
Abstract
The post-genomic era is characterized by a range of high throughput profiling methods capable of broadly characterizing gene expression levels, protein, and metabolite abundances. Application of these methods, enzyme profiling, and more recently, protein-metabolite interactions and flux analysis have alongside modeling approaches allowed us to refine our understanding of the regulation of metabolism even in the case of the canonical pathways of primary plant metabolism. Here we review recent insights obtained by using such methods in the context of our previous knowledge. In doing so, we hope to highlight the effectiveness of these methods and postulate that their application to less well-studied metabolic pathways will likely allow the elucidation of the hitherto unknown mechanism of metabolic regulation.
I. Introduction
Study of metabolic regulation this century – like that of most facets of plant biology – is greatly facilitated by genomic information. Since the landmark sequencing of Arabidopsis thaliana in 2000 (Arabidopsis Genome Initiative, Citation2000), genome sequencing has gathered pace with over 100 species now sequenced not to mention 1000s of accessions of certain species (Purugganan and Jackson, Citation2021). With regard to the understanding of metabolic regulation post-genomic technologies particularly those relying on mass spectrometry have been particularly informative. In this respect proteomics has facilitated the simultaneous determination of the levels of up to 18 000 proteins (corresponding to approximately 50% of the genes encoded in Arabidopsis (Mergner et al., Citation2020). In addition, mass spectra of peptide mixtures allow the quantification of post-translational modifications of amino acid residues including amongst others phosphorylation, acylation and ubiquitination, oftentimes providing direct determinants of the mechanism of functional regulation (van Wijk et al., Citation2021). Whilst experimental analysis of protein–protein interactions is slowly providing us with information concerning the consequences of protein aggregation and disaggregation. Similarly, whilst relatively poor in terms of current coverage (Alseekh and Fernie, Citation2018) allowing the quantification of only a minor percentage of the myriad of metabolites estimated to be present in the plant kingdom (200 000 to 1 million; (Fang et al., Citation2019; Wang et al., Citation2019)), metabolomics has provided some insight into metabolic regulation with early studies revealing co-regulation of primary metabolite levels in a manner consistent with the patterns of feedforward and feedback regulation of metabolism (which we describe below). Mass spectrometry (Allen and Young, Citation2020), alongside nuclear magnetic resonance spectroscopy (Kruger and Ratcliffe, Citation2021), has also been harnessed as a tool to investigate flux at a much higher resolution than typically attained by radiolabeling approaches (Fernie et al., Citation2005) – allowing the discrimination of flux through single enzymes and branched pathways in addition to the bulk fluxes that could be estimated previously. In parallel, robotization of enzyme extraction and measurement has offered a more rapid way of measuring multiple enzyme activities than possible a mere 20 years ago (Gibon et al., Citation2004). Finally, in recent years the adoption of multiple profiling techniques in so-called multi-omics approaches has proven a powerful route to gain functional information since for example changes in the post-translational modification of an enzyme can be correlated directly to changes in the pool size of certain metabolites (Zhang et al., Citation2021). In addition, in recent years a range of techniques to evaluate protein-metabolite interactions have been developed (Diether and Sauer, Citation2017; Venegas-Molina et al., Citation2021; Wagner et al., Citation2021), and it can be anticipated that their application will finally lead to the identification of a considerable number of metabolite receptors which have to date with the exception of nitrate and hormone receptors proven somewhat elusive in plants. Given the massive torrent of data that these profiling technologies have generated researchers are often content on evaluating it in isolation (see ). However, it is our belief that the data is much more valuable when viewed through the lens of what we already knew and as such we aim here to contextualize the novel insights obtained from sequencing data within the framework of our understanding of metabolism that was derived from the targeted study of single enzymes and or pathways in the last century.
A. The 20th century foundation of plant metabolic regulation
Whilst novel regulatory mechanisms are still being uncovered our understanding of the regulation of primary metabolism was already very well developed by the final decades of the last Century. As we will illustrate with examples from four canonical pathways of plant primary metabolism, namely the Calvin-Benson cycle, glycolysis, the TCA cycle and the sucrose to starch transition, the major mechanisms controlling flux through these pathways had already been established by the end of last Century (Miflin and Lea, Citation1982; Stitt, Citation1987; Plaxton, Citation1996; Schurmann and Jacquot, Citation2000; Fernie et al., Citation2004). Indeed, they have long become staples of plant metabolism textbooks. Here we will provide a brief overview of some of the seminal experiments and the key concepts that arose from their findings. Although more recent studies have indicated that transcriptional regulation is an occasional mode employed in the regulation of primary metabolism it is not a common mode of regulation since many enzymes or primary metabolism are already highly expressed (Gibon et al., Citation2004). We will therefore not discuss it in any detail here but rather limit ourselves to fine control by metabolites, pH and post-translational regulation of proteins.
The now irregularly used term fine control was frequently applied to metabolism at the end of the last century to define fast energetically, inexpensive, regulatory devices which modulate the activity of the preexisting enzyme molecule (Plaxton, Citation1997). In terms of the pathways mentioned above the following forms of metabolic control are particularly pertinent: alteration of substrate or co-substrate concentration, variation of pH, allosteric effectors or covalent modifications of proteins. The first method is applicable to all enzymes but particularly important for so-called rate-limiting enzymes which often have a large proportion of the control of flux divested in them (we will return to this latter when discussing ADP glucose pyrophosphorylase), but is best considered alongside allosteric regulation. We do so in the following paragraph by describing metabolic regulation of the fructose-2, 6-bisphosphate (Fru-2,6-P2) system below which integrates the pathways of glycolysis and the sucrose to starch transition as well as describing adenylate and redox control of the TCA cycle. The second method of fine control is undoubtedly best illustrated by the diurnal control of the Calvin-Benson cycle. We return to these canonical cycles to showcase examples of metabolic regulation by covalent modification of proteins.
In the 1980s, Fru-2,6-P2 was discovered in animals (Van Schaftingen and Hers, Citation1980), subsequently being found in fungi (Lederer et al., Citation1981), plants (Sabularse and Anderson, Citation1981) and protists (Van Schaftingen and Opperdoes, Citation1985). Fru-2,6-P2 was first discovered in C3 plants such as mung bean, spinach and pea (Sabularse and Anderson, Citation1981; Cseke et al., Citation1982) but has since been detected in C4 plants such as maize (Soll et al., Citation1985) and CAM plants such as Ananus comosus and Mesmbryanthum crystallinium (Fahrendorf et al., Citation1987). It is additionally present in both photosynthetic and nonphotosynthetic tissues, although its role in the latter remains unclear (Fernie et al., Citation2001). Nonaqueous fractionation studies revealed that Fru-2,6-P2 is present in the cytosol of leaves (Gerhardt et al., Citation1987), where its concentration is usually of the range 1-10uM, although it can be as much as ten times this in some CAM species (Fahrendorf et al., Citation1987).
In all systems, the cellular concentrations of Fru-2,6-P2 is a function of the relative activities of 6-phosphofructo, 2-kinase (which synthesizes it; (El-Maghrabi et al., Citation1981)) and fructose-2, 6-bisphosphatase (which degrades it; El-Maghrabi et al., Citation1982). In mammals, both activities are long-known to reside on a single bifunctional enzyme (El-Maghrabi et al., Citation1982) and have been thoroughly characterized. In plants, a bifunctional 6-phosphofructo, 2-kinase/fructose-2, 6-bisphosphatase was first isolated from spinach leaves (Cséke and Buchanan, Citation1983; Stitt et al., Citation1984) and was subsequently, unsurprisingly found to exhibit similar distribution at species, tissue and subcellular levels to Fru-2,6-P2. Indeed much characterization of the bifunctional enzyme as well as a plant monofunctional fructose-2, 6-bisphosphatase suggest that they operate similarly across species being subject to exquisite allosteric control but by contrast to the situation in animals there being no clear evidence of regulation by post-translational phosphorylation of the enzyme. To be more specific, the enzyme is clearly phosphorylated and binds 14-3-3 but the function of this modification has not been assigned. Spinach leaf 6-phosphofructo, 2-kinase was found to exhibit hyperbolic saturation curves with respect to ATP (Km in the presence of 5 mM Pi, is 0.25 mM) (Cséke and Buchanan, Citation1983), but a sigmoidal saturation curve for Fru-6-P (S0.5 is between 1.25 and 2.5 mM) (Stitt, Cseke, et al., Citation1984) particularly at low Pi (Cséke and Buchanan, Citation1983; Stitt et al., Citation1984; Larondelle et al., Citation1986). 6-phosphofructo, 2-kinase is activated by Pi, which increases the Vmax and the affinity for Fru-6-P (Cséke and Buchanan, Citation1983) but is inhibited by PPi, which acts competitively to ATP and induces sigmoidal ATP saturation kinetics (Larondelle et al., Citation1986; Macdonald et al., Citation1987). 6-phosphofructo, 2-kinase is inhibited by several 3-carbon phosphoesters, including 3-PGA, 2PGA, PEP, DHAP, and the 2-C intermediate glycollate-2-P (Cséke et al., Citation1983; Stitt, Cseke, et al., Citation1984). These metabolites all act antagonistically to Pi except for DHAP, which is a poor inhibitor unless high (10 mM) Pi is present (Stitt, Cseke, et al., Citation1984). As they have similar Ki values, the relative importance of these inhibitors in vivo depends on the cytosolic levels of these metabolites. (Stitt, Citation1990b) estimated the relative abundance of PEP: 3-PGA: 2-PGA to be 3:10:1, based on the equilibrium constants of enolase and phosphoglycerate mutase. This would suggest that 3-PGA is likely to be the most important physiological inhibitor of this activity – a fact that was supported by the results of pharmacological experiments in spinach (Ekkehard and Stitt, Citation1989).
Kinetic properties of the bifunctional fructose-2, 6-bisphosphatase from spinach mesophyll cells were also well characterized fructose 6-P is a potent noncompetitive inhibitor with a Ki of 0.2 mM, and Pi acts as a relatively weak competitive inhibitor (Stitt, Cseke, et al., Citation1984; Larondelle et al., Citation1986; Citation1989) In plants, as in animals, Pi decreases the sensitivity of the bifunctional Fru-2,6-P2 to inhibition by fructose 6-P (Herzog et al., Citation1984). Similarly, the kinetic properties of spinach mesophyll monofunctional fructose-2, 6-bisphosphatase reveal that it is also inhibited by fructose 6-P and Pi; however, the mechanisms of inhibition are distinct from those of the bifunctional enzyme. Fru-2,6-P2 acts as a weak mixed-type inhibitor of the spinach monofunctional fructose-2, 6-bisphosphatase; at 10 μM Fru-2,6-P2 4 mM fructose 6-only reduced activity by 30% (Macdonald et al., Citation1989) Pi, however, is a strong mixed-type inhibitor (Macdonald et al., Citation1989). The monofunctional fructose-2, 6-bisphosphatase is also competitively inhibited by fructose-1, 6-P2 (Larondelle et al., Citation1989), and by divalent cations including Mg2+, Ca2+ and Mn2+ at 25 μM Fru-2,6-P2 (Macdonald et al., Citation1989). These kinetic properties allowed estimation of the relative contribution of bi- and monofunctional fructose-2, 6-bisphosphatase to fructose-2, 6-bisphosphate hydrolysis with the general conclusion being that both played a physiological role (Larondelle et al., Citation1989; Stitt, Citation1990b).
Fru-2,6-P2 exerts a strong regulatory influence on the interconversion of Fru-6-P and fructose 1, 6-bisphosphate (Fru 1, 6-P2). In plants, it does this by affecting the activities of both cytosolic fructose 1, 6-bisphosphatase (FBPase) (Cseke et al., Citation1982; Kruger and Beevers, Citation1984) and pyrophosphate:fructose 6-phosphate 1-phosphotransferase (PFP) (Sabularse and Anderson, Citation1981; Cseke et al., Citation1982; Kruger et al., Citation1983; Kombrink and Kruger, Citation1984; Kruger and Beevers, Citation1984). In contrast to its effects in animal tissues and in fungi, Fru-2,6-P2 does not stimulate plant PFK (Sabularse and Anderson, Citation1981; Kruger and Scott, Citation1995).
Cytosolic FBPase was found to hydrolyze Fru-1,6-P2 to Fru-6-P in photosynthetic tissues such as spinach (Cseke et al., Citation1982) and wheat leaves (Stitt et al., Citation1982). The properties of the enzyme are generally conserved. In the absence of effectors, the Km for Fru-1, 6-P2 is about 2-3μM (Herzog et al., Citation1984; Kruger and Beevers, Citation1984) [and its saturation kinetics for Fru-1, 6-P2 are hyperbolic (Zimmermann et al., Citation1978; Stitt, Citation1987). Fru-2,6-P2 is a high-affinity inhibitor of cytosolic FBPase (Cseke et al., Citation1982) and changes the saturation curve for Fru-1, 6-P2 to sigmoidal (Herzog et al., Citation1984; Kruger and Beevers, Citation1984). This is achieved by approximately halving the Vmax and halving the affinity for Fru-1, 6-P2 in the presence of 2 μM Fru-2,6-P2 (Stitt and Heldt, Citation1985). AMP acts as a weak mixed inhibitor with respect to Fru-1, 6-P2 when present in the 50–200 μM range (Zimmermann et al., Citation1978; Stitt, Cseke, et al., Citation1984). However, it shows a marked synergistic inhibition in the presence of Fru-2,6-P2 (Kruger and Beevers, Citation1984). By contrast, the chloroplastic FBPase is not significantly inhibited by Fru-2,6-P2 (Cseke et al., Citation1982). Fru-2,6-P2 uptake into isolated chloroplasts has been reported (Smeekens et al., Citation1989). However, a range of evidence suggests that it is extremely unlikely that this is of physiological significance for several reasons. First, the reported rate of uptake is very slow (0.01 μM mgChl−1 h−1) and uptake is inhibited by physiological concentrations of PPi (Smeekens et al., Citation1989). Secondly, the inhibition of chloroplastic FBPase only occurs at very high concentrations of Fru-2,6-P2 at 0.8 mM Fru-1,6-P2 the Ki for Fru-2,6-P2 is 0.1 mM (Gottschalk et al., Citation1982). Concentrations of Fru-2,6-P2 are usually well below this in plants. Chloroplastic FBPase is insensitive to low Fru-2,6-P2 even in the presence of AMP (Cseke et al., Citation1982). Thirdly, nonaqueous fractionation studies have shown that despite the ability of isolated chloroplasts to take up this metabolite Fru-2,6-P2 is largely extra-chloroplastic (Stitt et al., Citation1982; Fahrendorf et al., Citation1987)
Although Fru-2,6-P2 has no effect on plant PFK, it does markedly stimulate pyrophosphate: fructose-6-phosphate phosphotransferase (PFP). PFP is located in the cytosol where it is thought to catalyze a near-equilibrium reaction in which Fru-6-P is converted to Fru-1, 6-P2utilizing PPi as the phosphoryl donor (Sabularse and Anderson, Citation1981; Carnal and Black, Citation1983; Stitt, Citation1998). PFP was first reported in plants by (Carnal and Black, Citation1983), subsequently being reported in a wide range of species, and in all instances PFP was found to be stimulated by Fru-2,6-P2. Kinetic studies revealed this stimulation occurs at nanomolar concentrations of Fru-2,6-P2 (Cseke et al., Citation1982; Kruger et al., Citation1983; Kombrink and Kruger, Citation1984), which increase the Vmax of the forward reaction with a Ka of 5 nM (Stitt, Citation1989) and increase the affinity for both Fru-6-P and Fru-1,6-P2 approximately 10-fold (Kombrink and Kruger, Citation1984; Stitt, Citation1989). Kinetic studies on potato tuber PFP (Trevanion and Kruger, Citation1991) suggest that the levels of Fru-2,6-P2 found in the cytosol of plant cells are such that PFP will be fully activated in vivo. This suggests that changes in the concentration of Fru-2,6-P2 will have no effect on the activity of PFP. However, three considerations argued against this suggestion. First, at low temperatures, the Ka of PFP is increased several fold from that at 25 °C. Therefore, at low temperatures at least, the Ka of PFP is in the range in which changes in the Fru-2,6-P2 concentration will influence PFP activity. Secondly, (Nielsen, Citation1995) has reassessed the concentration of free cytosolic Fru-2,6-P2 in relation to the number of allosteric binding sites of PFP. They suggested that despite the fact that the cytosolic concentration of Fru-2,6-P2 is typically estimated at between 1–10 μM, the amount of free Fru-2,6-P2 in barley leaves is insufficient to saturate all the allosteric binding sites of PFP, due to high concentrations of PFP in barley. Thirdly, the in vivo Ka (Fru-2,6-P2) is likely to be significantly higher than that estimated in vitro Ka, since metabolites such as Pi, Fru-1, 6-P2, and 3-PGA decrease the affinity for Fru-2,6-P2 (Kombrink and Kruger, Citation1984; Stitt et al., Citation1989). Thus changes in the concentration of free Fru-2,6-P2 will lead to changes in PFP activity. This is concordant with the finding that increasing the concentration of Fru-2, 6P2 in transgenic tobacco by expression of a mammalian 6PF2K activity leads to changes in metabolite concentrations and fluxes that are consistent with in vivo activation of PFP (Fernie et al., Citation2001).
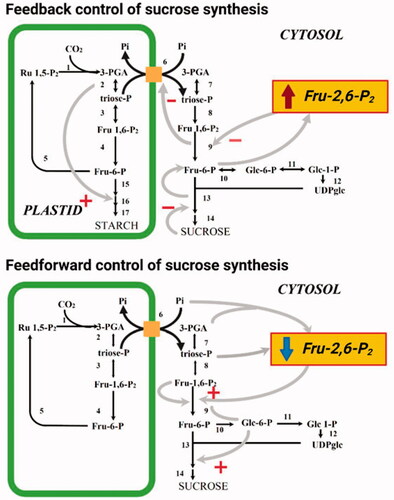
In plants, Fru-2,6-P2 plays an important role in two aspects of the regulation of sucrose metabolism in photosynthetic tissues (). One of these is feedforward control in which sucrose synthesis is stimulated in response to the increase in photosynthesis at the start of the photoperiod. The other is feedback control, whereby an accumulation of sucrose decreases the rate of sucrose synthesis and diverts photosynthates into starch production (Stitt and Paul Quick, Citation1989). At the start of the photoperiod, the rate of photosynthesis increases. This results in an increased cytosolic DHAP concentration due to a greater rate of export from the chloroplast via the triose-P translocator in exchange for Pi, and therefore the cytosolic 3-PGA/Pi ratio rises (Stitt et al., Citation1984; Neuhaus et al., Citation1989). These changes bring about an increase in the cytosolic concentration of its substrate, Fru-1,6-P2, which is nearly in equilibrium with triose-P. Simultaneously with the increase in the levels of Fru-1,6-P2 is a rapid drop in the Fru-2,6-P2 concentration, which relieves inhibition of cytosolic FBPase. A consequence of the increased flux through FBPase is an increase in the cytosolic concentration of Glc 6-P. This leads to an increased Glc 6-P/Pi ratio and causes potent allosteric activation of sucrose phosphate synthase (SPS) (Huber and Hanson, Citation1992). During the day, the rate of sucrose synthesis increases with the rate of photosynthesis. If the rate of sucrose production exceeds its rate of export from the cell, sucrose will accumulate. However, in response to feedback signals probably related to the absolute level of sucrose the rate of synthesis is decreased via inhibition of SPS (Stitt, Citation1990a). This inhibition leads to increased cytosolic levels of hexose-P, which result in a large increase in the Fru-2,6-P2 level leading to inhibition of cytosolic FBPase. The reduced rate of sucrose synthesis additionally prevents Pi cycling, which has consequently been shown to result in an accumulation of 3-PGA in isolated chloroplasts (Heldt et al., Citation1977). This is probably due to the fact that phosphoglycerate kinase is particularly sensitive to the falling concentrations of ATP that occur during these conditions. The elevated chloroplastic 3-PGA/Pi ratio stimulates starch synthesis by the activation of ADP-glucose pyrophosphorylase. As mentioned above, ADP-glucose pyrophosphorylase was classically considered as the rate-limiting step of starch biosynthesis it operates highly removed from equilibrium massively favoring the production, as opposed to degradation, of ADP-glucose (Stitt et al., Citation2010). In addition, the proposed role for Fru-2,6-P2 in photosynthetic tissue is also supported by studies in which the level of Fru-2,6-P2 was directly modified by the expression of a mammalian 6PF2K in tobacco plants (Scott et al., Citation1995). Leaves of these transgenic tobacco were demonstrated to exhibit a reduction in the rate of sucrose accumulation that is proportional to the increase in the level of Fru-2,6-P2 and a corresponding increase in the rate of starch accumulation (Scott et al., Citation1995). In summary, the rate of carbon export from the chloroplast depends on a balance between feedforward mechanisms which decrease Fru-2,6-P2 (and activate SPS) and feedback mechanisms which increase Fru-2,6-P2 (and inhibit SPS). Whilst this is the most exquisitely studied regulation of glycolysis and the sucrose-starch transition it is important to note that additional metabolite-regulated control of glycolysis (Plaxton, Citation1996) and metabolite-regulated control of the Calvin-Benson cycle are also well characterized (Raines, Citation2003).
Figure 1. System-level characterization of metabolic regulation (A) by multi-omics analysis of plant species, natural accessions, mutants, conditions, developmental stages, organs, tissues and organelles (B). Progress in chromatography, mass spectrometry, robotization, sequencing (among others) (C) has enabled the identification and quantification of genomes, transcriptomes, proteomes, and metabolomes (D). Computational analysis of the obtained data led to identifying novel interactions, improved annotation of metabolic pathways, and understanding of signaling and regulatory cascades upstream and downstream of metabolism (E). Figure was created using Biorender.com.
Figure 2. The role of Fru-2,6-P2 in feedback and feedforward control of sucrose synthesis. + represents allosteric activation, - represents allosteric inhibition. Reactions shown are catalyzed by the following enzymes (note in some instances multiple reactions are represented by a single arrow): 1, Rubisco; 2, chloroplastic PGK and chloroplastic TPI; 3, chloroplastic Fru-1-6-P2 aldolase; 4, chloroplastic FBPase; 5, transketolase, sedoheptolase-1,7-bisphosphatase aldolase, sedoheptolase-1,7-bisphosphatase, phosphopentoepimerase, phosphoriboisomerase and phosphoribulokinase; 6, triose phosphate transporter; 7, cytoslic PGK and cytosolic TPI; 8, cytosolic Fru-1-6-P2 aldolase; 9, cytosolic FBPase; 10, cytosolic PGI; 11, cytosolic PGlcM, 12 UGPase, 13 SPS, 14 sucrose phosphatase; 15, choroplastic PGI and chloroplstic PGlcM; 16, AGPase; 17, starch synthase and branching enzyme.
In common with the above pathways that of the tricarboxylic acid (TCA) cycle and its associated enzymes pyruvate dehydrogenase and NAD-malic enzyme has been well characterized at the metabolite level. Indeed the regulatory properties of the pyruvate dehydrogenase complex (PDC) was studied in some detail by Randall and coworkers being found to be subject to product inhibition by both NADH and acetyl CoA which act as strong competitive inhibitors of NAD+ and CoA, respectively (Budde et al., Citation1991). Sulfate ions, CoA and fumarate activate whilst chloride inhibits NAD-ME (Canellas and Wedding, Citation1984), whilst NADH and NADPH inhibit the NAD+-isocitrate dehydrogenase (McIntosh and Oliver, Citation1992), the inhibition by NADPH is not competitive with either NAD + or NADH suggesting its regulation may be allosteric. By contrast, to the enzyme from nonplant sources which are affected by ATP, ADP and AMP, the plant enzyme is insensitive to adenylates (McIntosh and Oliver, Citation1992). The 2-oxoglutarate dehydrogenase complex, similar to many other TCA cycle enzymes, is competitively inhibited by NADH (Wigge et al., Citation1993). Subsequent work this Century characterizing the kinetics of succinyl CoA ligase, and mitochondrial malate dehydrogenase and fumarase (Studart-Guimarães et al., Citation2005; Zubimendi et al., Citation2018) suggesting that these enzymes, in combination with those described above, are regulated in a manner that would allow a high cyclic flux in times when carbon is in rich supply and a reduced flux in times of carbon deficiency. Although, as we will describe later, flux through the TCA cycle is also greatly influenced by covalent modification.
We listed pH as a major form of metabolic regulation. Before discussing this in detail it is important to note that the literature is overflowing with lists of enzymes that have been noted to display altered kinetics on changing pH, however, the physiological relevance of these effects is often questionable. One needs to be sure that the pH range under which these alterations in in vitro kinetic measurements are observed are similar to those that the enzyme “sees” in its intracellular environment. The pH-mediated, light-dependent activation of several of the enzymes of the Calvin-Benson cycle provides an excellent example of where such control is relevant. Photosynthetic electron transport has long been characterized to be linked to H+ uptake into the thylakoid lumen (Plaxton, Citation1997). This establishes a lumen to stroma proton gradient, which is believed to drive photophosphorylation. Transport of H+ ions into the lumen results in a light-dependent increase of stromal pH from approximately 7.0 to approximately 8.0. Conversely, in the absence of light H+ ions leak back into the stroma, and the pH falls from 8.0 back to 7.0. Given that many of the Calvin-Benson cycle enzymes have a relatively sharp alkaline pH optima of 7.8-8.2 (Plaxton, Citation1997) this is a highly effective switch which ensures these enzymes, and consequently, this pathway, will only be fully operative in the light. Whilst this is arguably the most impressive “switch” activation of the key glycolytic enzyme pyruvate kinase has also been observed during anaerobiosis of germinating castor oil seeds (Podestá and Plaxton, Citation1991).
In addition to the ultra-rapid methods of fine control described above, covalent modification is a key area for the regulation of primary metabolism. Such regulation occurs via thermodynamically favorable enzyme catalyzed reactions which result in the formation of new covalent bonds. Interconversion can be very rapid, i.e. within minutes, and highly complete (up to 100% conversion). Nowadays, as we will discuss below, these changes can be followed directly by mass spectrometry (Lehmann et al., Citation2008), but in the last Century, radiolabeling and gel-shift assays were the methods of choice for their identification. Importantly, changes in enzyme conformation result in altered enzyme kinetics, including changes in Vmax, Km and nH. Last Century, 150 types of post-translational modification had already been documented to occur in vivo; however, very few appear to be important in enzyme regulation (Cohen, Citation1983). That said the response to external stimuli such as light, hormones or environmental stress were well known to modify the effects of several key enzymes (see for example, Ranjeva and Boudet, Citation1987; Buchanan, Citation1991; Poovaiah and Reddy, Citation1993) and a large number of these involved either dithiol-disulfide interconversion or phosphorylation.
Covalent modification by dithiol-disulfide interconversion involves reactions similar to those that occur on the formation of disulfide bonds and stabilize proteins with the exception that they must be accessible to reduction by external thiols (Ziegler, Citation1985). Thioredoxin is a heat-stable protein which in its reduced SH form can act as a protein disulfide reductase (Cohen, Citation1983). Dithiol-disulfide exchange appears to have massive regulatory significance in photosynthetic organisms being extremely important in linking photosynthetic electron flow to light regulation of several key chloroplastic enzymes of the Calvin-Benson cycle and associated metabolism (Buchanan, Citation1991; Citation1992). The thioredoxin system constitutes the proteins ferredoxin, ferredoxin-thioredoxin reductase and thioredoxin itself, with thioredoxin f and m being the key regulators. In the reduced state, thioredoxin f activates enzymes of carbohydrate synthase, whereas thioredoxin m activates NADP malate dehydrogenase and glucose 6-phosphate dehydrogenase, respectively (Buchanan, Citation1991; Citation1992). With some of these effects being synergistic to those of pH described above (Scheibe, Citation1991; Buchanan, Citation1992). Since these pivotal findings, the suite of thioredoxins in the plastid has been expanded, as has our understanding regarding the mechanism by which these enzymes are oxidized to form disulfide bonds in the dark (Geigenberger et al., Citation2017). Another key covalent reaction is phosphorylation/dephosphorylation, which occurs on serine, threonine and tyrosine residues. Both protein kinases and phosphatase can have wide or narrow substrate specificities, with the kinases often being under the control of signals including Ca2+ and a range of signal metabolites. The first plant enzyme demonstrated to be regulated by phosphorylation/dephosphorylation was the PDC from broccoli (Ranjeva and Boudet, Citation1987). As detailed below, this enzyme complex is interconverted between an activated dephosphorylated form and an activated phosphorylated form by the relative activities of an identified PDC kinase (Tovar-Méndez et al., Citation2003) and an only very recently identified phosphatase (see Proteomic identification of metabolic regulation below). Not only this key enzyme of the TCA cycle but also a wide range of other enzymes of primary metabolism have subsequently been identified to be under the control of phosphorylation/dephosphorylation, including the mitochondrial isoforms of malate dehydrogenase and aconitase.
B. Reverse genetic confirmation of aspects of metabolic regulation and the advent and adoption of broad profiling technologies
Given both the vastness of their scope and the fact that a number of comprehensive reviews have previously covered these studies (Stitt and Sonnewald, Citation1995; Raines, Citation2003; Nunes-Nesi et al., Citation2013) we will not provide any details here suffice to say that transgenic and mutagenic approaches in the 1980s through to the first decade of the 21st Century provided excellent in planta confirmation concerning the physiological importance of many of the above elements of metabolic regulation. The turn of the century also saw the widespread adoption of genomic, transcriptomic, proteomic and metabolomic approaches (Fiehn et al., Citation2001). Whilst the first of these was insightful in determining the sizes of the gene families involved in metabolism, which proved essential in the interpretation of results from, for example, single mutants it is proteomics and metabolomics that made the largest impact on our understanding of metabolic regulation (Sweetlove et al., Citation2014; Friso and van Wijk, Citation2015). Since the insight derived from metabolomics has also been covered in detail elsewhere, we will highlight only four examples (i) de novo amino acid biosynthesis, (ii) trehalose metabolism and its role in the regulation of starch metabolism, (iii) the identification of the 2-Carboxy-D-arabinitol 1-phosphate as an inhibitor of Rubisco and (iv) the suggestion that ABA interacts with Rubisco to regulate its activity which we detail in in the section below. The first of these came from a very early metabolomics experiment in potato which was carried out prior to genome sequencing or indeed even microarrays were available in this species. By evaluating a range of transgenics altered in sucrose mobilization Roessner et al were able to postulate the presence of an entire de novo network of amino acid biosynthesis in the tuber, contradicting the received wisdom at the time that these were all transported from the leaf (Roessner et al., Citation2001). In follow-up experiments incubating isolated tuber disks with glucose promoted the accumulation of amino acids and revealed that this enhanced the expression of the amino acid biosynthetic machinery (Roessner, Citation2001). Work on trehalose metabolism was also promoted by the sensitivity afforded by mass spectrometry allowing its detection in plants where it had previously been thought to be absent (Vogel et al., Citation2001). Following up on this work established that the trehalose/trehalose 6 phosphate system played important roles in the coordination of sucrose and starch metabolism as well as in orchestrating lipid metabolism (Zhai et al., Citation2018; Fichtner and Lunn, Citation2021). Similarly, 2-carboxyarabinitol 1-phosphate was identified by chromatographic properties alongside nuclear magnetic resonance spectroscopy (Andralojc et al., Citation1994). 2-carboxyarabinitol 1-phosphate binds to RuBisCO, preventing it from participating in chemical reactions. As the amount of light present increases, CA1P levels decrease, freeing RuBisCO's reactive ends, allowing more of the molecules to participate in chemical reactions. It can be broken down by the enzyme 2-carboxy-D-arabinitol-1-phosphatase into 2-carboxy-D-arabinitol. Subsequent work reveals that this inhibitor is derived from chloroplastic fructose 1,6-bisphosphate with transgenic plants containing decreased amounts of chloroplastic fructose 1,6-bisphosphate phosphatase containing increased amounts of fructose 1,6-bisphosphate and 2-carboxyarabinitol 1-phosphate and greatly increased amounts of the putative intermediates hamamelose and 2-carboxyarabinitol, which in some cases were as abundant as sucrose. In addition, French bean leaves in the light were shown to incorporate 14C from 14CO2 sequentially into fructose 1,6-bisphosphate, hamamelose bisphosphate, hamamelose monophosphate, hamamelose, and 2-carboxyarabinitol thereby defining the biosynthetic pathway of this regulator (Andralojc et al., Citation2002).
C. Proteomic identification of enzyme-enzyme interactions
Whilst stable protein–protein interactions in the form of multi-enzyme complexes are a long-known hallmark of metabolism, transient enzyme-enzyme interactions, whilst considerably harder to convincingly demonstrate, have been studied for almost half a Century (Srere, Citation1994; Fernie et al., Citation2018; Sweetlove and Fernie, Citation2018; Zhang and Fernie, Citation2021). Such interactions are often, erroneously, referred to as metabolons – a term coined by Srere in the 1980s (Srere et al., Citation1987); however, strictly speaking, metabolons should only be applied to the subset of such interactions that perform substrate channeling (Sweetlove and Fernie, Citation2018; Zhang and Fernie, Citation2021). The advent of proteomics and the parallel development of fluorescent marker proteins facilitated the identification of a protein’s partners within a complex with considerable research effort identifying protein–protein interactions in the pathways of glycolysis (Brandina et al., Citation2006; Graham et al., Citation2007; Zhang et al., Citation2020a), the TCA cycle (Zhang et al., Citation2017; Citation2018), the Calvin-Benson cycle (Scheibe et al., Citation2002) and photorespiration (discussed in (Zhang and Fernie, Citation2021)) as well as a range of pathways of secondary metabolism (Laursen et al., Citation2016; Fujino et al., Citation2018; Camagna et al., Citation2019; Mucha et al., Citation2019). However, with the exception of the TCA cycle and glycolysis and cyanogenic glucoside synthesis (Laursen et al., Citation2016; Zhang et al., Citation2020a), the evidence for any of these channeling substrates is weak. Evidence for these three pathways includes the normal demonstration of protein interaction as well as evidence that pathway intermediates do not mix with the bulk substrate but are rather channeled between the active sites of the constituent enzymes. Intriguingly, evaluation of the plant TCA cycle revealed both the canonical channel also found in animals and microbes but also a plant specific channel whilst that of glycolysis appears to have a moonlighting function with respect to positioning of organelles. That said, as evidenced by the fact that many of the interactions are in nonsequential enzymes (Zhang et al., Citation2018), or even act to deactivate competing enzymes the biological rationale for interaction need not only be channeling. In this vein recent studies on the mitochondrial interactome (Senkler et al., Citation2017) and even the cross-species comparison of the cellular interactome (McWhite et al., Citation2020; Zhang et al., Citation2020b) are highly informative. The former was able to catalog the rich number of interacting proteins within the mitochondria but not yet to directly link their occurrence with their biological function, whilst the latter provided an immense resource demonstrating phylogenetically conserved and restricted interactions. As such both studies provide rich data sources allowing the generation of numerable hypothesis which can be tested in future focused studies.
D. Proteomic identification of metabolic regulation
In addition to the identification of protein interaction, proteomics affords a direct survey of post-translational modifications. Early examples of the power of this approach include such surveys of putative thioredoxin-regulated enzymes of the Calvin-Benson and TCA cycles (Balmer et al., Citation2003, Citation2004). In the interim similar surveys have been performed for, amongst others, phosphorylation aceylation (Finkemeier et al., Citation2011), methylation (Alban et al., Citation2014) glycosylation (Nguema-Ona et al., Citation2014), ubiquitinylation (Vierstra, Citation2012) and sumolyation (Elrouby and Coupland, Citation2010). The insight gained from these studies has been comprehensively reviewed elsewhere (Friso and van Wijk, Citation2015), so we will not detail them here rather focusing on follow-up studies into the TCA cycle that builds on them. The metabolic regulation of the plant TCA cycle has been relatively recently reviewed by Nunes-Nesi et al. (Nunes-Nesi et al., Citation2013). By contrast to our understanding of the physiological role of the constituent enzymes as assessed by the evaluation of transgenic plants (Nunes-Nesi et al., Citation2011), however, this remains relatively fragmentary. At the time of this earlier review, phospho- and redox-regulation were known to occur at many of the steps as was the sensitivity of aconitase to oxidative stress and NO and the light-dependent regulation of 2-oxoglutarate dehydrogenase and succinyl CoA ligase by phytochrome (Daloso et al., Citation2015; Zhang et al., Citation2021). Moreover, whilst a wealth of metabolite effectors of the component enzymes had also characterized. Since this earlier review, four studies are of particular relevance – functional characterization of both phospho- and redox regulation of the TCA cycle, characterization of the hormonal control of succinate dehydrogenase (Gleason et al., Citation2011) and finally, the characterization of metabolite damage and metabolite repair in the cycle (Niehaus and Hillmann, Citation2020). We will discuss these chronologically thus beginning with the demonstration of an important link between SA levels and complex II function as mediated by succinate dehydrogenase which was beautifully demonstrated to play an important role in regulating respiration during avirulent bacterial infection (Gleason et al., Citation2011). Whilst as mentioned above previous screens had suggested several enzymes of the TCA cycle to be potential targets of thioredoxin mediated regulation studies of thioredoxin mutants combined with protein- based complementation demonstrated that thioredoxin clearly regulated the flux through this cycle -with the major regulation occurring at the cytosolic ATP-citrate lyase which subsequently resulted in an upregulated export of citrate from the mitochondria (Daloso et al., Citation2015). This was evidence by a combination of 13C feeding experiments and metabolite and enzyme measurements in thioredoxin mutants of Arabidopsis alongside complemented controls in which recombinant thioredoxin was provided in the assay mixture. Just this year a study of phosphatases previously identified as weakly interacting with TCA cycle enzymes (Zhang et al., Citation2018) were demonstrated to be involved in phosphoregulation of the TCA cycle.(Zhang et al., Citation2021). A range of phosphoproteomic, enzyme kinetics and metabolic flux studies demonstrated the presence of two proteases involved in metabolic regulation of the TCA cycle operating of several enzymes. These studies suggest that the TCA cycle is highly regulated, however, more studies will be required to study the hierarchy of these levels of regulation. A further complexity of the regulation of the cycle that has recently been uncovered is the characterization of metabolite damage and repair cycles that are linked to this cycle. Arguably the best characterized of these is that conserved across the kingdoms of life involving 2-hydroxyglutarate (Araújo et al., Citation2010; Maurino and Engqvist, Citation2015). However, the study of Niehaus and Hillmann identified further cycles – with the physiological function of these yet to be deciphered (Niehaus and Hillmann, Citation2020). The very recent publication of the Arabidopsis peptide atlas (van Wijk et al., Citation2021), will undoubtably serve as an important future resource. It currently houses 143 million tandem-MS datasets providing information concerning both splice variants and post-translational modifications and their experimental variance.
E. High-resolution flux analysis
As for many approaches in plant metabolic biology, early studies following innovations in mammalian and microbial sciences with the discovery of the canonical Calvin-Benson cycle being achieved in 1954 (Sharkey, Citation2019) and many of the evaluations of flux through glycolysis, the oxidative pentose phosphate pathway and the TCA cycle (Fernie et al., Citation2005; Nunes-Nesi et al., Citation2011) being based on seminal studies by Katz and coworkers in the 1980s (Cohen et al., Citation1981; Katz, Citation1985). These studies were beautiful in their simplicity, either involving only capturing the 14CO2 evolved from differentially positionally labeled substrates (Nunes-Nesi et al., Citation2005) or crude chemical fractionation of labeled tissues (Hill and Aprees, Citation1995a; Citation1995b), yet revealed important insight into the bulk fluxes through the pathways. Perhaps due to safety concerns, such experiments are no longer en vouge and, in some cases, no longer facile due to the discontinued commercial availability of several of the positional isotopomers. That said, as we detail below, they are still, on occasion, highly useful. Nowadays, approaches based on following the fate of heavy (but stably) labeled isotopes such as 13C and 15N via either NMR or GC/LC-MS is more commonly adopted (Allen and Young, Citation2020; Kruger and Ratcliffe, Citation2021). Such approaches allow far higher resolution of the fluxes – even down to the single reaction (Roscher et al., Citation1998; Szecowka et al., Citation2013; Abadie et al., Citation2021) and have allowed several important insights to be made. Firstly, the use of NMR revealed that as in microbes plants maintain the relative levels of flux through the core pathways of central metabolism and rather modulate flux through less central pathways in response to cellular circumstance (Rontein et al., Citation2002). Secondly, they provided much more facile approaches to assessing futile cycling in glycolysis that that achievable by chemical dissection (Hill and Aprees, Citation1995a; Citation1995b), allowing the demonstration that physiological changes in the level of fructose-2, 6-bisphosphate could indeed affect the activity of PFP (Fernie et al., Citation2001). Thirdly, they provided a quantification of the degree to which flux through the TCA cycle was downregulated in the light – being as much as 95% in illuminated French bean leaves (Tcherkez et al., Citation2005). Fourthly, it was intriguingly demonstrated that in seeds, the TCA cycle does not function in its cyclic mode and that the isocitrate dehydrogenase catalyzed reaction was operating reversibly in vivo (Schwender et al., Citation2006). Finally, the same group demonstrated that in seeds, Rubisco operates in the absence of the rest of the Calvin-Benson cycle to ensure more efficient lipid metabolism (Schwender et al., Citation2004). The use of mass spectrometry swiftly followed suit, allowing a considerably higher resolution of potato tuber metabolism (Roessner-Tunali et al., Citation2004) than earlier 14C based approaches. More recently, the approach has been adopted for testing the photosynthetic metabolism of Arabidopsis, maize and cassava (Szecowka et al., Citation2013; Arrivault et al., Citation2017). The first of these studies that of Szecowka et al., essentially benchmarked the method in Arabidopsis, demonstrating that the flux estimates derived were similar to those observed following classical methods. Thereafter application of this approach in maize revealed insight into the nature of the shuttles supporting the C4 syndrome (Weissmann et al., Citation2016; Arrivault et al., Citation2017) whilst in cassava it confirmed that, despite earlier claims to the contrary (El-Sharkawy and Cock, Citation1987), this species carried out C3 photosynthesis. It is important to note that to gain full utility of the data collected in such studies requires the generation of sophisticated mathematical models. However, the details of these are beyond the scope of this current review.
F. Multi-omics
Having detailed some highlights of the insights obtained on the use of the individual profiling methods in isolation, we feel it important to provide a few examples in which their use in tandem has provided quasi-mechanistic insight. In one of the earliest multiomics approaches Urbancycz-Wochniak and coworkers (Urbanczyk-Wochniak et al., Citation2003) simultaneously assessed the levels of mRNA and metabolites in an approach to identify targets for biotechnology. In similar studies, the groups of Rainer Höfgen and Kazuki Saito independently determined the gene-metabolite networks operating under Sulfur deficiency (Hirai et al., Citation2005; Nikiforova et al., Citation2005). Similarly, it was effectively used to look for metabolic signals in broadscale studies correlating transcript and metabolite levels across a wide range of experimental conditions in Arabidopsis (Hannah et al., Citation2010; Caldana et al., Citation2011, Citation2012). More recently, this approach has proven particularly effective in characterizing the interactions between primary metabolism and hormone signaling (Fabregas and Fernie, Citation2021) as well as in defining developmental transitions such as those observed on leaf maturation (Omidbakhshfard et al., Citation2021) or fruit ripening (Carrari et al., Citation2006). The studies on hormones have revealed a much more intimate association between primary metabolism and hormone signaling than was previously imagined, however, this has recently been reviewed elsewhere. There are a vast number of such multi-omics approaches which, as stated above, offer the advantage of potentially providing mechanistic insight. Two prominent insights underscore the developmental transitions occurring on leaf and fruit development. In the first, metabolomic analysis of the early leaf development identified an accumulation of jasmonate precursor and a signaling metabolite 12-oxophytodienoic acid coinciding with the meristem arrest. Obtained results implicate 12-OPDA in the regulation of cellular differentiation and consequently plant growth. In the second, differences in malate content were linked to tomato fruit starch content and ultimately to soluble solid content of ripe fruit (Centeno et al., Citation2011), leading researchers to question these changes (Batista-Silva et al., Citation2018). However, whilst these studies suggest possible regulatory mechanisms, they still, almost universally, remain to be validated in targeted studies.
G. Protein-metabolite interactions
A recently established frontier in the application of profiling methods to understand metabolic regulation is the search for metabolite protein interactions (Diether and Sauer, Citation2017; Venegas-Molina et al., Citation2021; Wagner et al., Citation2021). The nature of such interactions is manifold and includes, amongst others, regulation of protein activity, transcription factor binding and G protein-coupled receptors. The biological function of such interactions is similarly diverse with demonstrated roles in the regulation of enzyme activities (Zhai et al., Citation2018), affecting protein–protein interactions (Omini et al., Citation2021) including enzyme-enzyme complex (dis)association (Słabicki et al., Citation2020), and altering a protein's subcellular localization (Choi et al., Citation2015). For example, in bacteria the flux can be reversed from the glycolytic to gluconeogenic direction in under 30 s by the regulation of focal enzyme activities (Link et al., Citation2013). Coming back to a protein-metabolite interaction we mentioned above a recent study using a chemical proteomics approach based on an ABA mimetic probe was combined with in vitro assays, isothermal titration calorimetry (ITC), X-ray crystallography and in silico modeling to identify putative (+)-ABA binding-proteins in crude extracts of A. thaliana identifying Ribulose-1,5-bisphosphate carboxylase/oxygenase (Rubisco) as a potential ABA binding protein (Galka et al., Citation2015). Of course, covalent metabolite protein modifications have already been discussed above, however, those noncovalent modifications are more dynamic and at least as interesting. Indeed, it is expected that many more allosteric interactions are occurring than are known to date. Whilst early studies were based on protein-centric approaches such as tandem affinity purification which for example identified lipid ligands of approximately 20% of protein kinases in buddying yeast (Li et al., Citation2010). More recently, small-molecule centric approaches have begun to dominate, particularly those which measure changes in protein properties; several such methods exist. In one of these, stability of proteins from rates of oxidation (SPROX), where the protein-ligand complex is subjected to hydrogen peroxide alongside dose-dependent levels of a denaturing agent (West et al., Citation2008). By contrast, limited proteolysis with small-molecule mapping (Lip-SMap) and drug affinity responsive target stability (DARTS) approaches rely on resistance to proteolytic cleavage around the small-molecule binding site (Lomenick et al., Citation2009; Piazza et al., Citation2018). Finally, cellular thermal shift assay (CETSA) and thermal proteome profiling (TPP) determine shifts in protein thermal stability associated with a binding event (Martinez Molina et al., Citation2013; Savitski et al., Citation2014; Franken et al., Citation2015). Whilst powerful and relatively facile small-molecule centric approaches are by nature limited to known metabolites which are available as purified compounds. Nevertheless, as illustrated by a recent LiP-SMap study in Escherichia coli 1678 interaction could be detected for 20 metabolites ranging from one interactor for 3′, 5′-cAMP to hundreds for ATP (Piazza et al., Citation2018). Intriguingly, they delineated a set of core proteins which interacted with multiple metabolites and were less subject to transcriptional control. However, to date, this approach has not been applied in plants.
Another approach that has recently emerged as being highly informative is that of co-fractionation mass spectrometry. This approach which combines chromatographic separation of protein-metabolite complexes with subsequent mass spectral analyses has the great advantage of offering an untargeted approach, thereby greatly expanding the number of interactions that can be searched with studies in Arabidopsis, yeast and fungi alike reporting tens of annotated small-molecule interactors and hundreds of nonannotated interactors alike (Veyel et al., Citation2018; Li et al., Citation2021; Schlossarek et al., Citation2021) As well as re-finding known ligand–protein interactions, these studies identified several novel interactors – in the context of this review the most interesting of which were the interactions between the glycolytic enzymes glyceraldehyde 3 phosphate dehydrogenase in Arabidopsis (Moreno et al., Citation2021) and phosphoglycerate kinase in yeast (Luzarowski et al., Citation2021) and the proteinogenic dipeptides Tyr-Asp and Ser-Leu, respectively. In the case of the plant interaction, further research revealed that the dipeptide inhibits the activity of a glycolytic GAPDH, and redirects glucose toward the pentose phosphate pathway (PPP) and thereby NADPH production. In line with the metabolic data, Tyr-Asp supplementation improved the growth performance of both Arabidopsis and tobacco seedlings subjected to oxidative stress conditions. Obtained results open the intriguing possibility that proteogenic dipeptides act as evolutionarily conserved small-molecule regulators at the nexus of stress, protein degradation, and metabolism. However, considerable further research effort is required to fully assess the physiological significance of such interactions.
II. Conclusions and outlook
In this review, we have tried to provide a synthesis, which highlights how both established and nascent tools of experimental systems biology are adding to our already extensive knowledge of core central carbon metabolism in plants. There is a pervasive falsehood that we already understand primary metabolism. Interestingly with successive technological advances, we keep re-learning that this is not the case. This fact, notwithstanding, we do have extensive knowledge concerning a wide range of mechanisms by which the cell orchestrates the metabolic fluxes which sustain it and/or support cell division and growth or sustenance of other cell types. An important player in this process is the Target of Rapamycin (TOR) kinase pathway, a master regulator in converging nutrient status into growth and development programs to achieve metabolic homeostasis in plants. In yeast and mammals, large-scale TOR omics have provided new insights, expanding the list of upstream and downstream players, including the ones involved in the regulation of protein, nucleotide and lipids metabolism (Shimobayashi and Hall, Citation2014). Despite the recent advances in the plant field (Brunkard, Citation2020; Ingargiola et al., Citation2020; Burkart and Brandizzi, Citation2021), this knowledge lags behind, and such integrative approaches could shed light on how this kinase complex adjusts the metabolic network according to the nutrient availability and energetic requirement across distinct tissues and organs to control growth and development in plants. Similarly, there have been considerable developments in the role of trehalose 6-phosphate signaling in plants, however, these have largely followed more classical rather than genomics-based approaches (Fichtner and Lunn, Citation2021). That said the ability to measure trehalose 6-phosphate reliably was based on targeted mass spectrometry so this can at least in part be seen as an advance inspired by post-genomic technologies (Stitt et al., Citation2010). It is important to note that signatures of many of the previous classically determined mechanisms of metabolic regulation are visible in the vast datasets being generated in the post-genomic era. However, it is also important to note that despite multi-laboratory efforts to rectify this fact (Alseekh et al., Citation2021; van Wijk et al., Citation2021), the databasing of most of this data does not follow the excellent examples set by transcript profiling (Zimmermann et al., Citation2004). Thus whilst, as we demonstrate here, the identification of a vast number of (putative) regulatory mechanisms has been achieved by the adoption of profiling tools this century, enhancing availability and thereby interrogation of such datasets would likely augment this effort. We feel that the examples presented here suggest that identification of putative mechanisms of regulation is occurring aplenty. There is, however, an urgent need for accelerating the validation of these approaches in order to promote these from putative mechanisms to bona fide mechanisms by which cellular metabolism is physiologically regulated. Only once this challenge is more regularly met can we regard these novel insights as truly meriting discussion alongside the classical mechanism uncovered during the 20th Century.
| Abbreviations | ||
| TCA | = | tricarboxylic acid |
| Fru-2,6-P2 | = | fructose-2,6-bisphosphate |
| CAM | = | crassulacean acid metabolism |
| PFP | = | pyrophosphate dependent phosphofructokinase |
| PFK | = | phosphofructokinase |
| 3PGA | = | 3-phosphoglycerate |
| Pi | = | inorganic phosphate |
| DHAP | = | dihydroacetone phosphate |
| PDC | = | pyruvate dehydrogenase complex |
| 12ODPA | = | 12-oxophytodienoate |
| ABA | = | abscisic acid |
| LipSMap | = | small-molecule mapping |
| DARTS | = | drug affinity responsive target stability |
| CETSA | = | cellular thermal shift assay |
| TPP | = | thermal protein profiling |
References
- Abadie, C., Lalande, J., Limami, A. M., and Tcherkez, G. 2021. Non-targeted 13 C metabolite analysis demonstrates broad re-orchestration of leaf metabolism when gas exchange conditions vary. Plant Cell Environ. 44:445–457. doi:https://doi.org/10.1111/pce.13940
- Alban, C., Tardif, M., Mininno, M., Brugière, S., Gilgen, A., Ma, S., Mazzoleni, M., Gigarel, O., Martin-Laffon, J., Ferro, M., and Ravanel, S. 2014. Uncovering the protein lysine and arginine methylation network in Arabidopsis chloroplasts. PLoS One. 9:e95512. doi:https://doi.org/10.1371/journal.pone.0095512
- Allen, D. K., and Young, J. D. 2020. Tracing metabolic flux through time and space with isotope labeling experiments. Curr. Opin. Biotechnol. 64:92–100. doi:https://doi.org/10.1016/j.copbio.2019.11.003
- Alseekh, S., Aharoni, A., Brotman, Y., Contrepois, K., D'Auria, J., Ewald, J., C Ewald, J., Fraser, P. D., Giavalisco, P., Hall, R. D., Heinemann, M., Link, H., Luo, J., Neumann, S., Nielsen, J., Perez de Souza, L., Saito, K., Sauer, U., Schroeder, F. C., Schuster, S., Siuzdak, G., Skirycz, A., Sumner, L. W., Snyder, M. P., Tang, H., Tohge, T., Wang, Y., Wen, W., Wu, S., Xu, G., Zamboni, N., and Fernie, A. R. 2021. Mass spectrometry-based metabolomics: a guide for annotation, quantification and best reporting practices. Nat. Methods. 18:747–756.
- Alseekh, S., and Fernie, A. R. 2018. Metabolomics 20 years on: what have we learned and what hurdles remain? Plant J. 94:933–942. doi:https://doi.org/10.1111/tpj.13950
- Andralojc, P. J., Dawson, G. W., Parry, M. A., and Keys, A. J. 1994. Incorporation of carbon from photosynthetic products into 2-carboxyarabinitol-1-phosphate and 2-carboxyarabinitol. Biochem. J. 304:781–786. doi:https://doi.org/10.1042/bj3040781
- Andralojc, P. J., Keys, A. J., Kossmann, J., and Parry, M. A. J. 2002. Elucidating the biosynthesis of 2-carboxyarabinitol 1-phosphate through reduced expression of chloroplastic fructose 1,6-bisphosphate phosphatase and radiotracer studies with 14CO2. Proc. Natl. Acad. Sci. U. S. A. 99:4742–4747.
- Arabidopsis Genome Initiative 2000. Analysis of the genome sequence of the flowering plant Arabidopsis thaliana. Nature. 408:796–815.
- Araújo, W. L., Ishizaki, K., Nunes-Nesi, A., and Larson, T. R. 2010. Identification of the 2-hydroxyglutarate and isovaleryl-CoA dehydrogenases as alternative electron donors linking lysine catabolism to the electron transport chain of Arabidopsis mitochondria. Plant. 22:1549–1563. doi:https://doi.org/10.1105/tpc.110.075630
- Arrivault, S., Obata, T., Szecówka, M., Mengin, V., Guenther, M., Hoehne, M., Fernie, A. R., and Stitt, M. 2017. Metabolite pools and carbon flow during C4 photosynthesis in maize: 13CO2 labeling kinetics and cell type fractionation. J. Exp. Bot. 68:283–298. doi:https://doi.org/10.1093/jxb/erw414
- Balmer, Y., Koller, A., Val, G., del Manieri, W., Schürmann, P., and Buchanan, B. B. 2003. Proteomics gives insight into the regulatory function of chloroplast thioredoxins. Proc. Natl. Acad. Sci. U. S. A. 100:370–375. doi:https://doi.org/10.1073/pnas.232703799
- Balmer, Y., Vensel, W. H., Tanaka, C. K., Hurkman, W. J., Gelhaye, E., Rouhier, N., Jacquot, J.-P., Manieri, W., Schürmann, P., Droux, M., and Buchanan, B. B. 2004. Thioredoxin links redox to the regulation of fundamental processes of plant mitochondria. Proc. Natl. Acad. Sci. U. S. A. 101:2642–2647. doi:https://doi.org/10.1073/pnas.0308583101
- Batista-Silva, W., Nascimento, V. L., Medeiros, D. B., Nunes-Nesi, A., Ribeiro, D. M., Zsögön, A., and Araújo, W. L. 2018. Modifications in organic acid profiles during fruit development and ripening: correlation or causation? Front Plant Sci. 9:1689. doi:https://doi.org/10.3389/fpls.2018.01689
- Brandina, I., Graham, J., Lemaitre-Guillier, C., Entelis, N., Krasheninnikov, I., Sweetlove, L., Tarassov, I., and Martin, R. P. 2006. Enolase takes part in a macromolecular complex associated to mitochondria in yeast. Biochim. Biophys. Acta. 1757:1217–1228. doi:https://doi.org/10.1016/j.bbabio.2006.07.001
- Brunkard, J. O. 2020. Exaptive evolution of target of rapamycin signaling in multicellular eukaryotes. Dev. Cell. 54:142–155. doi:https://doi.org/10.1016/j.devcel.2020.06.022
- Buchanan, B. B. 1991. Regulation of CO2 assimilation in oxygenic photosynthesis: the ferredoxin/thioredoxin system. Perspective on its discovery, present status, and future development. Arch. Biochem. Biophys. 288:1–9.
- Buchanan, B. B. 1992. Carbon dioxide assimilation in oxygenic and anoxygenic photosynthesis. Photosynth. Res. 33:147–162. doi:https://doi.org/10.1007/BF00039177
- Budde, R. J., Fang, T. K., Randall, D. D., and Miernyk, J. A. 1991. Acetyl-coenzyme a can regulate activity of the mitochondrial pyruvate dehydrogenase complex in situ. Plant Physiol. 95:131–136. doi:https://doi.org/10.1104/pp.95.1.131
- Burkart, G. M., and Brandizzi, F. 2021. A tour of TOR complex signaling in plants. Trends Biochem. Sci. 46:417–428. doi:https://doi.org/10.1016/j.tibs.2020.11.004
- Caldana, C., Degenkolbe, T., Cuadros-Inostroza, A., Klie, S., Sulpice, R., Leisse, A., Steinhauser, D., Fernie, A. R., Willmitzer, L., and Hannah, M. A. 2011. High-density kinetic analysis of the metabolomic and transcriptomic response of Arabidopsis to eight environmental conditions. Plant J. 67:869–884. doi:https://doi.org/10.1111/j.1365-313X.2011.04640.x
- Caldana, C., Fernie, A. R., Willmitzer, L., and Steinhauser, D. 2012. Unraveling retrograde signaling pathways: finding candidate signaling molecules via metabolomics and systems biology driven approaches. Front. Plant Sci. 3:267. doi:https://doi.org/10.3389/fpls.2012.00267
- Camagna, M., Grundmann, A., Bär, C., Koschmieder, J., Beyer, P., and Welsch, R. 2019. Enzyme fusion removes competition for geranylgeranyl diphosphate in carotenogenesis. Plant Physiol. 179:1013–1027. doi:https://doi.org/10.1104/pp.18.01026
- Canellas, P. F., and Wedding, R. T. 1984. Kinetic properties of NAD malic enzyme from cauliflower. Arch. Biochem. Biophys. 229:414–425. doi:https://doi.org/10.1016/0003-9861(84)90171-1
- Carnal, N. W., and Black, C. C. 1983. Phosphofructokinase activities in photosynthetic organisms: the occurrence of pyrophosphate-dependent 6-phosphofructokinase in plants and algae. Plant Physiol. 71:150–155. doi:https://doi.org/10.1104/pp.71.1.150
- Carrari, F., Baxter, C., Usadel, B., Urbanczyk-Wochniak, E., Zanor, M.-I., Nunes-Nesi, A., Nikiforova, V., Centero, D., Ratzka, A., Pauly, M., Sweetlove, L. J., and Fernie, A. R. 2006. Integrated analysis of metabolite and transcript levels reveals the metabolic shifts that underlie tomato fruit development and highlight regulatory aspects of metabolic network behavior. Plant Physiol. 142:1380–1396. doi:https://doi.org/10.1104/pp.106.088534
- Centeno, D. C., Osorio, S., Nunes-Nesi, A., Bertolo, A. L. F., Carneiro, R. T., Araújo, W. L., Steinhauser, M.-C., Michalska, J., Rohrmann, J., Geigenberger, P., Oliver, S. N., Stitt, M., Carrari, F., Rose, J. K. C., and Fernie, A. R. 2011. Malate plays a crucial role in starch metabolism, ripening, and soluble solid content of tomato fruit and affects postharvest softening. Plant Cell. 23:162–184. doi:https://doi.org/10.1105/tpc.109.072231
- Choi, H. W., Tian, M., Manohar, M., Harraz, M. M., Park, S.-W., Schroeder, F. C., Snyder, S. H., and Klessig, D. F. 2015. Human GAPDH is a target of aspirin's primary metabolite salicylic acid and its derivatives. PLoS One. 10:e0143447. doi:https://doi.org/10.1371/journal.pone.0143447
- Cohen, P. 1983. Control of Enzyme Activity. Chapman and Hall, London and New York.
- Cohen, S. M., Rognstad, R., Shulman, R. G., and Katz, J. 1981. A comparison of 13C nuclear magnetic resonance and 14C tracer studies of hepatic metabolism. J. Biol. Chem. 256:3428–3432. doi:https://doi.org/10.1016/S0021-9258(19)69626-2
- Cséke, C., and Buchanan, B. B. 1983. An enzyme synthesizing fructose 2,6-bisphosphate occurs in leaves and is regulated by metabolite effectors. FEBS Lett. 155:139–142. doi:https://doi.org/10.1016/0014-5793(83)80226-9
- Cséke, C., Stitt, M., Balogh, Á., and Buchanan, B. B. 1983. A product-regulated fructose 2,6-bisphosphatase occurs in green leaves. FEBS Lett. 162:103–106. doi:https://doi.org/10.1016/0014-5793(83)81057-6
- Cseke, C., Weeden, N. F., Buchanan, B. B., and Uyeda, K. 1982. A special fructose bisphosphate functions as a cytoplasmic regulatory metabolite in green leaves. Proc. Natl. Acad. Sci. U. S. A. 79:4322–4326. doi:https://doi.org/10.1073/pnas.79.14.4322
- Daloso, D. M., Müller, K., Obata, T., Florian, A., Tohge, T., Bottcher, A., Riondet, C., Bariat, L., Carrari, F., Nunes-Nesi, A., Buchanan, B. B., Reichheld, J.-P., Araújo, W. L., and Fernie, A. R. 2015. Thioredoxin, a master regulator of the tricarboxylic acid cycle in plant mitochondria. Proc. Natl. Acad. Sci. U. S. A. 112:E1392–400.
- Diether, M., and Sauer, U. 2017. Towards detecting regulatory protein-metabolite interactions. Curr. Opin. Microbiol. 39:16–23. doi:https://doi.org/10.1016/j.mib.2017.07.006
- Ekkehard, H., and Stitt, M. 1989. Perturbation of photosynthesis in spinach leaf discs by low concentrations of methyl viologen: influence of increased thylakoid energisation on ATP synthesis, electron transport, energy dissipation, light-activation of the Calvin-cycle enzymes, and control of starch and sucrose synthesis. Planta. 179:51–60. doi:https://doi.org/10.1007/BF00395770
- El-Maghrabi, M. R., Claus, T. H., Pilkis, J., and Pilkis, S. J. 1981. Partial purification of a rat liver enzyme that catalyzes the formation of fructose 2,6-bisphosphate. Biochem. Biophys. Res. Commun. 101:1071–1077. doi:https://doi.org/10.1016/0006-291X(81)91858-1
- El-Maghrabi, M. R., Claus, T. H., Pilkis, J., Fox, E., and Pilkis, S. J. 1982. Regulation of rat liver fructose 2,6-bisphosphatase. J. Biol. Chem. 257:7603–7607.
- Elrouby, N., and Coupland, G. 2010. Proteome-wide screens for small ubiquitin-like modifier (SUMO) substrates identify Arabidopsis proteins implicated in diverse biological processes. Proc. Natl. Acad. Sci. U. S. A. 107:17415–17420. doi:https://doi.org/10.1073/pnas.1005452107
- El-Sharkawy, M. A., and Cock, J. H. 1987. C3-C 4 intermediate photosynthetic characteristics of cassava (Manihot esculenta Crantz): I. Gas exchange. Photosynth. Res. 12:219–235. doi:https://doi.org/10.1007/BF00055122
- Fabregas, N., and Fernie, A. R. 2021. The interface of central metabolism with hormone signaling in plants. Curr. Biol. 31:R1535–R1548. doi:https://doi.org/10.1016/j.cub.2021.09.070
- Fahrendorf, T., Holtum, J. A., Mukherjee, U., and Latzko, E. 1987. Fructose 2,6-bisphosphate, carbohydrate partitioning, and crassulacean Acid metabolism. Plant Physiol. 84:182–187.
- Fang, C., Fernie, A. R., and Luo, J. 2019. Exploring the diversity of plant metabolism. Trends Plant Sci. 24:83–98. doi:https://doi.org/10.1016/j.tplants.2018.09.006
- Fernie, A. R., Carrari, F., and Sweetlove, L. J. 2004. Respiratory metabolism: glycolysis, the TCA cycle and mitochondrial electron transport. Curr. Opin. Plant Biol. 7:254–261.
- Fernie, A. R., Geigenberger, P., and Stitt, M. 2005. Flux an important, but neglected, component of functional genomics. Curr. Opin. Plant Biol. 8:174–182.
- Fernie, A. R., Roscher, A., Ratcliffe, R. G., and Kruger, N. J. 2001. Fructose 2,6-bisphosphate activates pyrophosphate: fructose-6-phosphate 1-phosphotransferase and increases triose phosphate to hexose phosphate cycling in heterotrophic cells. Planta. 212:250–263.
- Fernie, A. R., Zhang, Y., and Sweetlove, L. J. 2018. Passing the baton: substrate channelling in respiratory metabolism. Research (Wash. D. C.). 2018:1539325. doi:https://doi.org/10.1155/2018/1539325
- Fichtner, F., and Lunn, J. E. 2021. The role of trehalose 6-phosphate (Tre6P) in plant metabolism and development. Annu. Rev. Plant Biol. 72:737–760. doi:https://doi.org/10.1146/annurev-arplant-050718-095929
- Fiehn, O., Kloska, S., and Altmann, T. 2001. Integrated studies on plant biology using multiparallel techniques. Curr. Opin. Biotechnol. 12:82–86. doi:https://doi.org/10.1016/S0958-1669(00)00165-8
- Finkemeier, I., Laxa, M., Miguet, L., Howden, A. J. M., and Sweetlove, L. J. 2011. Proteins of diverse function and subcellular location are lysine acetylated in Arabidopsis. Plant Physiol. 155:1779–1790. doi:https://doi.org/10.1104/pp.110.171595
- Franken, H., Mathieson, T., Childs, D., Sweetman, G. M. A., Werner, T., Tögel, I., Doce, C., Gade, S., Bantscheff, M., Drewes, G., Reinhard, F. B. M., Huber, W., and Savitski, M. M. 2015. Thermal proteome profiling for unbiased identification of direct and indirect drug targets using multiplexed quantitative mass spectrometry. Nat. Protoc. 10:1567–1593. doi:https://doi.org/10.1038/nprot.2015.101
- Friso, G., and van Wijk, K. J. 2015. Posttranslational protein modifications in plant metabolism. Plant Physiol. 169:1469–1487. doi:https://doi.org/10.1104/pp.15.01378
- Fujino, N., Tenma, N., Waki, T., Ito, K., Komatsuzaki, Y., Sugiyama, K., Yamazaki, T., Yoshida, S., Hatayama, M., Yamashita, S., Tanaka, Y., Motohashi, R., Denessiouk, K., Takahashi, S., and Nakayama, T. 2018. Physical interactions among flavonoid enzymes in snapdragon and torenia reveal the diversity in the flavonoid metabolon organization of different plant species. Plant J. 94:372–392. doi:https://doi.org/10.1111/tpj.13864
- Galka, M. M., Rajagopalan, N., Buhrow, L. M., Nelson, K. M., Switala, J., Cutler, A. J., Palmer, D. R. J., Loewen, P. C., Abrams, S. R., and Loewen, M. C. 2015. Identification of interactions between abscisic acid and ribulose-1,5-bisphosphate carboxylase/oxygenase. PLoS One. 10:e0133033.
- Geigenberger, P., Thormählen, I., Daloso, D. M., and Fernie, A. R. 2017. The unprecedented versatility of the plant thioredoxin system. Trends Plant Sci. 22:249–262. doi:https://doi.org/10.1016/j.tplants.2016.12.008
- Gerhardt, R., Stitt, M., and Heldt, H. W. 1987. Subcellular metabolite levels in spinach leaves: regulation of sucrose synthesis during diurnal alterations in photosynthetic partitioning. Plant Physiol. 83:399–407. doi:https://doi.org/10.1104/pp.83.2.399
- Gibon, Y., Blaesing, O. E., Hannemann, J., Carillo, P., Höhne, M., Hendriks, J. H. M., Palacios, N., Cross, J., Selbig, J., and Stitt, M. 2004. A robot-based platform to measure multiple enzyme activities in Arabidopsis using a set of cycling assays: comparison of changes of enzyme activities and transcript levels during diurnal cycles and in prolonged darkness [W]. Plant Cell. 16:3304–3325. ]. doi:https://doi.org/10.1105/tpc.104.025973
- Gleason, C., Huang, S., Thatcher, L. F., Foley, R. C., Anderson, C. R., Carroll, A. J., Millar, A. H., and Singh, K. B. 2011. Mitochondrial complex II has a key role in mitochondrial-derived reactive oxygen species influence on plant stress gene regulation and defense. Proc. Natl. Acad. Sci. U. S. A. 108:10768–10773. doi:https://doi.org/10.1073/pnas.1016060108
- Gottschalk, M. E., Chatterjee, T., Edelstein, I., and Marcus, F. 1982. Studies on the mechanism of interaction of fructose 2,6-bisphosphate with fructose-1,6-bisphosphatase. J. Biol. Chem. 257:8016–8020. doi:https://doi.org/10.1016/S0021-9258(18)34290-X
- Graham, J. W. A., Williams, T. C. R., Morgan, M., Fernie, A. R., Ratcliffe, R. G., and Sweetlove, L. J. 2007. Glycolytic enzymes associate dynamically with mitochondria in response to respiratory demand and support substrate channeling. Plant Cell. 19:3723–3738. doi:https://doi.org/10.1105/tpc.107.053371
- Hannah, M. A., Caldana, C., Steinhauser, D., Balbo, I., Fernie, A. R., and Willmitzer, L. 2010. Combined transcript and metabolite profiling of Arabidopsis grown under widely variant growth conditions facilitates the identification of novel metabolite-mediated regulation of gene expression. Plant Physiol. 152:2120–2129. doi:https://doi.org/10.1104/pp.109.147306
- Heldt, H. W., Chon, C. J., and Maronde, D. 1977. Role of orthophosphate and other factors in the regulation of starch formation in leaves and isolated chloroplasts. Plant Physiol. 59:1146–1155. doi:https://doi.org/10.1104/pp.59.6.1146
- Herzog, B., Stitt, M., and Heldt, H. W. 1984. Control of photosynthetic sucrose synthesis by fructose 2,6-bisphosphate: III. Properties of the cytosolic fructose 1,6-bisphosphatase. Plant Physiol. 75:561–565. doi:https://doi.org/10.1104/pp.75.3.561
- Hill, S. A., and Aprees, T. 1995a. The effect of glucose on the control of carbohydrate-metabolism in ripening bananas. Planta. 196:335–343. doi:https://doi.org/10.1007/BF00201393
- Hill, S. A., and Aprees, T. 1995b. The effect of hypoxia on the control of carbohydrate-metabolism in ripening bananas. Planta. 197:313–323. doi:https://doi.org/10.1007/BF00202653
- Hirai, M. Y., Klein, M., Fujikawa, Y., Yano, M., Goodenowe, D. B., Yamazaki, Y., Kanaya, S., Nakamura, Y., Kitayama, M., Suzuki, H., Sakurai, N., Shibata, D., Tokuhisa, J., Reichelt, M., Gershenzon, J., Papenbrock, J., and Saito, K. 2005. Elucidation of gene-to-gene and metabolite-to-gene networks in Arabidopsis by integration of metabolomics and transcriptomics. J. Biol. Chem. 280:25590–25595. doi:https://doi.org/10.1074/jbc.M502332200
- Huber, S. C., and Hanson, K. R. 1992. Carbon partitioning and growth of a starchless mutant of Nicotiana sylvestris. Plant Physiol. 99:1449–1454. doi:https://doi.org/10.1104/pp.99.4.1449
- Ingargiola, C., Turqueto Duarte, G., Robaglia, C., Leprince, A.-S., and Meyer, C. 2020. The plant target of rapamycin: a conductor of nutrition and metabolism in photosynthetic organisms. Genes. 11:1285. doi:https://doi.org/10.3390/genes11111285
- Katz, J. 1985. Determination of gluconeogenesis in vivo with 14C-labeled substrates. Am. J. Physiol-Regulat. Integrat. Comparat. Physiol. 248:R391–R399. doi:https://doi.org/10.1152/ajpregu.1985.248.4.R391
- Kombrink, E., and Kruger, N. J. 1984. Inhibition by metabolic intermediates of pyrophosphate: fructose 6-phosphate phosphotransferase from germinating castor bean endosperm. Z. Pflanzen. 114:443–453. doi:https://doi.org/10.1016/S0044-328X(84)80064-1
- Kruger, N. J., and Beevers, H. 1984. Effect of fructose 2,6-bisphosphate on the kinetic properties of cytoplasmic fructose 1,6-bisphosphatase from germinating castor bean endosperm. Plant Physiol. 76:49–54.
- Kruger, N. J., and Ratcliffe, R. G. 2021. Whither metabolic flux analysis in plants? J. Exp. Bot. 72: 7653–7657.
- Kruger, N. J., and Scott, P. 1995. Integration of cytosolic and plastidic carbon metabolism by fructose 2,6-bisphosphate. J. Exp. Bot. 46:1325–1333. doi:https://doi.org/10.1093/jxb/46.special_issue.1325
- Kruger, N. J., Bulpin, P. V., and Ap Rees, T. 1983. The extent of starch degradation in the light in pea leaves. Planta. 157:271–273. doi:https://doi.org/10.1007/BF00405193
- Larondelle, Y., Mertens, E., Schaftingen, E., and Hers, H.-G. 1986. Purification and properties of spinach leaf phosphofructokinase 2/fructose 2,6-bisphosphatase. Eur. J. Biochem. 161:351–357. doi:https://doi.org/10.1111/j.1432-1033.1986.tb10454.x
- Larondelle, Y., Mertens, E., Van Schaftingen, E., and Hers, H.-G. 1989. Fructose 2,6-bisphosphate hydrolyzing enzymes in higher plants. Plant Physiol. 90:827–834.
- Laursen, T., Borch, J., Knudsen, C., Bavishi, K., Torta, F., Martens, H. J., Silvestro, D., Hatzakis, N. S., Wenk, M. R., Dafforn, T. R., Olsen, C. E., Motawia, M. S., Hamberger, B., Møller, B. L., and Bassard, J.-E. 2016. Characterization of a dynamic metabolon producing the defense compound dhurrin in sorghum. Science. 354:890–893. doi:https://doi.org/10.1126/science.aag2347
- Lederer, B., Vissers, S., Van Schaftingen, E., and Hers, H. G. 1981. Fructose 2,6-bisphosphate in yeast. Biochem. Biophys. Res. Commun. 103:1281–1287.
- Lehmann, U., Wienkoop, S., Tschoep, H., and Weckwerth, W. 2008. If the antibody fails-a mass western approach. Plant J. 55:1039–1046. doi:https://doi.org/10.1111/j.1365-313X.2008.03554.x
- Li, X., Gianoulis, T. A., Yip, K. Y., Gerstein, M., and Snyder, M. 2010. Extensive in vivo metabolite-protein interactions revealed by large-scale systematic analyses. Cell. 143:639–650. doi:https://doi.org/10.1016/j.cell.2010.09.048
- Li, Y., Kuhn, M., Zukowska-Kasprzyk, J., Hennrich, M. L., Kastritis, P. L., O'Reilly, F. J., Phapale, P., Beck, M., Gavin, A.-C., and Bork, P. 2021. Coupling proteomics and metabolomics for the unsupervised identification of protein-metabolite interactions in Chaetomium thermophilum. PLoS One. 16:e0254429. doi:https://doi.org/10.1371/journal.pone.0254429
- Link, H., Kochanowski, K., and Sauer, U. 2013. Systematic identification of allosteric protein-metabolite interactions that control enzyme activity in vivo. Nat. Biotechnol. 31:357–361. doi:https://doi.org/10.1038/nbt.2489
- Lomenick, B., Hao, R., Jonai, N., Chin, R. M., Aghajan, M., Warburton, S., Wang, J., Wu, R. P., Gomez, F., Loo, J. A., Wohlschlegel, J. A., Vondriska, T. M., Pelletier, J., Herschman, H. R., Clardy, J., Clarke, C. F., and Huang, J. 2009. Target identification using drug affinity responsive target stability (DARTS). Proc. Natl. Acad. Sci. U. S. A. 106:21984–21989. doi:https://doi.org/10.1073/pnas.0910040106
- Luzarowski, M., Vicente, R., Kiselev, A., Wagner, M., Schlossarek, D., Erban, A., de Souza, L. P., Childs, D., Wojciechowska, I., Luzarowska, U., Górka, M., Sokołowska, E. M., Kosmacz, M., Moreno, J. C., Brzezińska, A., Vegesna, B., Kopka, J., Fernie, A. R., Willmitzer, L., Ewald, J. C., and Skirycz, A. 2021. Global mapping of protein-metabolite interactions in Saccharomyces cerevisiae reveals that Ser-Leu dipeptide regulates phosphoglycerate kinase activity. Commun. Biol. 4:181. doi:https://doi.org/10.1038/s42003-021-01684-3
- Macdonald, F. D., Chou, Q., and Buchanan, B. B. 1987. Ion-exchange chromatography separates activities synthesizing and degrading fructose 2,6-bisphosphate from C3 And C4 leaves but not from rat liver. Plant Physiol. 85:13–16. doi:https://doi.org/10.1104/pp.85.1.13
- Macdonald, F. D., Chou, Q., Buchanan, B. B., and Stitt, M. 1989. Purification and characterization of fructose-2,6-bisphosphatase, a substrate-specific cytosolic enzyme from leaves. J. Biol. Chem. 264:5540–5544. doi:https://doi.org/10.1016/S0021-9258(18)83579-7
- Martinez Molina, D., Jafari, R., Ignatushchenko, M., Seki, T., Larsson, E. A., Dan, C., Sreekumar, L., Cao, Y., and Nordlund, P. 2013. Monitoring drug target engagement in cells and tissues using the cellular thermal shift assay. Science. 341:84–87. doi:https://doi.org/10.1126/science.1233606
- Maurino, V. G., and Engqvist, M. K. M. 2015. 2-hydroxy acids in plant metabolism. Arabidopsis Book. 13:e0182. doi:https://doi.org/10.1199/tab.0182
- McIntosh, C. A., and Oliver, D. J. 1992. NAD-linked isocitrate dehydrogenase: isolation, purification, and characterization of the protein from pea mitochondria. Plant Physiol. 100:69–75.
- McWhite, C. D., Papoulas, O., Drew, K., Cox, R. M., June, V., Dong, O. X., Kwon, T., Wan, C., Salmi, M. L., Roux, S. J., Browning, K. S., Chen, Z. J., Ronald, P. C., and Marcotte, E. M. 2020. A pan-plant protein complex map reveals deep conservation and novel assemblies. Cell. 181:460–474.e14. doi:https://doi.org/10.1016/j.cell.2020.02.049
- Mergner, J., Frejno, M., List, M., Papacek, M., Chen, X., Chaudhary, A., Samaras, P., Richter, S., Shikata, H., Messerer, M., Lang, D., Altmann, S., Cyprys, P., Zolg, D. P., Mathieson, T., Bantscheff, M., Hazarika, R. R., Schmidt, T., Dawid, C., Dunkel, A., Hofmann, T., Sprunck, S., Falter-Braun, P., Johannes, F., Mayer, K. F. X., Jürgens, G., Wilhelm, M., Baumbach, J., Grill, E., Schneitz, K., Schwechheimer, C., and Kuster, B. 2020. Mass-spectrometry-based draft of the Arabidopsis proteome. Nature. 579:409–414. doi:https://doi.org/10.1038/s41586-020-2094-2
- Miflin, B. J., and Lea, P. J. 1982. Ammonia assimilation and amino acid metabolism. In Nucleic Acids and Proteins in Plants I: Structure, Biochemistry and Physiology of Proteins; Boulter, D. and Parthier, B., Eds. Springer Berlin Heidelberg: Berlin, Heidelberg, pp 5–64.
- Moreno, J. C., Rojas, B. E., Vicente, R., Gorka, M., Matz, T., Chodasiewicz, M., Peralta‐Ariza, J. S., Zhang, Y., Alseekh, S., Childs, D., Luzarowski, M., Nikoloski, Z., Zarivach, R., Walther, D., Hartman, M. D., Figueroa, C. M., Iglesias, A. A., Fernie, A. R., and Skirycz, A. 2021. Tyr‐Asp inhibition of glyceraldehyde 3‐phosphate dehydrogenase affects plant redox metabolism. EMBO J. 40: e106800. doi:https://doi.org/10.15252/embj.2020106800
- Mucha, S., Heinzlmeir, S., Kriechbaumer, V., Strickland, B., Kirchhelle, C., Choudhary, M., Kowalski, N., Eichmann, R., Hückelhoven, R., Grill, E., Kuster, B., and Glawischnig, E. 2019. The formation of a camalexin biosynthetic metabolon. Plant Cell. 31:2697–2710. doi:https://doi.org/10.1105/tpc.19.00403
- Neuhaus, H. E., Ekkehard Neuhaus, H., Kruckeberg, A. L., Feil, R., and Stitt, M. 1989. Reduced-activity mutants of phosphoglucose isomerase in the cytosol and chloroplast of Clarkia xantiana: II. Study of the mechanisms which regulate photosynthate partitioning. Planta. 178:110–122. doi:https://doi.org/10.1007/BF00392534
- Nguema-Ona, E., Vicré-Gibouin, M., Gotté, M., Plancot, B., Lerouge, P., Bardor, M., and Driouich, A. 2014. Cell wall O-glycoproteins and N-glycoproteins: aspects of biosynthesis and function. Front. Plant Sci. 5:499. doi:https://doi.org/10.3389/fpls.2014.00499
- Niehaus, T. D., and Hillmann, K. B. 2020. Enzyme promiscuity, metabolite damage, and metabolite damage control systems of the tricarboxylic acid cycle. FEBS J. 287:1343–1358.
- Nielsen, T. H. 1995. Fructose-1,6-bisphosphate is an allosteric activator of pyrophosphate:fructose-6-phosphate 1-phosphotransferase. Plant Physiol. 108:69–73.
- Nikiforova, V. J., Kopka, J., Tolstikov, V., Fiehn, O., Hopkins, L., Hawkesford, M. J., Hesse, H., and Hoefgen, R. 2005. Systems rebalancing of metabolism in response to sulfur deprivation, as revealed by metabolome analysis of Arabidopsis plants. Plant Physiol. 138:304–318.
- Nunes-Nesi, A., Araújo, W. L., and Fernie, A. R. 2011. Targeting mitochondrial metabolism and machinery as a means to enhance photosynthesis. Plant Physiol. 155:101–107. doi:https://doi.org/10.1104/pp.110.163816
- Nunes-Nesi, A., Araújo, W. L., Obata, T., and Fernie, A. R. 2013. Regulation of the mitochondrial tricarboxylic acid cycle. Curr. Opin. Plant Biol. 16:335–343. doi:https://doi.org/10.1016/j.pbi.2013.01.004
- Nunes-Nesi, A., Carrari, F., Lytovchenko, A., Smith, A. M. O., Loureiro, M. E., Ratcliffe, R. G., Sweetlove, L. J., and Fernie, A. R. 2005. Enhanced photosynthetic performance and growth as a consequence of decreasing mitochondrial malate dehydrogenase activity in transgenic tomato plants. Plant Physiol. 137:611–622. doi:https://doi.org/10.1104/pp.104.055566
- Omidbakhshfard, M. A., Sokolowska, E. M., Di Vittori, V., Souza, L. P., de Kuhalskaya, A., Brotman, Y., Alseekh, S., Fernie, A. R., and Skirycz, A. 2021. Multi-omics analysis of early leaf development in Arabidopsis thaliana. PATTER. 2: 100235.
- Omini, J., Wojciechowska, I., Skirycz, A., Moriyama, H., and Obata, T. 2021. Association of the malate dehydrogenase-citrate synthase metabolon is modulated by intermediates of the Krebs tricarboxylic acid cycle. Sci. Rep. 11:18770.
- Piazza, I., Kochanowski, K., Cappelletti, V., Fuhrer, T., Noor, E., Sauer, U., and Picotti, P. 2018. A map of protein-metabolite interactions reveals principles of chemical communication. Cell. 172:358–372.e23. doi:https://doi.org/10.1016/j.cell.2017.12.006
- Plaxton, W. C. 1996. The organization and regulation of plant glycolysis. Annu. Rev. Plant Physiol. Plant Mol. Biol. 47:185–214. doi:https://doi.org/10.1146/annurev.arplant.47.1.185
- Plaxton, W. C. 1997. Metabolic regulation. Plant Metabol. 50–68. New York: Longman.
- Podestá, F. E., and Plaxton, W. C. 1991. Association of phosphoenolpyruvate phosphatase activity with the cytosolic pyruvate kinase of germinating mung beans. Plant Physiol. 97:1329–1333. doi:https://doi.org/10.1104/pp.97.4.1329
- Poovaiah, B. W., and Reddy, A. S. N. 1993. Calcium and signal transduction in plants. CRC Crit. Rev. Plant Sci. Crit. Rev. Plant.12:185–185. doi:https://doi.org/10.1080/07352689309701901
- Purugganan, M. D., and Jackson, S. A. 2021. Advancing crop genomics from lab to field. Nat. Genet. 53:595–601. doi:https://doi.org/10.1038/s41588-021-00866-3
- Raines, C. A. 2003. The Calvin cycle revisited. Photosynth. Res. 75:1–10. doi:https://doi.org/10.1023/A:1022421515027
- Ranjeva, R., and Boudet, A. M. 1987. Phosphorylation of proteins in plants: regulatory effects and potential involvement in stimulus/response coupling. Annu. Rev. Plant Physiol. 38:73–94. doi:https://doi.org/10.1146/annurev.pp.38.060187.000445
- Roessner, U., Willmitzer, L., and Fernie, A. R. 2001. High-resolution metabolic phenotyping of genetically and environmentally diverse potato tuber systems. identification of phenocopies. Plant Physiol. 127:749–764. doi:https://doi.org/10.1104/pp.010316
- Roessner, U., Luedemann, A., Brust, D., Fiehn, O., Linke, T., Willmitzer, L., and Fernie, A. 2001. Metabolic profiling allows comprehensive phenotyping of genetically or environmentally modified plant systems. Plant Cell. 13:11–29. doi:https://doi.org/10.1105/tpc.13.1.11
- Roessner-Tunali, U., Liu, J., Leisse, A., Balbo, I., Perez-Melis, A., Willmitzer, L., and Fernie, A. R. 2004. Kinetics of labelling of organic and amino acids in potato tubers by gas chromatography-mass spectrometry following incubation in (13)C labelled isotopes. Plant J. 39:668–679. doi:https://doi.org/10.1111/j.1365-313X.2004.02157.x
- Rontein, D., Dieuaide-Noubhani, M., Dufourc, E. J., Raymond, P., and Rolin, D. 2002. The metabolic architecture of plant cells. Stability of central metabolism and flexibility of anabolic pathways during the growth cycle of tomato cells. J. Biol. Chem. 277:43948–43960. doi:https://doi.org/10.1074/jbc.M206366200
- Roscher, A., Emsley, L., Raymond, P., and Roby, C. 1998. Unidirectional steady state rates of central metabolism enzymes measured simultaneously in a living plant tissue. J. Biol. Chem. 273:25053–25061. doi:https://doi.org/10.1074/jbc.273.39.25053
- Sabularse, D. C., and Anderson, R. L. 1981. Inorganic pyrophosphate: D-fructose-6-phosphate 1-phosphotransferase in mung beans and its activation by D-fructose 1,6-bisphosphate and D-glucose 1, 6-bisphosphate. Biochem. Biophys. Res. Commun. 100:1423–1429.
- Savitski, M. M., Reinhard, F. B. M., Franken, H., Werner, T., Savitski, M. F., Eberhard, D., Martinez Molina, D., Jafari, R., Dovega, R. B., Klaeger, S., Kuster, B., Nordlund, P., Bantscheff, M., and Drewes, G. 2014. Tracking cancer drugs in living cells by thermal profiling of the proteome. Science. 346:1255784. doi:https://doi.org/10.1126/science.1255784
- Scheibe, R. 1991. Redox-modulation of chloroplast enzymes: a common principle for individual control. Plant Physiol. 96:1–3. doi:https://doi.org/10.1104/pp.96.1.1
- Scheibe, R., Wedel, N., Vetter, S., Emmerlich, V., and Sauermann, S.-M. 2002. Co-existence of two regulatory NADP-glyceraldehyde 3-P dehydrogenase complexes in higher plant chloroplasts. Eur. J. Biochem. 269:5617–5624. doi:https://doi.org/10.1046/j.1432-1033.2002.03269.x
- Schlossarek, D., Luzarowski, M., Sokołowska, E., Górka, M., Willmitzer, L., and Skirycz, A. 2021. PROMISed: a novel web-based tool to facilitate analysis and visualization of the molecular interaction networks from co-fractionation mass spectrometry (CF-MS) experiments. Comput. Struct. Biotechnol. J. 19:5117–5125. doi:https://doi.org/10.1016/j.csbj.2021.08.042
- Schurmann, P., and Jacquot, J.-P. 2000. Plant thioredoxin systems revisited. Annu. Rev. Plant Physiol. Plant Mol. Biol. 51:371–400. doi:https://doi.org/10.1146/annurev.arplant.51.1.371
- Schwender, J., Goffman, F., Ohlrogge, J. B., and Shachar-Hill, Y. 2004. Rubisco without the Calvin cycle improves the carbon efficiency of developing green seeds. Nature. 432:779–782. doi:https://doi.org/10.1038/nature03145
- Schwender, J., Shachar-Hill, Y., and Ohlrogge, J. B. 2006. Mitochondrial metabolism in developing embryos of Brassica napus. J. Biol. Chem. 281:34040–34047. doi:https://doi.org/10.1074/jbc.M606266200
- Scott, P., Lange, A. J., Pilkis, S. J., and Kruger, N. J. 1995. Carbon metabolism in leaves of transgenic tobacco (Nicotiana tabacum L.) containing elevated fructose 2,6-bisphosphate levels. Plant J. 7:461–469.
- Senkler, J., Senkler, M., Eubel, H., Hildebrandt, T., Lengwenus, C., Schertl, P., Schwarzländer, M., Wagner, S., Wittig, I., and Braun, H.-P. 2017. The mitochondrial complexome of Arabidopsis thaliana. Plant J. 89:1079–1092. doi:https://doi.org/10.1111/tpj.13448
- Sharkey, T. D. 2019. Discovery of the canonical Calvin-Benson cycle. Photosynth. Res. 140:235–252. doi:https://doi.org/10.1007/s11120-018-0600-2
- Shimobayashi, M., and Hall, M. N. 2014. Making new contacts: the mTOR network in metabolism and signalling crosstalk. Nat. Rev. Mol. Cell. Biol. 15:155–162. doi:https://doi.org/10.1038/nrm3757
- Słabicki, M., Yoon, H., Koeppel, J., Nitsch, L., Roy Burman, S. S., Di Genua, C., Donovan, K. A., Sperling, A. S., Hunkeler, M., Tsai, J. M., Sharma, R., Guirguis, A., Zou, C., Chudasama, P., Gasser, J. A., Miller, P. G., Scholl, C., Fröhling, S., Nowak, R. P., Fischer, E. S., and Ebert, B. L. 2020. Small-molecule-induced polymerization triggers degradation of BCL6. Nature. 588:164–168. doi:https://doi.org/10.1038/s41586-020-2925-1
- Smeekens, S., Macdonald, F. D., and Buchanan, B. B. 1989. Studies on the entry of fructose-2,6-bisphosphate into chloroplasts. Plant Physiol. 89:1270–1274.
- Soll, J., Wötzel, C., and Buchanan, B. B. 1985. Enzyme regulation in c(4) photosynthesis: identification and localization of activities catalyzing the synthesis and hydrolysis of fructose-2,6-bisphosphate in corn leaves. Plant Physiol. 77:999–1003. doi:https://doi.org/10.1104/pp.77.4.999
- Srere, P. 1994. Complexities of metabolic regulation. Trends Biochem. Sci. 19:519–520. doi:https://doi.org/10.1016/0968-0004(94)90048-5
- Srere, P. A., Sumegi, B., and Sherry, A. D. 1987. Organizational aspects of the citric acid cycle. Biochem. Soc. Symp. 54:173–178.
- Stitt, M., Cseke, C., Buchannen B.B. (1984). Regulation of fructose 2,6-bisphosphate concentration in spinach leaves . Eur. J. Blochem. 143:89–93.
- Stitt, M. 1987. Fructose 2,6-bisphosphate and plant carbohydrate metabolism. Plant Physiol. 84:201–204. doi:https://doi.org/10.1104/pp.84.2.201
- Stitt, M. 1989. Control of sucrose synthesis: estimation of free energy changes, investigation of the contribution of equilibrium and non-equilibrium reactions, and estimation of elasticities and flux control coefficients. Tech. New. Dev. Photosynth. Res. 323, 365–391.
- Stitt, M. 1990a. Fructose 2, 6-bisphosphate. Methods Plant Biochem. 3:87–92.
- Stitt, M. 1990b. Fructose-2,6-bisphosphate as a regulatory molecule in plants. Annu. Rev. Plant Physiol. Plant Mol. Biol. 41:153–185. doi:https://doi.org/10.1146/annurev.pp.41.060190.001101
- Stitt, M. 1998. Pyrophosphate as an energy donor in the cytosol of plant cells: an enigmatic alternative to ATP. Bot. Acta. 111:167–175. doi:https://doi.org/10.1111/j.1438-8677.1998.tb00692.x
- Stitt, M., and Heldt, H. W. 1985. Control of photosynthetic sucrose synthesis by fructose-2, 6-bisphosphate: intercellular metabolite distribution and properties of the cytosolic fructosebisphosphatase in leaves of Zea mays L. Planta. 164:179–188. doi:https://doi.org/10.1007/BF00396080
- Stitt, M., and Paul Quick, W. 1989. Photosynthetic carbon partitioning: its regulation and possibilities for manipulation. Physiol. Plant. 77:633–641. doi:https://doi.org/10.1111/j.1399-3054.1989.tb05402.x
- Stitt, M., and Sonnewald, U. 1995. Regulation of metabolism in transgenic plants. Annu. Rev. Plant Physiol. Plant Mol. Biol. 46:341–368. doi:https://doi.org/10.1146/annurev.pp.46.060195.002013
- Stitt, M., Herzog, B., and Heldt, H. W. 1984. Control of photosynthetic sucrose synthesis by fructose 2,6-bisphosphate: I. coordination of CO(2) fixation and sucrose synthesis. Plant Physiol. 75:548–553. doi:https://doi.org/10.1104/pp.75.3.548
- Stitt, M., Lilley, R. M., and Heldt, H. W. 1982. Adenine nucleotide levels in the cytosol, chloroplasts, and mitochondria of wheat leaf protoplasts. Plant Physiol. 70:971–977.
- Stitt, M., Lunn, J., and Usadel, B. 2010. Arabidopsis and primary photosynthetic metabolism - more than the icing on the cake. Plant J. 61:1067–1091. doi:https://doi.org/10.1111/j.1365-313X.2010.04142.x
- Stitt, M., Walker, D. A., and Osmond, C. B. 1989. Control analysis of photosynthetic sucrose synthesis: assignment of elasticity coefficients and flux-control coefficients to the cytosolic fructose 1,6-bisphosphatase and sucrose phosphate synthase. Philos. Trans. R. Soc. Lond. B Biol. Sci 323:327–338.
- Studart-Guimarães, C., Gibon, Y., Frankel, N., Wood, C. C., Zanor, M. I., Fernie, A. R., and Carrari, F. 2005. Identification and characterisation of the alpha and beta subunits of succinyl CoA ligase of tomato. Plant Mol. Biol. 59:781–791. doi:https://doi.org/10.1007/s11103-005-1004-1
- Sweetlove, L. J., Obata, T., and Fernie, A. R. 2014. Systems analysis of metabolic phenotypes: what have we learnt? Trends Plant Sci. 19:222–230. doi:https://doi.org/10.1016/j.tplants.2013.09.005
- Sweetlove, L. J., and Fernie, A. R. 2018. The role of dynamic enzyme assemblies and substrate channelling in metabolic regulation. Nat. Commun. 9:2136. doi:https://doi.org/10.1038/s41467-018-04543-8
- Szecowka, M., Heise, R., Tohge, T., Nunes-Nesi, A., Vosloh, D., Huege, J., Feil, R., Lunn, J., Nikoloski, Z., Stitt, M., Fernie, A. R., and Arrivault, S. 2013. Metabolic fluxes in an illuminated Arabidopsis rosette. Plant Cell. 25:694–714. doi:https://doi.org/10.1105/tpc.112.106989
- Tcherkez, G., Cornic, G., Bligny, R., Gout, E., and Ghashghaie, J. 2005. In vivo respiratory metabolism of illuminated leaves. Plant Physiol. 138:1596–1606. doi:https://doi.org/10.1104/pp.105.062141
- Tovar-Méndez, A., Miernyk, J. A., and Randall, D. D. 2003. Regulation of pyruvate dehydrogenase complex activity in plant cells. Eur. J. Biochem. 270:1043–1049. doi:https://doi.org/10.1046/j.1432-1033.2003.03469.x
- Trevanion, S. J., and Kruger, N. J. 1991. Effect of temperature on the kinetic properties of pyrophosphate: fructose 6-phosphate phosphotransferase from potato tuber. J. Plant Physiol. 137:753–759. doi:https://doi.org/10.1016/S0176-1617(11)81235-6
- Urbanczyk-Wochniak, E., Luedemann, A., Kopka, J., Selbig, J., Roessner-Tunali, U., Willmitzer, L., and Fernie, A. R. 2003. Parallel analysis of transcript and metabolic profiles: a new approach in systems biology. EMBO Rep. 4:989–993. doi:https://doi.org/10.1038/sj.embor.embor944
- Van Schaftingen, E., and Hers, H. G. 1980. Synthesis of a stimulator of phosphofructokinase, most likely fructose 2,6-bisphosphate, from phosphoric acid and fructose 6-phosphoric acid. Biochem. Biophys. Res. Commun. 96:1524–1531.
- Van Schaftingen, E., and Opperdoes, F. R., 1985. Stimulation of Trypanosoma brucei pyruvate kinase by ftuctose 2,6‐bisphosphate. Eur. J. Biochem. 153:403–406. doi:https://doi.org/10.1111/j.1432-1033.1985.tb09316.x
- Venegas-Molina, J., Molina-Hidalgo, F. J., Clicque, E., and Goossens, A. 2021. Why and how to dig into plant metabolite-protein interactions. Trends Plant Sci. 26:472–483. doi:https://doi.org/10.1016/j.tplants.2020.12.008
- Veyel, D., Sokolowska, E. M., Moreno, J. C., Kierszniowska, S., Cichon, J., Wojciechowska, I., Luzarowski, M., Kosmacz, M., Szlachetko, J., Gorka, M., Méret, M., Graf, A., Meyer, E. H., Willmitzer, L., and Skirycz, A. 2018. PROMIS, global analysis of PROtein–metabolite interactions using size separation in Arabidopsis thaliana. J. Biol. Chem. 293:12440–12453.
- Vierstra, R. D. 2012. The expanding universe of ubiquitin and ubiquitin-like modifiers. Plant Physiol. 160:2–14. doi:https://doi.org/10.1104/pp.112.200667
- Vogel, G., Fiehn, O., Jean‐Richard‐dit‐Bressel, L., Boller, T., Wiemken, A., Aeschbacher, R. A., and Wingler, A. 2001. Trehalose metabolism in Arabidopsis: occurrence of trehalose and molecular cloning and characterization of trehalose-6-phosphate synthase homologues . J. Exp. Bot. 52:1817–1826. doi:https://doi.org/10.1093/jexbot/52.362.1817
- Wagner, M., Zhang, B., Tauffenberger, A., Schroeder, F. C., and Skirycz, A. 2021. Experimental methods for dissecting the terra-incognita of protein-metabolite interactomes. Curr. Opin. Syst. Biol. 28:100403. doi:https://doi.org/10.1016/j.coisb.2021.100403
- Wang, S., Alseekh, S., Fernie, A. R., and Luo, J. 2019. The structure and function of major plant metabolite modifications. Mol. Plant. 12:899–919. doi:https://doi.org/10.1016/j.molp.2019.06.001
- Weissmann, S., Ma, F., Furuyama, K., Gierse, J., Berg, H., Shao, Y., Taniguchi, M., Allen, D. K., and Brutnell, T. P. 2016. Interactions of C4 subtype metabolic activities and transport in maize are revealed through the characterization of DCT2 mutants. Plant Cell. 28:466–484. doi:https://doi.org/10.1105/tpc.15.00497
- West, G. M., Tang, L., and Fitzgerald, M. C. 2008. Thermodynamic analysis of protein stability and ligand binding using a chemical modification- and mass spectrometry-based strategy. Anal. Chem. 80:4175–4185. doi:https://doi.org/10.1021/ac702610a
- Wigge, B., Kromer, S., and Gardestrom, P. 1993. The redox levels and subcellular distribution of pyridine nucleotides in illuminated barley leaf protoplasts studied by rapid fractionation. Physiol. Plant. 88:10–18. doi:https://doi.org/10.1111/j.1399-3054.1993.tb01754.x
- van Wijk, K. J., Leppert, T., Sun, Q., Boguraev, S. S., Sun, Z., Mendoza, L., and Deutsch, E. W. 2021. The Arabidopsis peptideatlas: harnessing worldwide proteomics data to create a comprehensive community proteomics resource. Plant Cell. 33:3421–3453. doi:https://doi.org/10.1093/plcell/koab211
- Zhai, Z., Keereetaweep, J., Liu, H., Feil, R., Lunn, J. E., and Shanklin, J. 2018. Trehalose 6-phosphate positively regulates fatty acid synthesis by stabilizing WRINKLED1. Plant Cell. 30:2616–2627. doi:https://doi.org/10.1105/tpc.18.00521
- Zhang, Y., and Fernie, A. R. 2021. Metabolons, enzyme–enzyme assemblies that mediate substrate channeling, and their roles in plant metabolism. Plant Commun. 2:100081.
- Zhang, Y., Beard, K. F. M., Swart, C., Bergmann, S., Krahnert, I., Nikoloski, Z., Graf, A., Ratcliffe, R. G., Sweetlove, L. J., Fernie, A. R., and Obata, T. 2017. Protein-protein interactions and metabolite channelling in the plant tricarboxylic acid cycle. Nat. Commun. 8:15212. doi:https://doi.org/10.1038/ncomms15212
- Zhang, Y., Giese, J., Kerbler, S. M., Siemiatkowska, B., Perez de Souza, L., Alpers, J., Medeiros, D. B., Hincha, D. K., Daloso, D. M., Stitt, M., Finkemeier, I., and Fernie, A. R. 2021. Two mitochondrial phosphatases, PP2c63 and Sal2, are required for posttranslational regulation of the TCA cycle in Arabidopsis. Mol. Plant. 14:1104–1118.
- Zhang, Y., Sampathkumar, A., Kerber, S. M.-L., Swart, C., Hille, C., Seerangan, K., Graf, A., Sweetlove, L., and Fernie, A. R. 2020a. A moonlighting role for enzymes of glycolysis in the co-localization of mitochondria and chloroplasts. Nat. Commun 11:1–15.
- Zhang, Y., Skirycz, A., and Fernie, A. R. 2020b. An abundance and interaction encyclopedia of plant protein function. Trends Plant Sci. 25:627–630. doi:https://doi.org/10.1016/j.tplants.2020.04.006
- Zhang, Y., Swart, C., Alseekh, S., Scossa, F., Jiang, L., Obata, T., Graf, A., and Fernie, A. R. 2018. The extra-pathway interactome of the TCA cycle: expected and unexpected metabolic interactions. Plant Physiol. 177:966–979. doi:https://doi.org/10.1104/pp.17.01687
- Ziegler, D. M. 1985. Role of reversible oxidation-reduction of enzyme thiols-disulfides in metabolic regulation. Annu. Rev. Biochem. 54:305–329. doi:https://doi.org/10.1146/annurev.bi.54.070185.001513
- Zimmermann, G., Kelly, G. J., and Latzko, E. 1978. Purification and properties of spinach leaf cytoplasmic fructose-1,6-bisphosphatase. J. Biol. Chem. 253:5952–5956.
- Zimmermann, P., Hirsch-Hoffmann, M., Hennig, L., and Gruissem, W. 2004. GENEVESTIGATOR. Arabidopsis microarray database and analysis toolbox. Plant Physiol. 136:2621–2632. doi:https://doi.org/10.1104/pp.104.046367
- Zubimendi, J. P., Martinatto, A., Valacco, M. P., Moreno, S., Andreo, C. S., Drincovich, M. F., and Tronconi, M. A. 2018. The complex allosteric and redox regulation of the fumarate hydratase and malate dehydratase reactions of Arabidopsis thaliana fumarase 1 and 2 gives clues for understanding the massive accumulation of fumarate. FEBS J. 285:2205–2224. doi:https://doi.org/10.1111/febs.14483