
Abstract
Several members of Alternaria section Alternaria are economically important plant-pathogenic fungi that cause disease on a wide range of host types and plant tissues. The production of Alternaria-derived mycotoxins can lead to significant post-harvest losses due to contamination of agricultural products. Multiple Alternaria species are listed as regulated organisms, which are monitored by international plant protection programmes. These taxa often share high levels of both morphological and phylogenetic similarity, and the establishment of molecular markers that are able to distinguish among them unambiguously has proven to be quite challenging. Previously, we examined fine-scale genome-wide phylogenetic patterns and proposed a list of candidate genes for development into informative markers that are diagnostic for the main section Alternaria lineages. Here, we design primer sets and sequence three new markers (ASA-05, ASA-10, ASA-19) in order to evaluate their diagnostic performance. Sequence data for 49 new independent test taxa were combined with existing data for 38 taxa, and phylogenetic analyses revealed that, in general, the new molecular markers consistently classified section Alternaria strains into one of the main lineages. A discrepancy among the three markers for lineage assignment was observed only for one strain. We also sequenced a commonly used marker in molecular phylogenetics of fungi (rpb2, RNA polymerase II second largest su bunit) and found it was outperformed by all three of the new markers. We suggest that an ASA marker presented here could form the basis of a convenient one-locus test for rapid and routine diagnostic screening of unknown section Alternaria strains.
Résumé
Plusieurs membres de la section Alternaria d’Alternaria sont des champignons phytopathogènes importants du point de vue économique qui causent des maladies chez une vaste gamme de types d’hôtes et de tissus végétaux. La production de mycotoxines dérivées d’Alternaria peut engendrer des pertes substantielles après récolte à cause de la contamination des produits agricoles. De multiples espèces d’Alternaria sont classées comme organismes réglementés qui sont suivis en vertu de programmes internationaux de protection des végétaux. Ces taxons partagent souvent des taux élevés de similarité morphologique et phylogénétique, et la conception de marqueurs moléculaires permettant de les différencier s’est révélée tout un défi. D’abord, nous avons examiné les schémas phylogénétiques de l’ensemble du génome à fine échelle, puis nous avons proposé une liste de gènes candidats pour en faire des marqueurs informatifs qui serviraient à reconnaître les principales lignées de la section Alternaria. À partir de cela, nous avons conçu des jeux d’amorces et séquencé trois nouveaux marqueurs (ASA-05, ASA-10, ASA-19) afin d’évaluer leur exactitude diagnostique. Les données de séquençage pour 49 nouveaux taxons utilisés dans des tests indépendants ont été combinées aux données connues pour 38 taxons, et les analyses phylogénétiques ont révélé que, en général, les nouveaux marqueurs moléculaires avaient systématiquement classé les souches de la section Alternaria dans une des principales lignées. Une différence parmi les trois marqueurs utilisés pour l’assignation des lignées a été observée pour une souche seulement. Nous avons également séquencé un marqueur utilisé couramment dans la phylogénétique moléculaire des champignons (rpb2, deuxième plus grosse sous-unité de l’ARN polymérase II) et avons constaté qu’il a été surclassé par les trois nouveaux marqueurs. Nous suggérons qu’un des marqueurs ASA présentés ici pourrait servir de base à un test pratique à un locus en vue d’examens rapides et routiniers de dépistage des souches inconnues de la section Alternaria.
Introduction
Alternaria is one of the most cosmopolitan and ubiquitous fungal genera, with species displaying the full range of saprobic, endophytic, and pathogenic lifestyles (Thomma Citation2003). The results of numerous recent metagenomic microbiome studies confirm that Alternaria can be commonly found in an astounding diversity of environmental niches and substrates (e.g., Barberán et al. Citation2015; Gonçalves et al. Citation2016; Luis et al. Citation2019). In regards to economically important pathogens, Alternaria is known to cause disease on hundreds of agricultural crops (Rotem Citation1994), affecting nearly every possible above-ground tissue or structure of a plant host. In addition, Alternaria causes significant post-harvest losses due to spoilage or contamination with Alternaria-derived mycotoxins such as alternariol, alternariol monomethyl ether, and tenuazonic acid (Meena et al. Citation2017). In particular, the accumulation of Alternaria mycotoxins in stored grains and consumer end-products has received renewed attention from food safety researchers and regulatory agencies (Tralamazza et al. Citation2018; Mujahid et al. Citation2020; Aichinger et al. Citation2021).
Due to significant pre- and post-harvest problems, several species of Alternaria are included on international regulated organism lists and may be screened for during phytosanitary certification of agricultural trade goods. Although the Canadian Food Inspection Agency (CFIA) regulates only two Alternaria species (A. gaisen and A. kikuchiana), a total of 22 described species (plus ‘Alternaria sp.’) are regulated by Canada, US, and Mexico collectively. As research continues to reveal the association between fungal species and mycotoxin profiles, the ability to accurately detect and diagnose Alternaria species will be essential for regulatory and quarantine purposes.
Alternaria section Alternaria (Lawrence et al. Citation2013; Woudenberg et al. Citation2015) contains many important plant pathogens including species that cause canker, spot, blotch, and blight diseases of hosts such as tomato, tobacco, apple, pear, citrus fruits, wheat, and potato. Six of the 22 aforementioned regulated species are within Alternaria section Alternaria, including the two CFIA-regulated species. This section also includes A. alternata, a species that has an extremely wide host range and is very commonly encountered in agricultural surveys, both in Canada and worldwide. Species in section Alternaria have no known sexual state and reproduce by asexual means only. Commonly known as a ‘small-spored’ section, species are notoriously difficult to distinguish by morphology alone (e.g. conidial shape, size; conidiophore structure). Much effort has been put towards the molecular systematics of this group (e.g. Andrew et al. Citation2009; Armitage et al. Citation2015, Citation2020; Woudenberg et al. Citation2015; Dettman and Eggertson Citation2021), but widely applicable markers that are diagnostic for focal species have been difficult to establish.
In recent work, we demonstrated that the genes commonly used in fungal phylogenetics are not optimally informative at the level of genetic divergence found within section Alternaria (Dettman and Eggertson Citation2021). While ‘housekeeping’ genes may be useful at more distant levels of divergence (e.g. between sections and greater), they typically possessed insufficient variation to positively distinguish the main phylogenetic lineages within the section. Here, we use the taxonomically neutral term ‘lineage’ to acknowledge that section Alternaria lineages may be composed of a single species, multiple species, or an unresolved species complex. In a phylogenomic analysis of 38 whole genomes from section Alternaria, we determined the fine-scale phylogenetic patterns for 6209 genes distributed along the entire genome, and compared these individual gene trees with the species tree. We provided a list of 20 genes that were located in large genomic regions of contiguous gene/species tree concordance, and would therefore be prime candidates for development into informative molecular markers that may be diagnostic for the main lineages. Here, we test how well three of these newly proposed markers perform when challenged by a wider range of section Alternaria strains, and test their diagnostic ability to clearly place new strains into a single lineage. Primer sets were designed to PCR-amplify and sequence the three candidate genes from an additional 40 strains, all of which were not included in our previous study. We also challenged the markers with an additional nine publicly available genomes from strains that were not available when the markers were initially developed. Phylogenetic analyses revealed that, in general, the new molecular markers were able to consistently classify the section Alternaria strains and outperformed loci that are commonly employed for molecular phylogenetics of fungi.
Materials and methods
Fungal strains and DNA extraction
Live strains or lyophilized tissue were obtained from Agriculture and Agri-Food Canada mycological research collections and the Canadian Collection of Fungal Cultures. When selecting strains for an independent test set, we chose those that were likely to be within section Alternaria based on culture and conidial morphologies or preliminary ITS sequences (data not shown). Preliminary species identifications made by previous investigators were noted, if available (). Live strains were grown on potato dextrose agar (potato extract, 4 g L−1; dextrose, 20 g L−1; agar 15 g L−1) at 25°C. Tissue was mixed with lysis buffer and 25 µL of Proteinase K in sterilized 2.0 mL XXTuff Reinforced o-ring tubes containing 0.25 mL of 1-mm zirconia beads (both from BioSpec Products, Bartlesville, OK). Homogenization was done using a Precellys 24 homogenizer (6000 rpm for 40s; Bertin Instruments, Montigny-le-Bretonneux, France). Genomic DNA was extracted using the Macherey-Nagel NucleoMag 96 Trace kit (Macherey Nagel GmbH & Co. KG, Düren, Germany) and a KingFisher Flex magnetic particle processor (Thermo Fisher Scientific Oy, Vantaa, Finland) following the manufacturer’s suggested protocols.
Table 1. Information on strains, genomes, and lineage assignment.
Publicly available genome assemblies
Information on the 38 previously analyzed section Alternaria genomes is presented in Supplementary Table 1 and Dettman and Eggertson (Citation2021). After our initial study, genome sequences of nine additional section Alternaria strains became publicly available and their assemblies were downloaded from the NCBI database (accessed September 17, 2021). Sequences for target loci were BLASTn-searched (2.10) with relaxed settings (-perc_identity 80 -evalue 0.00001) against a local custom BLAST database. Target loci were confirmed to be single-copy, then corresponding sequences were extracted for each locus from each genome.
Primer design and Sanger sequencing
New primers were designed based on sequences of candidate loci from the 38 previously analyzed section Alternaria genomes. Highly conserved regions in alignments were targeted to avoid the need for degenerate primers, and to increase the probability of amplification success across all target taxa. New primer sets were designed to amplify gene fragments 300–600 bp long, allowing for standard Sanger sequencing methods. For rpb2, we used existing primers from Liu et al. (Citation1999) but also designed new primers. All primer sequences are listed in . PCR methods were as described in Robideau et al. (Citation2011). Thermocycler profiles for amplification with newly designed primer sets were as follows: initial denaturation at 94°C for 2 min, followed by 35 cycles of 94°C for 30s, annealing for 30s (58°C for ASA-05 and ASA-10, 55°C for ASA-19, 53°C for rpb2), extension at 72°C for 1 min, with a final extension at 72°C for 10 min. The thermocycler profile for the rpb2 primers from Liu et al. (Citation1999) was denaturation at 95°C for 2 min, followed by 40 cycles of 95°C for 1 min, annealing at 55°C for 2 min, extension at 72°C for 2 min, with a final extension at 72°C for 10 min. Amplification of PCR products for sequencing was done using ABI BigDye™ Terminator v3.1 Cycle Sequencing Kits, with an initial denaturation of 95°C for 3 min, followed by 40 cycles of 95°C for 30s, annealing for 20s at 55°C (58°C for ASA-10 and 50° for all rpb2 primers), with an extension for 60°C for 2 min. DNA sequence data was generated using an Applied Biosciences Prism® 3130xl Genetic Analyser (Life Technologies™, Carlsbad, CA). Geneious (11.1.5; Biomatters, Auckland, New Zealand) was used to edit data and determine consensus sequences.
Table 2. Information on loci, primer sets, and alignments.
Phylogenetic analyses
Sequences were aligned with MUSCLE (3.8; Edgar Citation2004) using default parameters and verified by visual inspection. Ends of alignments were trimmed to remove primer binding sites and regions with excessive missing data, so final alignments may be slightly shorter than the original PCR fragment size. Alignment statistics were calculated with AMAS (Borowiec Citation2016). Substitution model selection and construction of maximum likelihood (ML) phylogenies were performed with IQ-TREE (2.0, Nguyen et al. Citation2015). The MODELFINDER module implemented by IQ-TREE was used to determine the best substitution model for each locus, and the optimal partitioning scheme for multilocus datasets (-m MFP+MERGE). ML tree construction was performed under best-fit substitution models and partitions. Branch support was assessed using 1000 non-parametric bootstrap replicates (-b 1000). Bayesian phylogenetic inference was performed with MrBayes (3.2.6, Ronquist and Huelsenbeck Citation2003) with a GTR substitution model with gamma-distributed rate variation across sites and a proportion of invariable sites (nst = 6, rates = invgamma, ploidy = haploid). For combined analyses of the three loci, each locus was treated as a separate partition with independent parameter estimation. Default priors were used for all analyses and the starting trees were random (starttree = random). For each dataset, two runs with four chains each were run for one or two million generations (ngen = 1 000 000 [single locus] or 2 000 000 [combined]) with a sampling frequency of every 100 generations (samplefreq = 100). The first 25% of sampled trees were discarded as burn-in, resulting in 15 thousand (single locus) or 30 thousand (combined) credible trees for calculation of posterior probabilities. Multi-species coalescent analysis was implemented by ASTRAL-III (5.7.3, Zhang et al. Citation2018) using the 3000 ML bootstrap trees as input (1000 non-parametric bootstrap replicates from each locus). The strain CBS 107.38 (A. burnsii) was used for rooting of trees, based on the relative branching order of taxa in Nishikawa et al. (Citation2020).
Results
Independent test taxa
The performance of the putatively diagnostic molecular markers was evaluated on a panel of 49 untested strains or genomes from Alternaria section Alternaria (). The 40 included strains were collected from a diversity of hosts and substrates (e.g. cereals, grasses, vegetables, and fruits), 75% of which were of Canadian origin (). Genomic DNA was successfully extracted from all 40 strains. The recently released genome sequences of nine additional strains from international sources were added to the panel to increase potential strain diversity even further. None of these 49 taxa were included in the development of the candidate loci, so they represent an independent test set.
Characterization of new candidate loci
Here, we investigate three candidate diagnostic loci proposed in our previous study: ASA-05, ASA-10, and ASA-19 (ASA = Alternaria Section Alternaria, Dettman and Eggertson Citation2021). ASA-05 encodes a hypothetical protein with a best protein match (81%) to a phosphatidylserine decarboxylase in Stemphylium lycopersici, and ASA-10 encodes a hypothetical protein containing an F-box domain (protein-protein interaction motif). ASA-19 encodes a histone-fold-containing protein and is a putative histone-like transcription factor. New primer sets () were designed for the candidate loci and, when applied to DNA extracted from the 40 previously untested strains, we obtained successful PCR-amplification of 98.3% of primer-by-sample combinations. Good quality Sanger sequences were obtained for 39 (ASA-05), 39 (ASA-10), and 40 (ASA-19) strains. The sequences for these three loci were also extracted from the assemblies of the nine newly available genomes, and from the 38 genome assemblies upon which candidate marker development was originally based. Sequence data from all 87 taxa were aligned and summary statistics for each locus are shown in . The ASA loci were quite variable and contained a high percentage of phylogenetically informative sites (6.3–8.9%). This amount of informative variation is likely to be sufficient for diagnostic purposes, as long as it is distributed appropriately between the main lineages.
Diagnostic utility of new candidate loci
Maximum likelihood (ML) trees constructed from the single-locus alignments of the three new loci are shown in . Using the previously analyzed strains as verified representatives, each of the three new loci recovered the four main phylogenetic lineages (clades) in section Alternaria: alternata, arborescens, gaisen, and longipes. These lineages were identified and well supported by previous phylogenomic analyses, however, a lineage may contain one species or a group of species, depending on the current state of taxonomy (e.g. the arborescens and longipes lineages include multiple species). For the new locus ASA-19, the alternata lineage was non-monophyletic and divided into two closely related clades, but not all branches received significant bootstrap support. The smaller alternata clade was composed entirely of new strains (n = 7) that were not included in our previous analysis.
Fig. 1 Maximum likelihood trees constructed from each of the three new candidate markers ASA-05, ASA-10, and ASA-19. Phylogenetic tree branches are colour-coded by main lineage (blue = alternata, red = longipes, green = arborescens, grey = gaisen). Taxa from the independent test set have taxon labels in bold and black. Taxa that were included in our previous phylogenomic analyses have colour-coded taxon labels, and strain names are followed by lineage names. Taxon labels with asterisks indicate strains that fell outside of the four main lineages, or have conflicting placement among the three loci. Branch support values are indicated above (ML bootstrap percentage) or below (Bayesian posterior probability) branches.
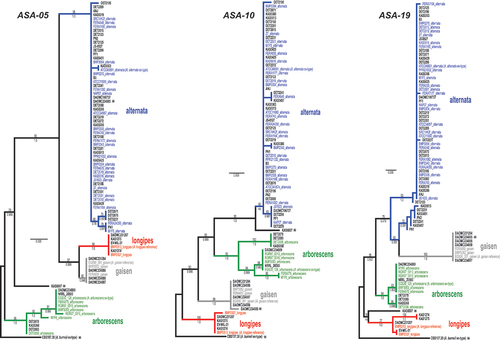
Based on the gene trees, 46 of the 49 test taxa were assigned to one of the four main lineages, with full agreement between all three ASA loci (, ). The exceptions were:
(1) Two strains (KAS6097 and CBS 107.38) were consistently placed outside of the four main lineages: KAS6097 is an uncharacterized strain and CBS 107.38 is the ex-type strain for A. burnsii. No representatives of A. burnsii were included in the previous phylogenomic analyses, highlighting a shortcoming of the limited breadth of available genomic diversity. For both of these strains, their branches were relatively long and their relationships with the four main lineages differed between the three gene trees.
(2) A discrepancy among the three loci for lineage assignment was observed for only one strain: DAOMC 234885 (EGS 37.132), previously identified as A. gaisen, was confidently assigned to the gaisen lineage by ASA-10 and ASA-19, but to the alternata lineage by ASA-05. As this was the only case of disagreement between loci, the ASA-05 locus was re-sequenced from DAOMC 234885 and the conflict was confirmed.
Combined analyses of new candidate loci
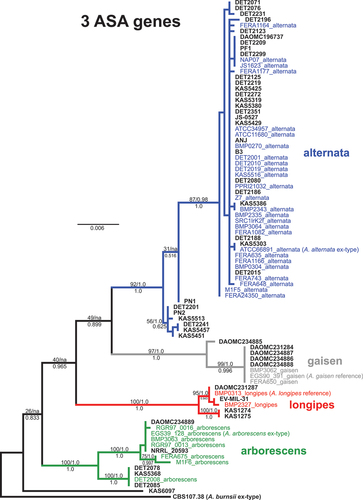
Data for the three candidate loci were combined and an ML tree was constructed under the best-fit partition model (). The main lineages all received significant support from ML bootstrapping (92–100%), Bayesian (posterior probability of 1.0), and multi-species coalescent analysis (posterior probability of 1.0). The alternata lineage was divided into two main clades, likely caused by the ASA-19 locus. The phylogenetic relationships between the main lineages were poorly resolved. A sister-relationship between alternata and gaisen lineages was recovered, in agreement with some previous phylogenomic analyses (Dettman and Eggertson Citation2021). This affinity, however, may be driven by the one strain that was assigned to both alternata and gaisen lineages (DAOMC 234885). When this strain was removed from the dataset and ML analysis was repeated, a different sister-relationship (longipes-gaisen) was significantly supported (results not shown). Note that these ASA markers were chosen for their potential ability to discriminate lineages from each other, not for investigating the phylogenetic relationships between the well-defined lineages.
Fig. 2 Maximum likelihood tree constructed from the three new candidate markers combined (ASA-05, ASA-10, and ASA-19). Phylogenetic tree branches are colour-coded by main lineage (blue = alternata, red = longipes, green = arborescens, grey = gaisen). Taxa from the independent test set have taxon labels in bold and black. Taxa that were included in our previous phylogenomic analyses have colour-coded taxon labels, and strain names are followed by lineage names. Branch support values are indicated above (ML bootstrap percentage/multispecies coalescent posterior probability) or below (Bayesian posterior probability) branches. Values of ‘na’ indicate the branch was not present in the respective analysis.
Comparison to a standard housekeeping gene
For comparison to an existing housekeeping gene commonly used in the molecular systematics of Ascomycetes, we investigated the RNA polymerase II second largest subunit gene (rpb2). We sequenced a portion of rpb2 from all 40 untested strains, and extracted homologous sequences from the nine new and 38 previously studied genome assemblies. Alignment statistics and the ML tree are shown in and , respectively.
Fig. 3 Maximum likelihood tree constructed from the RNA polymerase II second largest subunit gene (rpb2). Phylogenetic tree branches are colour-coded by main lineage (red = longipes, green = arborescens, grey = gaisen). Taxa from the independent test set have taxon labels in bold and black. Taxa that were included in our previous phylogenomic analyses have colour-coded taxon labels, and strain names are followed by lineage names. Branch support values are indicated above (ML bootstrap percentage) or below (Bayesian posterior probability) branches.
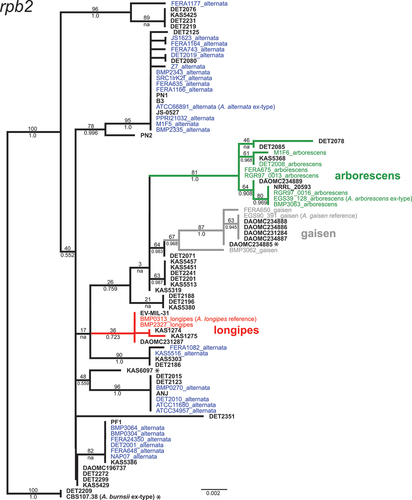
The rpb2 gene tree recovered three of the four main lineages (arborescens, gaisen, longipes), but the arborescens branch was the only one that received significant support. The alternata lineage was highly polyphyletic and was broken into several distinct subclades. KAS6097, which was divergent and placed outside of the main lineages in all three ASA gene trees, was not distinguished from the alternata lineage by rpb2. Furthermore, the divergent strain CBS 107.38 (A. burnsii) shared an identical rpb2 allele with DET2209, a strain that was confidently placed within the alternata lineage by all ASA loci. The only strain with conflicting placement among ASA loci (DAOMC 234885) was assigned to the gaisen lineage by rpb2, in agreement with ASA-10 and ASA-19. Despite the poorly defined alternata lineage, the assignment of strains to arborescens, longipes, and gaisen lineages matched between rpb2 and the ASA loci (). Given the lack of unique sequence polymorphisms shared by all alternata strains, and the discrepancies noted above, the rpb2 gene does not appear to be well suited for diagnosing the alternata lineage.
Nomenclature and taxonomy
Seventeen of the 49 test taxa had original species identifications available (). In four cases, these species identifications did not match the assignments inferred from our multi-locus sequence data.
(1) DAOMC 196737 was originally determined to be A. longipes (longipes lineage) but all four gene sequences assigned it to the alternata lineage. Alternaria longipes represents the ‘tobacco pathotype’ with presumed host-specificity, so this strain was likely named A. longipes simply because it was sampled from tobacco.
(2) DAOMC 234889 (EGS 90.131) was originally determined to be A. gaisen (gaisen lineage) but all four gene sequences assigned it to the arborescens lineage. Alternaria gaisen was long considered to be specifically pathogenic and restricted to Japanese pear, so this strain was likely named A. gaisen due to its host association with Japanese pear.
(3) EV-MIL-31 was described as a ‘tangerine pathotype’ of A. alternata (alternata lineage), however, all four loci consistently placed it within the longipes lineage. The longipes lineage includes A. longipes and a second, closely related species A. gossypina, which contains some strains that are pathogenic to tangerines. Based on this shared host affinity, EV-MIL-31 may belong to A. gossypina but further within-lineage analyses would be required for confirmation.
(4) ANJ was originally named A. tenuissima but was assigned here to the alternata lineage. This discrepancy is easily reconciled because A. tenuissima has been synonymized into A. alternata (Woudenberg et al. Citation2015) and is therefore within the alternata lineage.
Discussion
Which markers should be used for molecular systematics and diagnostics? Often, the most variable loci are chosen as top candidates for diagnostic marker development, as they provide the most information. However, the other important questions are how the variation is distributed within and between lineages, and whether or not the evolutionary history of the marker is consistent with the history of the organisms? These concepts were explored for section Alternaria in our previous study and form the basis for selecting the new markers presented here. The premise of ‘good’ diagnostic markers relies on the assumption that our taxonomic framework of well-defined lineages is biologically accurate. We view our current framework as the best possible estimate as it was inferred from as much data as possible (i.e. whole genomes).
The complexity and cost of whole genome sequencing still precludes its application as a routine diagnostic tool for large numbers of unidentified strains (e.g. pathology surveys, high-throughput culturing). Protocols based on PCR and Sanger sequencing are still routinely employed in diagnostic laboratories, often because they are cost-effective and easily transferable between laboratories around the world. Our goal was to characterize molecular markers that were informative and specific enough to fulfill that role for the main lineages of section Alternaria. Although not formally quantified, our experience with isolation attempts from Canadian substrates suggests that the alternata lineage is the most common, followed by arborescens. This finding is consistent with the high frequency of A. alternata (and synonym A. tenuissima) reported from various crops in other countries (da Cruz Cabral et al. Citation2017; Somma et al. Citation2019; Gao et al. Citation2021). As such, our aim was to establish molecular markers that were diagnostic for all lineages, but with special emphasis on the alternata lineage.
Sequence alignment-based phylogenetic approaches for fungal identification, as employed here, are recommended as more accurate alternatives to the simple similarity or BLAST-based methods (Lücking et al. Citation2020). Although the fungal research community has agreed upon a handful of commonly used genes for molecular systematics, the resolving power of these and any markers must be explored empirically and verified for the taxonomic group in question. The alternata lineage appears to be geographically widespread and to have a large population size, resulting in high levels of genetic diversity (Dettman and Eggertson Citation2021). Finding molecular markers that contain sequence variation that is unique to, and shared by all strains, in this lineage has proven to be quite challenging (Armitage et al. Citation2015; Woudenberg et al. Citation2015). Some loci contain lineage-specific variation for all lineages except alternata, so designation to the alternata lineage would occur in the absence of that variation. This approach may produce erroneous results if the unknown strains actually belong to a new, undiscovered species or lineage. Here, the goal was to design new markers that reflect the monophyly of the alternata lineage, and can diagnose all lineages based on exclusive presence of molecular polymorphisms.
Overall, the three new ASA markers performed well when challenged by the panel of 49 independent test taxa from section Alternaria: 46 of the taxa were unambiguously assigned to one of the main lineages, with full agreement between all three ASA loci. Two additional taxa consistently fell outside of the four main lineages, representing lineages that were not characterized in our previous phylogenomic study. One of these divergent strains (CBS 107.38) belongs to A. burnsii, whereas the other (KAS6097) may be from a previously undocumented lineage. Further work is required to assess the taxonomic placement of KAS6097 and genome sequencing of this strain is currently underway. The focus of this study was solely on section Alternaria, so additional work would be required to assess the utility of these markers for other sections in the genus.
Despite the predominance of agreement among the three ASA markers, a discrepancy in lineage assignment was observed for a single strain (DAOMC 234885). The sequences of the alleles possessed by DAOMC 234885 were identical to other taxa in the same lineage for ASA-05 and ASA-19 (gaisen), and differed by only one nucleotide for ASA-10 (alternata). This low divergence and shared alleles with confident placement within lineages suggest that this strain is an alternata-gaisen hybrid. DAOMC 234885 (also known as EGS 37.132 or Tottori 15A) has been studied intensively as A. gaisen and is a toxicity standard for black spot disease of Japanese pear (Simmons and Roberts Citation1993). Interestingly, a previous study noted that this strain displays a sporulation pattern that is non-typical of A. gaisen and could not be chosen as ‘definitive representative’ of the species (Simmons Citation1993). Our previous phylogenomic analyses found that, for some gene regions whose history did not match the rest of the genome, the gaisen and alternata lineages could share similar alleles (Dettman and Eggertson Citation2021). Future work should investigate how common this putative hybridization is and how it affects the resulting allelic composition along the genome.
For direct comparison with our ASA markers, we assessed the performance of a commonly sequenced ‘housekeeping’ gene to assign the same panel of 49 independent test taxa to main lineages. We chose the RNA polymerase II second largest subunit gene (rpb2) because it has been used for studying Alternaria section Alternaria, and appears to be one of the most informative housekeeping genes available (Woudenberg et al. Citation2015). Our preliminary analyses of other housekeeping genes, such as glyceraldehyde-3-phosphate dehydrogenase (gapdh) and translation elongation factor 1-alpha (tef1), showed much less promise for diagnostic purposes and were not characterized any further (data not shown).
Phylogenetic analyses revealed that the rpb2 gene has limited diagnostic power. It was the least variable and least informative (proportionally) of the four studied loci, and provided unequivocal support for only one main lineage (arborescens). The alternata lineage, which has the greatest need for new diagnostic markers, was divided into multiple clades that were interspersed among the other main lineages. Furthermore, the rpb2 tree failed to distinguish the two divergent strains (KAS6097 and CBS 107.38) from all other lineages, in contrast to the ASA markers. The rpb2 tree also demonstrates how relying on the best hits from a BLAST-based similarity approach could provide potentially misleading results when trying to identify unknown strains. These findings highlight the reasons why we chose to develop new markers for stable and consistent diagnostics for section Alternaria.
In addition to the sequence alignment-based phylogenetic approach described here, the polymorphisms identified within the ASA markers could be exploited for the future development of new lineage-specific detection tools (e.g. qPCR, RT-PCR, loop-mediated isothermal amplification). Such molecular tools would be useful for direct screening of substrates or samples for the presence of infection by section Alternaria lineages. These diagnostic tools would need to be comprehensively tested on other closely related section Alternaria lineages that may not be represented here. Furthermore, testing and validation should also include lineages from other sections, to ensure that off-target detection does not influence the assay results.
The independent test strains were treated blindly and their previous species determinations had no effect on our lineage assignment methods. Many of these original determinations were made prior to the advent of molecular systematics and were informed by morphology and host association. As morphological variation is typically insufficient to identify species within section Alternaria (Andrew et al. Citation2009; Lawrence et al. Citation2013; Armitage et al. Citation2015; Woudenberg et al. Citation2015), host association is often the deciding factor. Based on our results, and those of other studies, we now know that many mis-identifications have been caused by false assumptions of exclusive host specificity. Some patterns of host specificity have broken down when challenged by more rigorous, molecular-based species identifications, and many pathotypes have been revealed to be polyphyletic (Peever et al. Citation2004; Rotondo et al. Citation2012; Woudenberg et al. Citation2015; Armitage et al. Citation2020; Dettman and Eggerston Citation2021). We recommend that molecular data form the primary basis for lineage/species assignment in section Alternaria, and that host association is used as supporting information only.
Species nomenclature has a direct impact on quarantine and regulatory processes, as the species name determines whether or not an organism is regulated under international phytosanitary practices. For example, the species A. gaisen causes black spot of pear and is regulated by CFIA and several other countries. Pear-associated strains that are mis-identified as A. gaisen (e.g. DAOMC 234889) could cause unnecessary trade issues. Species nomenclature also matters for transferability of research findings between species. For example, the EV-MIL-31 strain has a history of study as A. alternata causing citrus brown spot (Lin et al. Citation2009, Citation2010), and has been involved in much work on oxidative stress tolerance. All sequence data, however, suggests that EV-MIL-31 is not A. alternata, but rather is within the longipes lineage (putatively A. gossypina).
Overall, we predict that these new diagnostic markers will perform well when challenged by the majority of section Alternaria strains sampled from agricultural sources. As the majority of our independent test strains were of Canadian origin, we may have missed some important biodiversity that is present in other international settings. Additional work is needed to test how these markers will perform with rarer section Alternaria species that were not able to be represented here. Continued efforts towards the genome sequencing of multiple reference strains for all section Alternaria species will support the marker refinement process.
Data availability
The 168 newly generated nucleotide sequences are available under NCBI GenBank accessions OL451963-OL452001 for ASA-05, OL469328-OL469366 for ASA-10, OL469367-OL469406 for ASA-19, and OL469407-OL469446 for rpb2 (Supplementary Table 2).
TABLE_SUPP_2_for_TEXT.xlsx
Download MS Excel (11.4 KB)TABLE_SUPP_1_for_TEXT.xlsx
Download MS Excel (13.5 KB)Acknowledgements
We thank Allen Xue and Yuanhong Chen for providing access to grain samples for fungal isolation; Keith Seifert, Stephen Brière, and staff at the Canadian Collection of Fungal Cultures for providing fungal strains. We also thank Elizabeth Sears for DNA extraction work and the staff at the AAFC Molecular Technologies Laboratory for sequencing support.
Disclosure statement
No potential conflict of interest was reported by the author(s).
Supplementary material
Supplemental data for this article can be accessed online here: https://doi.org/10.1080/07060661.2022.2061605
Additional information
Funding
References
- Aichinger G, Del Favero G, Warth B, Marko D. 2021. Alternaria toxins—still emerging? Compr Rev Food Sci Food Saf. 20:4390–4406. doi:10.1111/1541-4337.12803.
- Andrew M, Peever TL, Pryor BM. 2009. An expanded multilocus phylogeny does not resolve morphological species within the small-spored Alternaria species complex. Mycologia. 101:95–109. doi:10.3852/08-135.
- Armitage AD, Barbara DJ, Harrison RJ, Lane CR, Sreenivasaprasad S, Woodhall JW, Clarkson JP. 2015. Discrete lineages within Alternaria alternata species group: identification using new highly variable loci and support from morphological characters. Fungal Biol. 119:994–1006. doi:10.1016/j.funbio.2015.06.012.
- Armitage AD, Cockerton HM, Sreenivasaprasad S, Woodhall J, Lane CR, Harrison RJ, Clarkson JP. 2020. Genomics evolutionary history and diagnostics of the Alternaria alternata species group including apple and Asian pear pathotypes. Front Microbiol. 10:3124. doi:10.3389/fmicb.2019.03124.
- Barberán A, Ladau J, Leff JW, Pollard KS, Menninger HL, Dunn RR, Fierer N. 2015. Continental-scale distributions of dust-associated bacteria and fungi. Proc Natl Acad Sci USA. 112:5756–5761. doi:10.1073/pnas.1420815112.
- Borowiec ML. 2016. AMAS: a fast tool for alignment manipulation and computing of summary statistics. PeerJ. 4:e1660. doi:10.7717/peerj.1660.
- da Cruz Cabral L, Rodriguero M, Stenglein S, Fog Nielsen K, Patriarca A. 2017. Characterization of small-spored Alternaria from Argentinean crops through a polyphasic approach. Int J Food Microbiol. 257:206–215. doi:10.1016/j.ijfoodmicro.2017.06.026.
- Dettman JR, Eggertson Q. 2021. Phylogenomic analyses of Alternaria section Alternaria: a high-resolution, genome-wide study of lineage sorting and gene tree discordance. Mycologia. 113:1218–1232. doi:10.1080/00275514.2021.1950456.
- Edgar RC. 2004. MUSCLE: multiple sequence alignment with high accuracy and high throughput. Nucleic Acids Res. 32:1792–1797. doi:10.1093/nar/gkh340.
- Gao J, Yang MJ, Xie Z, Lu BH, Hsiang T, Liu LP. 2021. Morphological and molecular identification and pathogenicity of Alternaria spp. associated with ginseng in Jilin province, China. Can J Plant Pathol. 43:537–550. doi:10.1080/07060661.2020.1858167.
- Gonçalves VN, Cantrell CL, Wedge DE, Ferreira MC, Soares MA, Jacob MR, Oliveira FS, Galante D, Rodrigues F, Alves TMA, et al. 2016. Fungi associated with rocks of the Atacama Desert: taxonomy, distribution, diversity, ecology and bioprospection for bioactive compounds. Environ Microbiol. 18:232–245. doi:10.1111/1462-2920.13005.
- Lawrence DP, Gannibal PB, Peever TL, Pryor BM. 2013. The sections of Alternaria: formalizing species-group concepts. Mycologia. 105:530–546. doi:10.3852/12-249.
- Lin CH, Chung KR. 2010. Specialized and shared functions of the histidine kinase- and HOG1 MAP kinase-mediated signaling pathways in Alternaria alternata, a filamentous fungal pathogen of citrus. Fungal Genet Biol. 47:818–827. doi:10.1016/j.fgb.2010.06.009.
- Lin C-H, Yang SL, Chung K-R. 2009. The YAP1 homolog–mediated oxidative stress tolerance is crucial for pathogenicity of the necrotrophic fungus Alternaria alternata in citrus. Mol Plant-Microbe Interact. 22:942–952. doi:10.1094/MPMI-22-8-0942.
- Liu YJ, Whelen S, Hall BD. 1999. Phylogenetic relationships among ascomycetes: evidence from an RNA polymerse II subunit. Mol Biol Evol. 12:1799–1808. doi:10.1093/oxfordjournals.molbev.a026092.
- Lücking R, Aime MC, Robbertse B, Miller AN, Ariyawansa HA, Aoki T, Cardinali G, Crous PW, Druzhinina IS, Geiser DM, et al. 2020. Unambiguous identification of fungi: where do we stand and how accurate and precise is fungal DNA barcoding? IMA Fungus. 11:14. doi:10.1186/s43008-020-00033-z.
- Luis P, Saint-Genis G, Vallon L, Bourgeois C, Bruto M, Marchand C, Record E, Hugoni M. 2019. Contrasted ecological niches shape fungal and prokaryotic community structure in mangroves sediments. Environ Microbiol. 21:1407–1424. doi:10.1111/1462-2920.14571.
- Meena M, Gupta SK, Swapnil P, Zehra A, Dubey MK, Upadhyay RS. 2017. Alternaria toxins: potential virulence factors and genes related to pathogenesis. Front Microbiol. 8:1451. doi:10.3389/fmicb.2017.01451.
- Mujahid C, Savoy MC, Baslé Q, Woo PM, Ee ECY, Mottier P, Bessaire T. 2020. Levels of Alternaria toxins in selected food commodities including green coffee. Toxins (Basel). 12:595. doi:10.3390/toxins12090595.
- Nguyen LT, Schmidt HA, Von Haeseler A, Minh BQ. 2015. IQ-TREE: a fast and effective stochastic algorithm for estimating maximum-likelihood phylogenies. Mol Biol Evol. 32:268–274. doi:10.1093/molbev/msu300.
- Nishikawa J, Nakashima C. 2020. Japanese species of Alternaria and their species boundaries based on host range. Fungal Syst Evol. 5:197–282. doi:10.3114/fuse.2020.05.13.
- Peever TL, Su G, Carpenter-Boggs L, Timmer LW. 2004. Molecular systematics of citrus-associated Alternaria species. Mycologia. 96:119–134. doi:10.2307/3761993.
- Robideau GP, De Cock AW, Coffey MD, Voglmayr H, Brouwer H, Bala K, Chitty DW, Désaulniers N, Eggertson QA, Gachon CM, et al. 2011. DNA barcoding of oomycetes with cytochrome c oxidase subunit I and internal transcribed spacer. Mol Ecol Resour. 6:1002–1011. doi:10.1111/j.1755-0998.2011.03041.x.
- Ronquist F, Huelsenbeck JP. 2003. MrBayes 3: bayesian phylogenetic inference under mixed models. Bioinformatics. 19:1572–1574. doi:10.1093/bioinformatics/btg180.
- Rotem J. 1994. The genus Alternaria. Biology, epidemiology and pathogenicity. St. Paul (MN): APS Press.
- Rotondo F, Collina M, Brunelli A, Pryor BM. 2012. Comparison of Alternaria spp. collected in Italy from apple with A. Mali and other AM-toxin producing strains. Phytopathology. 102:1130–1142. doi:10.1094/PHYTO-04-12-0076-R.
- Simmons EGS. 1993. Alternaria themes and variations (63-72). Mycotaxon. 47:91–107.
- Simmons EGS, Roberts R. 1993. Alternaria themes and variations (73). Mycotaxon. 47:109–140.
- Somma S, Amatulli MT, Masiello M, Moretti A, Logrieco AF. 2019. Alternaria species associated to wheat black point identified through a multilocus sequence approach. Int J Food Microbiol. 293:34–43. doi:10.1016/j.ijfoodmicro.2019.01.001.
- Thomma BPHJ. 2003. Alternaria spp.: from general saprophyte to specific parasite. Mol Plant Pathol. 4:225–236. doi:10.1046/j.1364-3703.2003.00173.x.
- Tralamazza SM, Piacentini KC, Iwase CHT, Rocha LDO. 2018. Toxigenic Alternaria species: impact in cereals worldwide. Curr Opin Food Sci. 23:57–63. doi:10.1016/j.cofs.2018.05.002.
- Woudenberg JHC, Seidl MF, Groenewald JZ, de Vries M, Stielow JB, Thomma BPHJ, Crous PW. 2015. Alternaria section Alternaria: species, formae speciales or pathotypes? Stud Mycol. 82:1–21. doi:10.1016/j.simyco.2015.07.001.
- Zhang C, Rabiee M, Sayyari E, Mirarab S. 2018. ASTRAL-III: polynomial time species tree reconstruction from partially resolved gene trees. BMC Bioinform. 19(Suppl 6):153. doi:10.1186/s12859-018-2129-y.