Cercospora beticola Sacc.). Mol Plant Pathol. 5:157–166.
- Wilcoxon F, Katti S, Wilcox RA. 1963. Critical values and probability levels for the Wilcoxon rank sum test and the Wilcoxon signed rank test. Pearl River (NY): American Cyanamid Company.
- Wilkinson G, Rogers C. 1973. Symbolic description of factorial models for analysis of variance. J Roy Stat Soc Ser C (Appl Stat). 22:392–399.
- Ye J, Coulouris G, Zaretskaya I, Cutcutache I, Rozen S, Madden TL. 2012. Primer-BLAST: a tool to design target-specific primers for polymerase chain reaction. BMC Bioinformatics. 13:134.
- Zhang Z, Schwartz S, Wagner L, Miller W. 2000. A greedy algorithm for aligning DNA sequences. J Comput Biol. 7:203–214.