
ABSTRACT
Vascular targeted therapies (VTTs) are agents that target tumor vasculature and can be classified into two categories: those that inhibit angiogenesis and those that directly interfere with established tumor vasculature. Although both the anti-angiogenic agents (AAs) and the vascular disrupting agents (VDAs) target tumor vasculature, they differ in their mechanism of action and therapeutic application. Combining these two agents may realize the full potential of VTT and produce an effective therapeutic regimen. Here, we review AAs and VDAs (monotherapy and in combination with conventional therapies). We also discuss the rationale of combined VTT and its potential to treat cancer.
Introduction
A functioning vascular supply is not only essential for the growth of solid tumors but also plays a critical role in metastatic spread (Citation1, 2). Early in their development, tumors utilize the vasculature of the host tissue in which the tumor arises. Once tumors reach a few millimeters in size, this host vasculature becomes inadequate in meeting the oxygen and nutrient demands of the growing tumor mass (Citation3). At this point, the tumor requires new tumor-specific blood vessels, which are formed primarily from the host vasculature via angiogenesis (Citation2). The tumor neovasculature that results is very different from vessels found in normal tissues (Citation3, 4): it is structurally chaotic with abnormal vascular density, contour irregularities, haphazard vessel interconnection, and loss of hierarchy. The tumor blood vessels are very primitive, with incomplete or missing basement membranes, and lack innervation and pharmacological receptors. Additionally, they are deficient in smooth muscle and pericytes and are, therefore, more reliant on endothelial cells to maintain the flat shape of vessel wall (Citation5). These attributes result in highly variable blood flow through the tumor vessels.
As tumor vasculature is essential for the development and progressive growth of tumors, this makes it a key anticancer target. Two important approaches in anticancer treatment have developed (Citation6–8): (1) the use of agents that inhibit the development of new vasculature (anti-angiogenesis and antivasculogenesis); and (2) treatments that destroy existing blood vessel networks (vascular disruption). Although both the anti-angiogenic agents (AAs; also known as angiogenesis inhibitors) and the vascular disrupting agents (VDAs) target tumor vasculature, they differ markedly in their mechanism of action and therapeutic application. These differences give rise to the possibility that AAs and VDAs in combination may produce an effective therapeutic regimen. Although there are older previously published reviews assessing vascular targeted therapies (VTTs) in oncology (Citation8–10), here we conduct a broader review of the VTT field, with an update of clinical trials. In this review, we appraise both AAs and VDAs and focus on the rationale of combined VTT and its potential to treat cancer.
Anti-angiogenic agents
Tumoral angiogenesis was first suggested as a therapeutic target in the early 1970s (Citation1) and, to date, many AAs have been identified and characterized. The detailed steps of angiogenesis are not included here as they have been reviewed elsewhere (Citation11). AAs inhibit the tumor-initiated angiogenic process by interrupting essential aspects of angiogenesis to prevent further development or expansion of the tumor neovascular network (). Consequently, these agents can be effective at inhibiting tumor progression (Citation2, 12, 13). Most AAs target the angiogenic factors secreted by tumor cells (Citation14) and suppress tumor growth by inhibiting the migration and proliferation of endothelial cells (Citation15, 16).
Figure 1. Solid tumor undergoing treatment with combined vascular targeted therapies (VTT). The top portion of the figure illustrates anti-angiogenic agent (AA)-induced inhibition of tumor progression. The lower portion of the figure illustrates vascular disrupting agent (VDA)-induced necrosis of the central tumor cells (with a peripheral rim of viable tumor cells spared). The right-hand side of the figure illustrates the complementary effects of VDAs and AAs in combination (combined VTT): the destruction of blood vessels in the interior of the tumor and the prevention of new tumor vasculature formation, which together have negative effects on the tumor's growth and survival. ©2017 Link Health Group, LLC; Illustrated by Laura Maaske, Medimagery LLC.
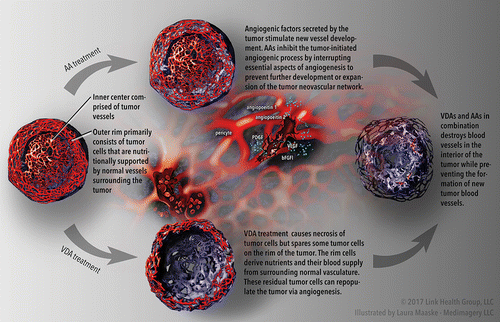
In tumor development, angiogenic factors are upregulated by a variety of mechanisms, including loss of tumor suppressor gene function, oncogene activation, and hypoxic microenvironments (Citation14, 17). Vascular endothelial growth factor (VEGF) is considered the major driver of angiogenesis in solid tumors as it is essential for endothelial cell proliferation and vessel formation. VEGF induces vascular permeability and is vital to endothelial cell survival in new vessels. Thus, the inhibition of VEGF, or other components of the angiogenic pathway, is key in anti-angiogenic cancer treatment. There are three major approaches to achieve VEGF blockade including targeting the VEGF ligand with agents such as bevacizumab (Citation18) (humanized anti-VEGF antibody), targeting VEGF signal transduction with tyrosine kinase inhibitors (e.g., cediranib) (Citation19), and using soluble recombinant fusion proteins that act as decoys for VEGF receptors (e.g., aflibercept) (Citation20). Other important pro-angiogenic mediators, which may also serve as targets for anti-angiogenic therapies (Citation21), include platelet-derived growth factor (Citation22), fibroblast growth factor (Citation23), and angiopoietins (Citation24).
AAs can inhibit the growth of newly formed blood vessels in tumors by targeting either the endothelial cells directly or by targeting other tumor-associated stromal cells (Citation25, 26). In 2004, the US Food and Drug Administration (FDA) approved the first AA, bevacizumab, for the treatment of colorectal cancer (Citation27). Since then, the FDA has approved bevacizumab for several other indications, including cervical cancer, renal cell carcinoma, recurrent glioblastoma, non-small cell lung cancer (NSCLC), and platinum-resistant ovarian cancer. A number of other AAs have also been FDA-approved, as shown in . These agents are currently being investigated in a variety of tumor types, while a number of new AAs are in clinical development ().
Table 1. Select anti-angiogenic agents that are FDA-approved for cancer treatment.
Table 2. Vascular targeted therapies in active clinical development.
AAs as monotherapy
There are a small number of AAs with FDA-approval as single-agent therapy in cancer treatment. Bevacizumab is approved as monotherapy for patients with recurrent glioblastoma following prior cancer treatment. The approval was based on the durable objective response rates observed in two single-arm trials (NCT00345163 and NCI 06-C-0064E) (Citation28–30). Ramucirumab is approved for use as a single agent for the treatment of patients with advanced or metastatic gastric or gastroesophageal junction adenocarcinoma, whose disease has progressed during or after prior treatment with fluoropyrimidine- or platinum-containing chemotherapy (NCT00917384). (Citation31, 32).
Due to the complexity of the pathways available for neovascularization, it is unlikely that inhibiting only a single mechanism (or pathway) of angiogenesis will be sufficient to control a tumor (Citation13). AA monotherapy is unlikely to be curative even after prolonged administration at high doses; however, it has clinical utility when used as maintenance therapy in cancer patients previously treated with first- or second-line chemotherapy in order to extend progression-free survival (PFS) (Citation33, 34).
AAs in combination with conventional therapies
AAs are more effective when combined with other conventional therapies, such as chemotherapy or radiotherapy, and may be added to treatment regimens to increase their efficacy or to reduce developing therapeutic resistance. The effectiveness of AAs in combination therapy may be due to the inhibition of potential tumor regeneration arising from residual cells that remain following initial anticancer treatment. While AAs in combination with chemotherapy have been extensively reviewed previously (Citation35–38), it is important to highlight that the majority of studies showed an increased benefit with this combination (Citation39, 40), with only a small number showing no additional benefit (Citation41–45). The sequencing schedule of AAs in combination with chemotherapy can be neoadjuvant, concurrent, (Citation46, 47) or adjuvant depending on the cancer type and which AA and chemotherapeutic agents are used (Citation48, 49).
Radiotherapy failures occur due to local tumor progression and/or tumor cell dissemination to distant sites (Citation50). Thus, inhibition of progression or spread can result in improvement of radiation treatment outcome. Numerous preclinical studies have investigated the therapeutic potential of combining AAs with radiation treatments (Citation40–42). There are relatively few clinical studies focusing on AAs in combination with radiotherapy; however, it is generally accepted that the combination of AAs and radiation therapy is superior to either treatment alone (Citation51–53). There have been a number of Phase I (NCT00307736), II (NCT00847119 and NCT00281840) and III (NCT00943826 and RTOG 0825) clinical trials that have investigated bevacizumab in combination with chemoradiation (Citation54–61) and have shown an increased benefit. However, in a Phase II trial (NCIC CTG BR.20) with vandetanib (ZD6474) in combination with chemoradiation in small cell lung cancer patients, vandetanib failed to show any efficacy (Citation62). The importance of sequencing between AAs and radiation has been investigated in both preclinical and clinical studies, although due to the lack of standardization for drug doses, treatment times and combination schedules that are applied with radiation, it is difficult to draw definitive conclusions. However, most studies have demonstrated that an adjuvant schedule may be more effective than concomitant or neoadjuvant administration (Citation63–65).
Early clinical studies have shown promising results for immunotherapy and AA combination therapy. The majority of these investigations utilized the AA bevacizumab. A Phase I study (NCT00790010) combining ipilimumab, a monoclonal antibody that activates the immune system by targeting CTLA-4, and bevacizumab in advanced melanoma patients yielded a 19.6% objective response rate and a median survival of 25.1 months (N = 46) – an outcome nearly twice that expected for ipilimumab alone (Citation46). High-grade colitis was observed in two patients, but this was not unexpected as colitis is an adverse effect of ipilimumab treatment. Results of a Phase Ib study (NCT01633970) presented at the 2015 Genitourinary Cancers Symposium, which combined atezolizumab, a monoclonal antibody against the protein programmed cell death-ligand 1 (PD-L1), with bevacizumab in clear cell metastatic renal cell carcinoma patients showed that the combination led to immune modulation in the tumor microenvironment, was well-tolerated, and offered promising clinical activity (Citation47).
Vascular disrupting agents
The pivotal role of tumor vasculature and its selective destruction were first highlighted by Woglum in 1923 (Citation66). While the feasibility of the VDA concept was suggested by experience with colchicine in the 1930s (Citation67), it wasn't until more recently, that the effects of VDAs on tumor pathophysiology began to be extensively studied and described (Citation51, 68–74). A number of other reviews of the VDA field have been published recently (Citation75–77). These agents damage the established tumor neovasculature, thus depriving tumors of oxygen and essential nutrients, resulting in rapid and widespread ischemia and necrosis in tumors, while leaving the blood flow through normal tissues unaffected (Citation68, 78–81). Although VDA treatment leads to extensive necrosis in the inner regions of tumors, typically a rim of viable tumor cells remains at the tumor periphery in areas where tumor and normal tissues interface (Citation69, 82–88) (). Tumor cells survive in these regions and serve as a source for tumor regrowth because the nearby normal tissue vasculature that supports their nutritional needs is unaffected by VDA treatment. Hence, the potential value of VDAs requires the addition of other therapies or agents, as covered in this review.
Small-molecule VDAs exploit the unique features of the tumor endothelium to induce selective vascular dysfunction (Citation68, 79). Many of these agents are tubulin-binding VDAs and include first-generation VDAs, such as combretastatin A4-phosphate (CA4P or fosbretabulin), and second-generation VDAs, such as OXi4503 (combretastatin A1-diphosphate or CA1P). Newer compounds have been evaluated in vitro/in vivo (Citation89, 90) but face the same shortcomings as other VDAs. The principle mechanism of action of tubulin-binding VDAs is believed to be the selective disruption of the cytoskeleton of proliferating endothelial cells, thereby changing the shape of the cells (Citation91). Binding to tubulin causes microtubule depolymerization, cytoskeletal rearrangements, and activation of actin stress fibers in endothelial cells, leading to changes in cell morphology (Citation69, 85, 91, 92); this, in turn, leads to the rounding up and detachment of the affected endothelial cells (Citation84, 93). In addition, leakage of plasma proteins results in increased interstitial fluid pressure (IFP), which is sufficient to contribute to vascular occlusion via a direct increase in resistance to blood flow. Loss of cell-to-cell contact also results in the exposure of abnormal basement membrane, which activates platelets and induces the coagulation cascade to form thrombi. Taken together, the damage to the expanding tumor vascular network leads to an occlusion of blood flow, and the subsequent compromised supply of oxygen and nutrients induces necrosis of tumor cells downstream (Citation68, 78–81). Although several VDAs are in clinical development (Citation94) (), CA4P is the most extensively studied tubulin-binding VDA. Robust evidence of tumor blood flow reductions has been reported in both preclinical and clinical investigations with this agent (Citation74, 78, 88, 95–97). For most other VDAs, published clinical evidence of meaningful blood flow reduction is limited (Citation97–107).
Synthetic flavonoids, particularly flavone acetic acid and its derivative DMXAA (also known as ASA404), represent another series of small-molecule VDAs that have been investigated extensively. The main action of flavonoids on endothelial cells in the tumor vasculature is thought to involve a cascade of direct and indirect events, which include release of vasoactive agents and cytokines, leading to induction of hemorrhagic necrosis (Citation99, 108). ASA404 is the only flavonoid agent to advance to Phase III clinical trials. While it failed in NSCLC (NCT00738387) (Citation109), it did show evidence of tumor blood flow reduction (Citation110–112).
In addition to the small-molecule VDAs, certain chemotherapeutic drugs, such as vinca alkaloids and arsenic trioxide, can directly damage the tumor vasculature (Citation113, 114). Furthermore, ligand-based VDA therapies use antibodies, peptides, or growth factors to deliver toxins, pro-coagulants, or pro-apoptotic effectors to the tumor vessels (Citation9, 68, 78, 79, 81, 115, 116).
It is also important to note that there are a small number of alternative tumor vasculature damaging methods that may be effective in tumor treatment such as hyperthermia (Citation51, 117), photodynamic therapy (Citation51, 118), focused ultrasound (Citation119, 120), and gold nanoparticles (Citation121).
Tumor imaging provides a platform for non-invasively assessing the damage induced by VDAs (Citation122, 123). As such, tumor imaging is a valuable tool in VDA development.
VDAs as monotherapy
In preclinical trials, VDA monotherapies have been shown to induce significant necrosis in solid tumors; however, limited effects on tumor growth or size are usually observed. Clinically, there is also limited evidence of tumor shrinkage using VDAs as single agents, although a complete response was observed in a patient with anaplastic thyroid cancer (ATC) treated in a Phase I trial (NCT00003768) with CA4P (Citation102). The VDA BNC105 is a tubulin polymerization inhibitor that preclinically demonstrated an ability to inhibit tumor growth (Citation124), but following a Phase II trial (no trial registration number) it was deemed ineffective as a monotherapy when used in mesothelioma (Citation125). Furthermore, as discussed above, VDA monotherapy is unlikely to be curative as these agents are not able to eliminate residual cells at the tumor's edge (Citation68, 71). Conceptually, two approaches could slow or prevent this regrowth: the first is targeting the remaining tumor cells using conventional chemotherapy or radiotherapy; the second is by inhibiting the process of angiogenesis initiated by the surviving tumor cells.
VDAs in combination with conventional therapies
Combining VDAs with conventional anticancer therapies has the potential to eradicate tumors. Radiotherapy and chemotherapy alone exhibit reduced efficacy against cancer cells in tumor areas of low vascular density because these regions contain either radiation-resistant hypoxic cells or have inadequate vasculature, impairing drug delivery. As a tumor grows, the pressures exerted on its vascular support network become increasingly severe. When the resultant physiological stresses are coupled with an increasing neoplastic cell burden, it is perhaps not surprising that bulky advanced tumors typically prove particularly difficult to control with conventional anticancer therapies such as radiation treatment (Citation11, 126, 127), chemotherapy and surgery (Citation50, 122, 128–130). In solid tumors, increases in size are often associated with increases in hypoxic cell fractions (Citation122, 130–135) likely due to size-dependent reductions in tumor blood flow (Citation132, 133). These hypoxic and acidic regions can cause radiation resistance (Citation136–138) and impair chemotherapeutic agent delivery and effectiveness (Citation139). By contrast, VDA investigations in a variety of rodent and human tumor models have demonstrated a reverse tumor size dependency; that is, unlike conventional anticancer treatments, VDAs are selectively more effective against large tumor masses when assessed by clonogenic cell survival or tumor growth delay () (Citation82, 140–143).
Figure 2. Tumor surviving fractions in rodent (a) and human (b) tumor models treated at various sizes with a single dose of ZD6126 150 mg/kg. Survival assays were performed 24 hours after treatment. Data points represent individual animals, and results for each tumor model were fit by linear regression. Data from Siemann and Rojiani, 2005 (Citation12); with permission.
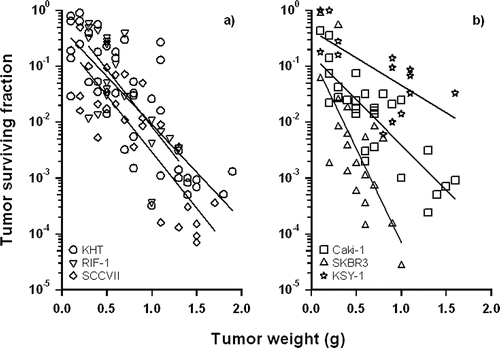
The nature of the mechanisms underlying the enhanced efficacy of VDAs against advanced disease is less clear. VDAs may be more effective in larger than smaller tumors due to the smaller masses having more significant portions supported by vessels located within the surrounding normal tissues. Since these normal vessels are unaffected by VDA treatment (Citation82, 85, 86, 142, 144), the portion of a tumor mass at risk of incurring ischemic damage from VDA treatment would increase along with its size. An additional reason that larger tumors become more sensitive to VDA treatment is because, at these sizes, the tumor vasculature might be particularly susceptible to occlusion due to elevated IFPs. Such pressures are known to be associated with advanced tumor masses (Citation145).
The observed tumor size-dependent VDA efficacy supports the rationale that combining conventional therapies with VDAs will lead to enhanced tumor responses based on the complementary nature of the antitumor action of these treatment modalities. The lack of VDA effect in the periphery is likely due to this region being primarily supplied by normal host vessels that are unresponsive to VDAs. The presence of normal vessels should not hinder drug delivery to the tumor rim and will make hypoxia less likely; thus, tumor cells in the periphery are more likely to be more sensitive to systemically administered agents and radiation (Citation71, 88). By combining VDAs with conventional therapies, it should be feasible to utilize the advantages of one to overcome the shortcomings of the other. Such a combination may be particularly effective when applied to advanced tumors. For example, in the study by Siemann and Rojiani (Citation12), which combined a VDA and radiation therapy, it was observed that the smallest tumors were most radiation-sensitive while the largest tumors were consistently most responsive to VDA treatment. However, when the VDA and radiation were combined, treatment efficacy was enhanced for all tumor sizes, but most significantly for larger tumors.
The preclinical findings outlined above highlight that the therapeutic benefits of VDAs in combination with other anticancer treatments may be more evident in the treatment of larger tumor masses. There is now emerging evidence that this may also occur in the clinic (Citation146–148). In a randomized Phase II study of plinabulin and docetaxel in patients with advanced NSCLC, although the primary objective of overall survival prolongation was not met, patients with large tumors were shown to have improved outcomes Citation127.
VDA treatment in combination with chemotherapy has been investigated previously, showing enhanced responses compared with VDA treatment alone (Citation9, 51, 149). The increased effect of VDAs when combined with chemotherapy is most likely due to the two drugs targeting different cell populations. This is possibly also the case when VDAs and radiation are combined, since preclinical studies generally report an additive effect of the two treatments (Citation9, 51, 150).
A Phase II/III trial (FACT; NCT00507429) showed positive results for ATC patients (N = 80) treated with CA4P plus the chemotherapy combination of carboplatin and paclitaxel, or carboplatin and paclitaxel alone. Although not statistically significant, the median overall survival of the patients receiving CA4P plus chemotherapy was 5.1 months versus 4.1 months for chemotherapy alone (hazard ratio [HR] 0.71 [95% CI 0.42–1.22]) (Citation151). A Phase II trial (CA4P-UKCTC-207) also showed positive results for platinum-resistant ovarian cancer patients who received CA4P plus carboplatin, and paclitaxel. The combination of CA4P and chemotherapy was generally well-tolerated and appears to produce a higher response rate in patients than if the chemotherapy is given alone (Citation152).
The tumor vascular effect of combining radiotherapy with CA4P was evaluated in patients with advanced NSCLC using volumetric dynamic contrast-enhanced computed tomography (Citation153). The study showed that after CA4P, the increase in permeability after radiotherapy and reduction in tumor blood volume were greater at the rim of the tumor and were sustained for up to 72 hours. These findings suggest that radiotherapy enhanced the tumor antivascular activity of CA4P and resulted in sustained tumor vascular shutdown – a result that could lead to greater than additive effects.
Rationale for combined VTT
As already outlined, AAs and VDAs induce vascular effects by different mechanisms, resulting in distinct antitumor effects; it is possible that these differences could be exploited by combining their potentially complementary mechanism to produce an effective antivascular therapeutic regimen. As both the initiation of new vessel formation and the integrity of the existing blood vessel network are critical to the growth and survival of a tumor, a two-pronged attack comprising a combination of anti-angiogenic and vascular disrupting strategies would seem logical. Such a combination therapy has the potential to have negative consequences for a tumor's blood vessel support network and, hence, its growth and survival.
There are additional reasons for considering a combination of these two classes of agents. Firstly, treatment duration: AA treatment is a long-term therapy, noting that on treatment cessation, the remaining dormant tumor cells have the capacity to reinitiate pro-angiogenic activities and progress (Citation154), while the effects of VDA exposure manifest themselves within hours after treatment and these agents can be given intermittently (Citation155). Secondly, the limitations of AAs (disabling a single angiogenic target via an AA will ultimately be insufficient to fully impair angiogenesis) and VDAs (residual, viable tumor cells surviving at the periphery) may be minimized or overcome by combining the two therapies. Given their disparate mechanisms of action, the combined application of AAs and VDAs (combined VTT) is likely to lead to complementary antitumor effects (). Finally, tumor stage and size: AA therapy is better suited for treating early-stage tumors of micrometastatic disease (Citation156–158), while the efficacy of VDAs in large tumor masses can be significant (Citation82, 140–142).
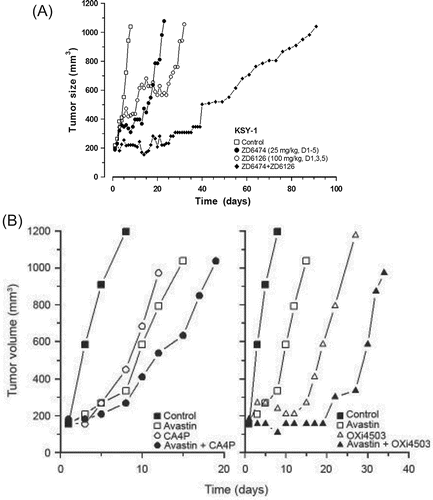
Preclinical evaluations of the combination of VDAs and AAs have demonstrated that the application of an AA in conjunction with one that disrupts established tumor blood vessels leads to complementary antitumor effects. This strategy was first examined in several human tumor xenograft models (Citation115, 159) in a setting which combined ZD6474, a selective inhibitor of VEGFR2-associated tyrosine kinase, with the microtubulin-disrupting agent ZD6126. In these studies, the AA was administered on a daily basis, whereas the VDA was given intermittently during the course of treatment. The results showed that, although each agent alone was capable of inducing a tumor response, when ZD6474 and ZD6126 were combined, the resultant tumor growth delay was markedly increased (). In a Kaposi's sarcoma model (KSY-1), this combination also significantly increased host survival and resulted in long-term tumor control (Citation115). Similar outcomes were noted when VDAs were combined with VEGF ligand inhibitors. For example, combining bevacizumab with the VDAs CA4P or OXi4503 (combretastatin A1-diphosphate or CA1P) resulted in significantly greater tumor responses in the human clear cell renal carcinoma model (Caki-1) than could be achieved with single-agent treatments () (Citation160). The tumor response and subsequent parallel regrowth of Caki-1 xenografts treated with an AA and VDA () likely reflects additive anti-tumor activity resulting from the combination of the two agents. However, the response of the KSY-1 xenografts to combined AA-VDA therapy suggests effects beyond an additive anti-tumor response based on the apparent increase in tumor doubling time following the combined treatment.
Figure 3. (A). The response of KSY-1 xenografts in mice treated with ZD6474 and ZD6126 either alone or in combination. ZD6474 was administered on a daily basis for 5 days starting when the tumors reached 200 mm3. ZD6126 was given on Days (D) 1, 3, and 5. In the combination group, on days when both agents were administered, ZD6126 preceded ZD6474 by 1 hour. The results show the growth of the median tumors of groups of 8 mice. In the anti-angiogenic agent plus vascular disrupting agent combination, 3 of 8 tumors were controlled. Data from Siemann and Shi, 2004 (Citation115); with permission. (B). Response of Caki-1 tumors to Avastin® (2 mg/kg, twice weekly for 2 weeks), CA4P, or Oxi4503 (100 mg/kg or 25 mg/kg, respectively, thrice weekly for 2 weeks), or the combination of anti-angiogenic agent and vascular disrupting agent. Data shown represent median tumor responses of groups of 8 to 10 mice. Data from Siemann and Shi, 2008 (Citation160); with permission.
The combined VTT may provide benefit not only in solid tumors but also in leukemia, in which blood vessels serve as a protective sanctuary for acute myeloid leukemia (AML). Recent studies have investigated this combined treatment approach in xenotransplant animal models of human AML chloromas and primary AML. The results showed that OXi4503 alone was capable of regressing AML by a multi-targeted mechanism and that the addition of bevacizumab mitigated the resultant reactive angiogenesis (Citation161).
A likely explanation for the effectiveness of combined VTT is that, while the VDA significantly reduces the tumor mass, the AA impairs the rapid tumor cell regrowth from the surviving viable rim by interfering with the re-establishment of the tumor vasculature. The latter is in keeping with reported increased vascularity and VEGF levels in the viable rim following VDA treatment, suggesting increased angiogenic activity in those parts of the tumor that survive VDA therapy (Citation70, 162). There is also evidence that the enhanced therapeutic effects observed when VDAs and AAs are combined may be due, in part, to the ability of AAs to suppress the recruitment of endothelial progenitor cells to tumor regions that survive VDA treatment (Citation163).
Clinical evaluation of the combined VTT has focused primarily on the use of VDAs in combination with bevacizumab. The combination of ombrabulin and bevacizumab has been clinically evaluated in a phase I trial (NCT01193595) and has been reported to be well tolerated with early evidence of clinical activity (Citation164). Clinical data from completed phase I and II trials and ongoing phase II/III trials has shown CA4P in combination with bevacizumab to be well tolerated with effective clinical activity.
A Phase I study (NCT00395434) investigated CA4P and bevacizumab combination therapy in patients with advanced solid malignancies. A total of 15 patients enrolled in this study, receiving 45 mg/m2 (3 patients), 54 mg/m2 (4 patients), or 63 mg/m2 (8 patients) of CA4P on Day 1, Day 8, and then every 14 days, with 10 mg/kg bevacizumab on Day 8 and at subsequent cycles 4 hours after CA4P. Results showed that this combination was well-tolerated with hypertension being the most common dose-limiting toxicity. Importantly, this study found that reductions in tumor perfusion (assessed using dynamic contrast-enhanced magnetic resonance imaging) after CA4P were more sustained when bevacizumab was used in combination (Citation107).
A Phase II study (GOG-0186I; NCT01305213) investigated CA4P and bevacizumab combination therapy in patients with recurrent ovarian carcinoma. A total of 107 patients, previously treated with three or fewer regimens, enrolled in the study. Patients were randomized to receive CA4P (60 mg/m2) in combination with bevacizumab (15 mg/kg) or bevacizumab alone once every 3 weeks, until progression or toxicity. The results from this study showed that median PFS was 7.3 months for CA4P with bevacizumab versus 4.8 months for bevacizumab alone (HR 0.69 [90% CI 0.47–1.00]; one-sided P = 0.049). The study met its primary outcome measure, showing that combined VTT (CA4P with bevacizumab) significantly improved PFS compared with bevacizumab alone in recurrent ovarian carcinoma (Citation165).
Triple combination therapy
A number of recent clinical trials have investigated potential triple therapy combining VDAs and AAs with chemotherapy. A randomized Phase II study (the FALCON trial; NCT00653939) evaluated the safety of CA4P when combined with carboplatin, paclitaxel, and bevacizumab in chemotherapy-naiïve subjects with advanced non-squamous NSCLC (Citation166). The overall incidence of treatment-emergent adverse events was similar between groups, with increased neutropenia, leukopenia, and hypertension in the CA4P group. Deaths, serious adverse events, and early discontinuations from treatment were comparable between the randomized treatment groups. The overall tumor response rate with CA4P was 50% versus 32% in controls. Overall and PFS rates were also comparable between the groups. This study demonstrated that treatment with CA4P plus carboplatin, paclitaxel, and bevacizumab appears to be a tolerable regimen with an acceptable toxicity profile. The FOCUS study (NCT02641639), an ongoing, randomized, double-blind Phase II/III study, is investigating physician's choice chemotherapy (PCC; paclitaxel or pegylated liposomal doxorubicin) plus bevacizumab and CA4P compared with PCC plus bevacizumab and placebo in platinum-resistant ovarian cancer (Citation167). Study enrollment began in June 2016; interim results will be available in 2017.
Conclusions
It is well-recognized that a functional network of blood vessels is essential for the progressive growth and dissemination of cancer. Therapeutic interventions targeting either the process of tumor-induced new vessel formation or the established tumor vasculature can provide significant antitumor efficacy. However, a combination of the two classes may be required to realize the full antitumor potential of VTT. Preclinical and initial clinical data support the therapeutic potential of combining VDAs with AAs. Combining such VTT with conventional anticancer therapies offers a promising future opportunity for cancer treatment, particularly for patients with more advanced disease.
Declaration of interest
We have read and understood Cancer Investigations policy on declaration of interests and declare the following potential conflict of interest: Chaplin D.J. is currently employed by Mateon Therapeutics. DWS and MRH report no declarations of interest.
Dr. Susan J. Cravero from Link Health Group, LLC, provided editorial support, funded by Mateon Therapeutics.
This article is not under consideration for publication elsewhere, and its publication is approved by all authors.
References
- Folkman J. Tumor angiogenesis: therapeutic implications. N Engl J Med 1971;285(21):1182–1186.
- Folkman J. Fundamental concepts of the angiogenic process. Curr Mol Med 2003;3(7):643–651.
- Horsman MR, Vaupel P. Pathophysiological basis for the formation of the tumor microenvironment. Front Oncol 2016;6:66.
- Vaupel P, Kallinowski F, Okunieff P. Blood flow, oxygen and nutrient supply, and metabolic microenvironment of human tumors: A review. Cancer Res 1989;49(23):6449–6465.
- McDonald DM, Choyke PL. Imaging of angiogenesis: From microscope to clinic. Nat Med 2003;9(6):713–725.
- Denekamp J. Review article: angiogenesis, neovascular proliferation and vascular pathophysiology as targets for cancer therapy. Br J Radiol 1993;66(783):181–196.
- Folkman J. Seminars in Medicine of the Beth Israel Hospital, Boston. Clinical applications of research on angiogenesis. N Engl J Med 1995;333(26):1757–1763.
- Siemann DW, Bibby MC, Dark GG, Dicker AP, Eskens FA, Horsman MR, et al. Differentiation and definition of vascular-targeted therapies. Clin Cancer Res 2005;11(2Pt 1):416–420.
- Siemann DW, Horsman MR. Vascular targeted therapies in oncology. Cell Tissue Res 2009;335(1):241–248.
- Siemann DW. Tumor vasculature: A target for anticancer therapies. Chichester (England): John Wiley & Sons, Ltd 2006;pp 1–8.
- Yadav L, Puri N, Rastogi V, Satpute P, Sharma V. Tumour angiogenesis and angiogenic inhibitors: A review. J Clin Diagn Res 2015;9(6):Xe01–xe05.
- Siemann DW, Rojiani AM. The vascular disrupting agent ZD6126 shows increased antitumor efficacy and enhanced radiation response in large, advanced tumors. Int J Radiat Oncol Biol Phys 2005;62(3):846–853.
- Ellis LM, Takahashi Y, Liu W, Shaheen RM. Vascular endothelial growth factor in human colon cancer: Biology and therapeutic implications. Oncologist 2000;5(Suppl 1):11–15.
- Ferrara N, Gerber HP, LeCouter J. The biology of VEGF and its receptors. Nat Med 20039(6):669–676.
- Ferrara N, Kerbel RS. Angiogenesis as a therapeutic target. Nature 2005;438(7070):967–974.
- Al-Husein B, Abdalla M, Trepte M, Deremer DL, Somanath PR. Antiangiogenic therapy for cancer: An update. Pharmacotherapy 2012;32(12), 1095–111.
- Pugh CW, Ratcliffe PJ. Regulation of angiogenesis by hypoxia: Role of the HIF system. Nat Med 2003;9(6):677–684.
- Willett CG, Boucher Y, di Tomaso E, Duda DG, Munn LL, Tong RT, et al.. Direct evidence that the VEGF-specific antibody bevacizumab has antivascular effects in human rectal cancer. Nat Med 200410(2):145147.
- Wedge SR, KendrewJ, Hennequin LF, Valentine PJ, Barry ST, Brave SR, et al. AZD2171: a highly potent, orally bioavailable, vascular endothelial growth factor receptor-2 tyrosine kinase inhibitor for the treatment of cancer. Cancer Res 2005;65(10):4389–400.
- Holash J, Davis S, Papadopoulos N, Croll SD, Ho L, Russell M, et al. VEGF-Trap: a VEGF blocker with potent antitumor effects. Proc Natl Acad Sci U S A 2002;99(17):11393–11398.
- Bouis D, Kusumanto Y, Meijer C, Mulder NH, Hospers GA. A review on pro- and anti-angiogenic factors as targets of clinical intervention. Pharmacol Res 2006;53(2):89–103.
- Raica M, Cimpean AM. Platelet-derived growth factor (PDGF)/PDGF receptors (PDGFR) axis as target for antitumor and antiangiogenic therapy. Pharmaceuticals (Basel) 20103(3):572–599.
- Korc M, Friesel RE. The role of fibroblast growth factors in tumor growth. Curr Cancer Drug Targets 2009;9(5):639–651.
- Holash J, Maisonpierre PC, Compton D, Boland P, Alexander CR, Zagzag D, et al. Vessel cooption, regression, and growth in tumors mediated by angiopoietins and VEGF. Science 1999;284(5422):1994–1998.
- Folkman J. Angiogenesis: An organizing principle for drug discovery? Nat Rev Drug Discov 2007;6(4):273–286.
- Kerbel R, Folkman J. Clinical translation of angiogenesis inhibitors. Nat Rev Cancer 2002;2(10):727–739.
- Cohen MH, Gootenberg J, Keegan P, Pazdur R. FDA drug approval summary: bevacizumab plus FOLFOX4 as second-line treatment of colorectal cancer. Oncologist 2007;12(3):356–361.
- Friedman HS, Prados MD, Wen PY, Mikkelsen T, Schiff D, Abrey LE, et al. Bevacizumab alone and in combination with irinotecan in recurrent glioblastoma. J Clin Oncol 2009;27(28):4733–4740.
- Kreisl TN, Kim L, Moore K, Duic P, Royce C, Stroud I, et al. Phase II trial of single-agent bevacizumab followed by bevacizumab plus irinotecan at tumor progression in recurrent glioblastoma. J Clin Oncol 2009;27(5):740–745.
- NIH. Website: Bevacizumab: second-line treatment of glioblastoma. Available at https://www.cancer.gov/about-cancer/treatment/drugs/fda-bevacizumab–Anchor-Glioblastoma (accessed July 2017).
- Fuchs CS, Tomasek J, Yong CJ, Dumitru F, Passalacqua R, Goswami C, et al. Ramucirumab monotherapy for previously treated advanced gastric or gastro-oesophageal junction adenocarcinoma (REGARD): An international, randomised, multicentre, placebo-controlled, phase 3 trial. Lancet 2014;383(9911):31–39.
- NIH. Website: Ramucirumab: Approved for gastric or gastroesophageal junction adenocarcinoma. Available at https://www.cancer.gov/about-cancer/treatment/drugs/fda-ramucirumab–anchor-single (accessed July 2017).
- Fabi A, Russillo M, Ferretti G, Metro G, Nistico C, Papaldo P, et al. Maintenance bevacizumab beyond first-line paclitaxel plus bevacizumab in patients with Her2-negative hormone receptor-positive metastatic breast cancer: Efficacy in combination with hormonal therapy. BMC Cancer 2012;12:482.
- Ledermann JA, Hackshaw A, Kaye S, Jayson G, Gabra H, McNeish I, et al. Randomized phase II placebo-controlled trial of maintenance therapy using the oral triple angiokinase inhibitor BIBF 1120 after chemotherapy for relapsed ovarian cancer. J Clin Oncol 2011;29(28):3798–804.
- Aravantinos G, Pectasides D. Bevacizumab in combination with chemotherapy for the treatment of advanced ovarian cancer: A systematic review. J Ovarian Res 2014;7:57.
- Horn L, Sandler A. Chemotherapy and antiangiogenic agents in non-small-cell lung cancer. Clin Lung Cancer 2007;8(Suppl 2);S68–73.
- Konda B, Shum H, Rajdev L. Anti-angiogenic agents in metastatic colorectal cancer. World J Gastrointest Oncol 2015;7(7):71–86.
- Teng LS, Jin KT, He KF, Wang HH, Cao J, Yu DC. Advances in combination of antiangiogenic agents targeting VEGF-binding and conventional chemotherapy and radiation for cancer treatment. J Chin Med Assoc 2010;73(6):281–288.
- Hurwitz H, Fehrenbacher L, Novotny W, Cartwright T, Hainsworth J, Heim W, et al. Bevacizumab plus irinotecan, fluorouracil, and leucovorin for metastatic colorectal cancer. N Engl J Med 2004;350(23):2335–2342.
- Van Cutsem E, Tabernero J, Lakomy R, Prenen H, Prausova J, Macarulla T, et al. Addition of aflibercept to fluorouracil, leucovorin, and irinotecan improves survival in a phase III randomized trial in patients with metastatic colorectal cancer previously treated with an oxaliplatin-based regimen. J Clin Oncol 2012;30(28):3499–3506.
- Miller K, Wang M, Gralow J, Dickler M, Cobleigh M, Perez EA, et al. Paclitaxel plus bevacizumab versus paclitaxel alone for metastatic breast cancer. N Engl J Med 2007;357(26):2666–2676.
- Miller KD, Chap LI, Holmes FA, Cobleigh MA, Marcom PK, Fehrenbacher L, et al. Randomized phase III trial of capecitabine compared with bevacizumab plus capecitabine in patients with previously treated metastatic breast cancer. J Clin Oncol 2005;23(4):792–799.
- Ohtsu A, Shah MA, Van Cutsem E, Rha SY, Sawaki A, Park SR, et al. Bevacizumab in combination with chemotherapy as first-line therapy in advanced gastric cancer: A randomized, double-blind, placebo-controlled phase III study. J Clin Oncol 2011;29(30):3968–3976.
- Hecht JR, Trarbach T, Hainsworth JD, Major P, Jager E, Wolff RA, et al. Randomized, placebo-controlled, phase III study of first-line oxaliplatin-based chemotherapy plus PTK787/ZK 222584, an oral vascular endothelial growth factor receptor inhibitor, in patients with metastatic colorectal adenocarcinoma. J Clin Oncol 2011;29(15):1997–2003.
- Van Cutsem E, Bajetta E, ValleJ, Kohne CH, Hecht JR, Moore M, et al. Randomized, placebo-controlled, phase III study of oxaliplatin, fluorouracil, and leucovorin with or without PTK787/ZK 222584 in patients with previously treated metastatic colorectal adenocarcinoma. J Clin Oncol 2011;29(15):2004–2010.
- Hodi FS, LawrenceD, Lezcano C, Wu X, Zhou J, Sasada T, Zeng W, et al. Bevacizumab plus ipilimumab in patients with metastatic melanoma. Cancer Immunol Res 2014;2(7):632–642.
- Sznol MM, DF, Jones SF, Mier JW, Waterkamp D, Rossi C, Wallin J, et al. Phase Ib evaluation of MPDL3280A (anti-PDL1) in combination with bevacizumab (bev) in patients (pts) with metastatic renal cell carcinoma (mRCC). J Clin Oncol 2015;33:(suppl 7):410–410.
- Li D, Williams JI, Pietras RJ. Squalamine and cisplatin block angiogenesis and growth of human ovarian cancer cells with or without HER-2 gene overexpression. Oncogene 2002;21;(18):28052814.
- Ma J, Waxman DJ. Dominant effect of antiangiogenesis in combination therapy involving cyclophosphamide and axitinib. Clin Cancer Res 2009;15(2):578–588.
- DeVita VT, Hellman S, Rosenberg SA. Cancer: Principles and practice of oncology. Lippincott-Raven: Philadelphia 1997, (5th Edition).
- Horsman MR, Siemann DW. Pathophysiologic effects of vascular-targeting agents and the implications for combination with conventional therapies. Cancer Res 2006;66(24):11520–11539.
- Mazeron R, Anderson B, Supiot S, Paris F, Deutsch E. Current state of knowledge regarding the use of antiangiogenic agents with radiation therapy. Cancer Treat Rev 2011;37(6):476–486.
- Kleibeuker EA, Griffioen AW, Verheul HM, Slotman BJ, Thijssen VL. Combining angiogenesis inhibition and radiotherapy: A double-edged sword. Drug Resist Updat 2012;15(3):173–182.
- Chinot OL, Wick W, Mason W, Henriksson R, Saran F, Nishikawa R, et al. Bevacizumab plus radiotherapy-temozolomide for newly diagnosed glioblastoma. N Engl J Med 2014;370(8):709–722.
- Cubillo A, Hernando-Requejo O, Garcia-Garcia E, Rodriguez-Pascual J, De Vicente E, Morelli, P, et al.. A prospective pilot study of target-guided personalized chemotherapy with intensity-modulated radiotherapy in patients with early rectal cancer. Am J Clin Oncol 2014;37(2);117–121.
- Czito BG, Bendell JC, Willett CG, Morse MA, Blobe GC, Tyler DS, et al. Bevacizumab, oxaliplatin, and capecitabine with radiation therapy in rectal cancer: Phase I trial results. Int J Radiat Oncol Biol Phys 200768(2):472–428.
- Gasparini G, Torino F, Ueno T, Cascinu S, Troiani T, Ballestrero A, et al. A phase II study of neoadjuvant bevacizumab plus capecitabine and concomitant radiotherapy in patients with locally advanced rectal cancer. Angiogenesis 2012;15(1):141–150.
- Gilbert MR, Dignam JJ, Armstrong TS, Wefel JS, Blumenthal DT, Vogelbaum MA, et al. A randomized trial of bevacizumab for newly diagnosed glioblastoma. N Engl J Med 2014370(8)699–708.
- Morganti AG, Mignogna S, Caravatta L, Deodato F, Macchia G, Plantamura NM, et al. FOLFIRI-bevacizumab and concurrent low-dose radiotherapy in metastatic colorectal cancer: preliminary results of a phase I-II study. J Chemother 2014;26(6):353–358.
- Yao M, Galanopoulos N, Lavertu P, Fu P, Gibson M, Argiris A, et al. Phase II study of bevacizumab in combination with docetaxel and radiation in locally advanced squamous cell carcinoma of the head and neck. Head Neck 2015;37(11):1665–1671.
- Blaszkowsky LS, Ryan DP, Szymonifka J, Borger DR, Zhu AX, Clark JW, et al. Phase I/II study of neoadjuvant bevacizumab, erlotinib and 5-fluorouracil with concurrent external beam radiation therapy in locally advanced rectal cancer. Ann Oncol 2014;25(1):121–126.
- Arnold AM, Seymour L, Smylie M, Ding K, Ung Y, Findlay B, et al. Phase II study of vandetanib or placebo in small-cell lung cancer patients after complete or partial response to induction chemotherapy with or without radiation therapy: National cancer institute of Canada clinical trials group study BR.20. J Clin Oncol 2007;25(27):4278–4284.
- Huber PE, Bischof M, Jenne J, Heiland S, Peschke P, Saffrich R, et al. Trimodal cancer treatment: beneficial effects of combined antiangiogenesis, radiation, and chemotherapy. Cancer Res 2005;65(9):3643–3655.
- Williams KJ, Telfer BA, Brave S, Kendrew J, Whittaker L, Stratford IJ, et al. ZD6474, a potent inhibitor of vascular endothelial growth factor signaling, combined with radiotherapy: Schedule-dependent enhancement of antitumor activity. Clin Cancer Res 2004;10(24):8587–8593.
- Zips D, Krause M, Hessel F, Westphal J, Bruchner K, Eicheler W, et al. Experimental study on different combination schedules of VEGF-receptor inhibitor PTK787/ZK222584 and fractionated irradiation. Anticancer Res 2003;23(5a):3869–3876.
- Woglom WH. A critique of tumour resistance. J Cancer Res 1923;7:283–311.
- Boyland E, Boyland ME. Studies in tissue metabolism: The action of colchicine and B. typhosus extract. Biochem J 1937;31(3):454–460.
- Siemann DW. Therapeutic strategies that selectively target and disrupt established tumor vasculature. Hematol Oncol Clin North Am 2004;18(5):1023–1037, viii.
- Tozer GM, Kanthou C, Baguley BC. Disrupting tumour blood vessels. Nat Rev Cancer 2005;5(6):423–435.
- Salmon BA, Siemann DW. Characterizing the tumor response to treatment with combretastatin A4 phosphate. Int J Radiat Oncol Biol Phys 2007;68:(1):211–217.
- Siemann DW. The unique characteristics of tumor vasculature and preclinical evidence for its selective disruption by Tumor-Vascular Disrupting Agents. Cancer Treat Rev 2011;37(1):63–74.
- Siemann DW, Horsman MR. Modulation of the tumor vasculature and oxygenation to improve therapy. Pharmacol Ther 2015;153:107–124.
- Chaplin DJ, Pettit GR, Parkins CS, Hill SA. Antivascular approaches to solid tumour therapy: Evaluation of tubulin binding agents. Br J Cancer Suppl 1996;27:S86–88.
- Dark GG, Hill SA, Prise VE, Tozer GM, Pettit GR, Chaplin DJ. Combretastatin A-4, an agent that displays potent and selective toxicity toward tumor vasculature. Cancer Res 1997;57(10):1829–1834.
- Clémenson C, Chargari C, Deutsch E. Combination of vascular disrupting agents and ionizing radiation. Crit Rev Oncol / Hematol 2013;86(2):143–160.
- Nepali K, Ojha R, Lee HY, Liou JP. Early investigational tubulin inhibitors as novel cancer therapeutics. Expert Opin Investigational Drugs 2016;25(8):917–936.
- Wu XY, Ma W, Gurung K, Guo CH. Mechanisms of tumor resistance to small-molecule vascular disrupting agents: Treatment and rationale of combination therapy. J Formosan Med Assoc 2013;112(3):115–124.
- Chaplin DJ, Dougherty GJ. Tumour vasculature as a target for cancer therapy. Br J Cancer 1999;80(Suppl 1):57–64.
- Chaplin DJ, Horsman MR, Siemann DW. Current development status of small-molecule vascular disrupting agents. Curr Opin Investig Drugs 2006;7(6):522–528.
- Siemann DW, Horsman MR. Small molecule vascular distrupting agents in cancer therapy. Ellis, L. M., Teicher, G. Eds. Towaha: Humana 2008;pp 297–310.
- Thorpe PE. Vascular targeting agents as cancer therapeutics. Clin Cancer Res 2004;10(2):415–427.
- Siemann DW, Rojiani AM. Antitumor efficacy of conventional anticancer drugs is enhanced by the vascular targeting agent ZD6126. Int J Radiat Oncol Biol Phys 2002;54(5):1512–1517.
- Siemann DW. Vascular targeting agents. Horizons in Cancer Therapeutics: From Bench to Bedside 2002;3(2):4–15.
- Blakey DC, Ashton SE, Westwood FR, Walker M, Ryan AJ. ZD6126: A novel small molecule vascular targeting agent. Int J Radiat Oncol Biol Phys 2002;54(5):1497–1502.
- Davis PD, Dougherty GJ, Blakey DC, Galbraith SM, Tozer GM, Holder AL, et al. ZD6126: a novel vascular-targeting agent that causes selective destruction of tumor vasculature. Cancer Res 2002;62(24):7247–7253.
- Goto H, Yano S, Zhang H, Matsumori Y, Ogawa H, Blakey DC, et al. Activity of a new vascular targeting agent, ZD6126, in pulmonary metastases by human lung adenocarcinoma in nude mice. Cancer Res 2002;62(13):3711–3715.
- Grosios K, Holwell SE, McGown AT, Pettit GR, Bibby MC. In vivo and in vitro evaluation of combretastatin A-4 and its sodium phosphate prodrug. Br J Cancer 1999;81(8):1318–1327.
- Siemann DW, Chaplin DJ, Horsman MR. Vascular-targeting therapies for treatment of malignant disease. Cancer 2004;100(12):2491–2499.
- Strecker TE, Odutola SO, Lopez R, Cooper MS, Tidmore JK, Charlton-Sevcik AK, et al. The vascular disrupting activity of OXi8006 in endothelial cells and its phosphate prodrug OXi8007 in breast tumor xenografts. Cancer Lett 2015;369(1):229–241.
- Yao N, Gao M, Ren K, Jiang X, Li Y, Jiang C, et al. PD806: A novel oral vascular disrupting agent shows antitumor and antivascular effects in vitro and in vivo. Anticancer Drugs 2015;26(2):148–159.
- Galbraith SM, Chaplin DJ, Lee F, Stratford MR, Locke RJ, Vojnovic B, et al. Effects of combretastatin A4 phosphate on endothelial cell morphology in vitro and relationship to tumour vascular targeting activity in vivo. Anticancer Res 2001;21(1a):93–102.
- Hori K, Saito S. Microvascular mechanisms by which the combretastatin A-4 derivative AC7700 (AVE8062) induces tumour blood flow stasis. Br J Cancer 2003;89(7):1334–1344.
- Griggs J, Metcalfe JC, Hesketh R. Targeting tumour vasculature: The development of combretastatin A4. Lancet Oncol 2001;2(2):82–87.
- Chase DM, Chaplin DJ, Monk BJ. The development and use of vascular targeted therapy in ovarian cancer. Gynecol Oncol 2017;145(2):393–406.
- Beauregard DA, Thelwall PE, Chaplin DJ, Hill SA, Adams GE, Brindle KM. Magnetic resonance imaging and spectroscopy of combretastatin A4 prodrug-induced disruption of tumour perfusion and energetic status. Br J Cancer 1998;77(11):1761–1767.
- Horsman MR, Ehrnrooth E, Ladekarl M, Overgaard J. The effect of combretastatin A-4 disodium phosphate in a C3H mouse mammary carcinoma and a variety of murine spontaneous tumors. Int J Radiat Oncol Biol Phys 1998;42(4);895–898.
- Anderson HL, Yap JT, Miller MP, Robbins A, Jones T, Price PM. Assessment of pharmacodynamic vascular response in a phase I trial of combretastatin A4 phosphate. J Clin Oncol 2003;21(15):2823–2830.
- Tozer GM, Prise VE, Wilson J, Locke RJ, Vojnovic B, Stratford MR, et al. Combretastatin A-4 phosphate as a tumor vascular-targeting agent: Early effects in tumors and normal tissues. Cancer Res 1999;59(7):1626–1634.
- Baguley BC, Siemann DW. Temporal aspects of the action of ASA404 (vadimezan; DMXAA). Expert Opin Investig Drugs 2010;19(11):1413–1425.
- Kim S, Peshkin L, Mitchison TJ. Vascular disrupting agent drug classes differ in effects on the cytoskeleton. PLoS One 2012;7(7):e40177.
- Siemann DW, Chaplin DJ, Walicke PA. A review and update of the current status of the vasculature-disabling agent combretastatin-A4 phosphate (CA4P). Expert Opin Investig Drugs 2009;18(2):189–197.
- Dowlati A, Robertson K, Cooney M, Petros WP, Stratford M, Jesberger J, et al. A phase I pharmacokinetic and translational study of the novel vascular targeting agent combretastatin a-4 phosphate on a single-dose intravenous schedule in patients with advanced cancer. Cancer Res 2002;62(12):3408–3416.
- Stevenson JP, Rosen M, Sun W, Gallagher M, Haller DG, Vaughn D, et al. Phase I trial of the antivascular agent combretastatin A4 phosphate on a 5-day schedule to patients with cancer: Magnetic resonance imaging evidence for altered tumor blood flow. J Clin Oncol 2003;21(23):4428–4438.
- Rustin GJ, Galbraith SM, Anderson H, Stratford M, Folkes LK, et al. Phase I clinical trial of weekly combretastatin A4 phosphate: Clinical and pharmacokinetic results. J Clin Oncol 2003;21(15):2815–2822.
- Akerly W, Schabel M, Morrell G. A randomized phase 2 trial of combretatstatin A4 phosphate (CA4P) in combination with paclitaxel and carboplatin to evaluate safety and efficacy in subjects with advanced imageable malignancies. Am Soc Clin Oncol Annu Meeting 2007;25:14060–14060.
- Galbraith SM, Maxwell RJ, Lodge MA, Tozer GM, Wilson J, Taylor NJ, et al. Combretastatin A4 phosphate has tumor antivascular activity in rat and man as demonstrated by dynamic magnetic resonance imaging. J Clin Oncol 2003;21(15):2831–2842.
- Nathan P, Zweifel M, Padhani AR, Koh DM, Ng M, Collins DJ, et al. Phase I trial of combretastatin A4 phosphate (CA4P) in combination with bevacizumab in patients with advanced cancer. Clin Cancer Res 2012;18(12):3428–3439.
- Baguley BC. Antivascular therapy of cancer: DMXAA. Lancet Oncol 2003;4(3):141–148.
- Lara PN Jr, Douillard JY, Nakagawa K, von PawelJ, McKeage MJ, et al. Randomized phase III placebo-controlled trial of carboplatin and paclitaxel with or without the vascular disrupting agent vadimezan (ASA404) in advanced non-small-cell lung cancer. J Clin Oncol 2011;29(22):2965–2971.
- Ching LM, Cao Z, Kieda C, Zwain S, Jameson MB, Baguley BC. Induction of endothelial cell apoptosis by the antivascular agent 5,6-Dimethylxanthenone-4-acetic acid. Br J Cancer 2002;86(12):1937–1942.
- Ching LM, Zwain S, Baguley BC. Relationship between tumour endothelial cell apoptosis and tumour blood flow shutdown following treatment with the antivascular agent DMXAA in mice. Br J Cancer 2004;90(4):906–910.
- Zhao L, Ching LM, Kestell P, Kelland LR, Baguley BC. Mechanisms of tumor vascular shutdown induced by 5,6-dimethylxanthenone-4-acetic acid (DMXAA): Increased tumor vascular permeability. Int J Cancer 2005;116(2):322–326.
- Sandler HM, Curran WJ Jr, Turrisi AT. 3rd The influence of tumor size and pre-treatment staging on outcome following radiation therapy alone for stage I non-small cell lung cancer. Int J Radiat Oncol Biol Phys 1990;19(1);9–13.
- Brown SL, Kolozsvary A, Kim JH. Vascular targeting therapies for treatment of malignant disease. Cancer 2005;104(1):216–217; author reply 217.
- Siemann DW, Shi W. Efficacy of combined antiangiogenic and vascular disrupting agents in treatment of solid tumors. Int J Radiat Oncol Biol Phys 2004;60(4):1233–1240.
- Huang X, Molema G, King S, Watkins L, Edgington TS, Thorpe PE. Tumor infarction in mice by antibody-directed targeting of tissue factor to tumor vasculature. Science 1997;275(5299):547–550.
- Horsman MR. Tissue physiology and the response to heat. Int J Hyperthermia 2006;22(3):197–203.
- Denekamp J, Hill S. Angiogenic attack as a therapeutic strategy for cancer. Radiother Oncol 1991;20(Suppl 1):103–112.
- Wang J, Zhao Z, Shen S, Zhang C, Guo S, Lu Y, et al. Selective depletion of tumor neovasculature by microbubble destruction with appropriate ultrasound pressure. Int J Cancer 2015;137(10):2478–2491.
- Tempany CM, McDannold NJ, Hynynen K, Jolesz FA. Focused ultrasound surgery in oncology: Overview and principles. Radiology 2011;259(1):39–56.
- Berbeco RI, Detappe A, Tsiamas P, Parsons D, Yewondwossen M, Robar J. Low Z target switching to increase tumor endothelial cell dose enhancement during gold nanoparticle-aided radiation therapy. Med Phys 2016;43(1):436–442.
- Mason RP, Antich PP, Babcock EE, Constantinescu A, Peschke P, Hahn EW. Non-invasive determination of tumor oxygen tension and local variation with growth. Int J Radiat Oncol Biol Phys 1994;29(1):95–103.
- Zhao D, Chang CH, Kim JG, Liu H, Mason RP. In vivo near-infrared spectroscopy and MRI monitoring of tumor response to Combretastatin A-4-phosphate correlated with therapeutic outcome. Int J Radiat Oncol Biol Phys 2011;80(2):574––581.
- Flynn BL, Gill GS, Grobelny DW, Chaplin JH, Paul D, Leske AF, et al. Discovery of 7-hydroxy-6-methoxy-2-methyl-3-(3,4,5-trimethoxybenzoyl)benzo[b]furan (BNC105), a tubulin polymerization inhibitor with potent antiproliferative and tumor vascular disrupting properties. J Med Chem 2011;54(17):6014–6027.
- Nowak AK, Brown C, Millward MJ, Creaney J, Byrne MJ, Hughes B, et al. A phase II clinical trial of the vascular disrupting agent BNC105P as second line chemotherapy for advanced Malignant Pleural Mesothelioma. Lung Cancer 2013;81(3):422–427.
- Nervi C, Arcangeli G, Badaracco G, Cortese M, Morelli M, Starace G. The relevance of tumor size and cell kinetics as predictors of radiation response in head and neck cancer. A randomized study on the effect of intraarterial chemotherapy followed by radiotherapy. Cancer 1978;41(3):900–906.
- Lee CC, Ho HC, Su YC, Lee MS, Hsiao SH, Hwang JH, et al. Bidimensional measurement of nasopharyngeal carcinoma: A simple method to predict outcomes. Clin Otolaryngol 2009;34(1):26–33.
- Gerber DE, Dahlberg SE, Sandler AB, Ahn DH, Schiller JH, et al. Baseline tumour measurements predict survival in advanced non-small cell lung cancer. Br J Cancer 2013;109(6):14761481.
- Li C, Oh SJ, Kim S, Hyung WJ, Yan M, Zhu ZG, et al. Risk factors of survival and surgical treatment for advanced gastric cancer with large tumor size. J Gastrointest Surg 2009;13(5):881–885.
- Baldwin NJ, Ng TC. Oxygenation and metabolic status of KHT tumors as measured simultaneously by 19F magnetic resonance imaging and 31P magnetic resonance spectroscopy. Magn Reson Imaging 1996;14(5):541–551.
- Bentzen L, Keiding S, Horsman MR, Gronroos T, Hansen SB, OvergaardJ. Assessment of hypoxia in experimental mice tumours by [18F]fluoromisonidazole PET and pO2 electrode measurements. Influence of tumour volume and carbogen breathing. Acta Oncol 2002;41(3);304–312.
- Hill RP. An appraisal of in vivo assays of excised tumours. Br J Cancer Suppl 1980;4:230–239.
- Kallman RF, DeNardo GL, Stasch MJ. Blood flow in irradiated mouse sarcoma as determined by the clearance of xenon-133. Cancer Res 1972;32(3):483–490.
- Pallavicini MG, Lalande ME, Miller RG, Hill RP. Cell cycle distribution of chronically hypoxic cells and determination of the clonogenic potential of cells accumulated in G2 + M phases after irradiation of a solid tumor in vivo. Cancer Res 1979;39(6Pt 1):1891–1897.
- Siemann DW, Johansen IM, Horsman MR. Radiobiological hypoxia in the KHT sarcoma: Predictions using the Eppendorf histograph. Int J Radiat Oncol Biol Phys 199840(5):1171–1176.
- Brizel DM, Dodge RK, Clough RW, Dewhirst MW. Oxygenation of head and neck cancer: Changes during radiotherapy and impact on treatment outcome. Radiother Oncol 1999;53(2):113–117.
- Hockel M, Knoop C, Schlenger K, Vorndran B, Baussmann E, Mitze M, et al. Intratumoral pO2 predicts survival in advanced cancer of the uterine cervix. Radiother Oncol 1993;26(1):45–50.
- Nordsmark M, Overgaard M, Overgaard J. Pretreatment oxygenation predicts radiation response in advanced squamous cell carcinoma of the head and neck. Radiother Oncol 1996;41(1):31–39.
- Durand RE. Keynote address: The influence of microenvironmental factors on the activity of radiation and drugs. Int J Radiat Oncol Biol Phys 1991;20(2):253–258.
- Landuyt W, Ahmed B, Nuyts S, Theys J, Op de Beeck M, Rijnders A, et al. In vivo antitumor effect of vascular targeting combined with either ionizing radiation or anti-angiogenesis treatment. Int J Radiat Oncol Biol Phys 2001;49(2);443–450.
- Nielsen T, Murata R, Maxwell RJ, Stodkilde-Jorgensen H, Ostergaard L, Ley CD, et al. Non-invasive imaging of combretastatin activity in two tumor models: Association with invasive estimates. Acta Oncol 2010;49(7):906–913.
- Siemann DW, Rojiani AM. Enhancement of radiation therapy by the novel vascular targeting agent ZD6126. Int J Radiat Oncol Biol Phys 2002;53(1):164–171.
- Landuyt W, Verdoes O, Darius DO, Drijkoningen M, Nuyts S, Theys J, et al. Vascular targeting of solid tumours: A major ‘inverse’ volume-response relationship following combretastatin A-4 phosphate treatment of rat rhabdomyosarcomas. Eur J Cancer 2000;36(14):1833–1843.
- Blakey DC, Westwood FR, Walker M, Hughes GD, Davis PD, Ashton SE, et al. Antitumor activity of the novel vascular targeting agent ZD6126 in a panel of tumor models. Clin Cancer Res 2002;8(6):1974–1983.
- Jain RK. Transport of molecules in the tumor interstitium: A review. Cancer Res 1987;47(12):3039–3051.
- Heist R, Aren OS, Mita AC, Polikoff J, Bazhenova L, Lloyd GK, et al. Randomized phase 2 trial of plinabulin (NPI-2358) plus docetaxel in patients with advanced non-small cell lung cancer (NSCLC). ASCO Annu Meeting 2014;32(5s), (J Clin Oncol suppl; abstr 8054).
- Tewari KS, Abrouk NED, Coleman RL, Aghajanian C, Couchenour R, Nelson J, et al. Improved progression-free survival among women with measurable recurrent ovarian carcinoma treated with CA4P plus bevacizumab: A post-hoc analysis of GOG-0186I. IGCS Lisbon, Portugal, 2016; Lisbon, Portugal, 2016.
- Bazhenova L. Randomized Phase 2 study of plinabulin and docetaxel in patients with advanced non-small cell lung cancer – mechanism-based efficacy analyses. 16th World Conference on Lung Cancer, Denver, Colorado, USA, 2015; Denver, Colorado, USA, 2015; (abstr. P3.01-057).
- Jorgensen TJ, Tian H, Joseph IB, Menon K, Frost D. Chemosensitization and radiosensitization of human lung and colon cancers by antimitotic agent, ABT-751, in athymic murine xenograft models of subcutaneous tumor growth. Cancer Chemother Pharmacol 2007;59(6):725–732.
- Siemann DW, Warrington KH, Horsman MR. Targeting tumor blood vessels: An adjuvant strategy for radiation therapy. Radiother Oncol 2000;57(1):5–12.
- Sosa JA, Elisei R, Jarzab B, Balkissoon J, Lu SP, Bal C, et al. Randomized safety and efficacy study of fosbretabulin with paclitaxel/carboplatin against anaplastic thyroid carcinoma. Thyroid 2014;24(2):232–240.
- Zweifel M, Jayson GC, Reed NS, Osborne R, Hassan B, Ledermann J, et al. Phase II trial of combretastatin A4 phosphate, carboplatin, and paclitaxel in patients with platinum-resistant ovarian cancer. Ann Oncol 201122(9):2036–2041.
- Ng QS, Mandeville H, Goh V, Alonzi R, Milner J, Carnell D, et al. Phase Ib trial of radiotherapy in combination with combretastatin-A4-phosphate in patients with non-small-cell lung cancer, prostate adenocarcinoma, and squamous cell carcinoma of the head and neck. Ann Oncol 2012;23(1):231–227.
- Hensel JA, Flaig TW, Theodorescu D. Clinical opportunities and challenges in targeting tumour dormancy. Nat Rev Clin Oncol 2013;10(1):41–51.
- Salmon HW, Siemann DW. Effect of the second-generation vascular disrupting agent OXi4503 on tumor vascularity. Clin Cancer Res 2006;12(13):4090–4094.
- LiaoF, Li Y, O'Connor W, Zanetta L, Bassi R, Santiago A, et al. Monoclonal antibody to vascular endothelial-cadherin is a potent inhibitor of angiogenesis, tumor growth, and metastasis. Cancer Res 2000;60(24):6805–6810.
- O'Reilly MS, Holmgren L, Shing Y, Chen C, Rosenthal RA, Moses M, et al. Angiostatin: a novel angiogenesis inhibitor that mediates the suppression of metastases by a Lewis lung carcinoma. Cell 1994;79(2):315–328.
- Yoon SS, Eto H, Lin CM, Nakamura H, Pawlik TM, Song SU, et al. Mouse endostatin inhibits the formation of lung and liver metastases. Cancer Res 1999;59(24):6251–6256.
- Wedge SRK, J, Ogilvie DJ, Hennequin LF, Brave AJ, Ryan AJ, Ashton SE, et al. Combination of the VEGF receptor tyrosine kinase inhibitor ZD6474 and vascular-targeting agent ZD6126 produces an enhanced anti-tumor response. Proc Am Assoc Cancer Res 2002(43):1081.
- Siemann DW, Shi W. Dual targeting of tumor vasculature: combining Avastin and vascular disrupting agents (CA4P or OXi4503). Anticancer Res 2008;28(4b):20272031.
- Madlambayan GJ, Meacham AM, Hosaka K, Mir S, Jorgensen M, Scott EW, et al. Leukemia regression by vascular disruption and antiangiogenic therapy. Blood 2010;116(9):1539–1547.
- Sheng Y, Hua J, Pinney KG, Garner CM, Kane RR, Prezioso JA, et al. Combretastatin family member OXI4503 induces tumor vascular collapse through the induction of endothelial apoptosis. Int J Cancer 2004;111(4):604–610.
- Shaked Y, Ciarrocchi A, Franco M, Lee CR, Man S, Cheung AM, et al. Therapy-induced acute recruitment of circulating endothelial progenitor cells to tumors. Science 2006;313(5794):1785–1787.
- Del Conte G, Bahleda R, Moeno V, Damian S, Perotti A, Lassau N, et al. A phase I study of ombrabulin (O) combined with bevacizumab (B) in patients with advanced solid tumors (NCT01193595). American Society of Clinical Oncology Annual Meeting, Chicago, IL, 2012; Chicago, IL 2012; Abstract 3080.
- Monk BJ, Sill MW, Walker JL, Darus CJ, Sutton G, Tewari KS, et al. Randomized Phase II evaluation of bevacizumab versus bevacizumab plus fosbretabulin in recurrent ovarian, tubal, or peritoneal carcinoma: An NRG Oncology/Gynecologic oncology group study. J Clin Oncol 2016;34(19):2279–2286.
- Garon EB, Neidhart JD, Gabrail NY, de Oliveira M, Balkissoon J, Kabbinavar F. A randomized Phase II trial of the tumor vascular disrupting agent ca4P (fosbretabulin tromethamine) with carboplatin, paclitaxel, and bevacizumab in advanced nonsquamous non-small-cell lung cancer. Onco Targets Therapy 2016;(9):1–9.
- Monk BJ, Herzog T, Alvarez R, Chan J, Chase D, Couchenour R, et al. Focus study: Physician's choice chemotherapy (PCC) plus bevacizumab and CA4P versus PCC plus bevacizumab and placebo in platinum-resistant ovarian cancer. ICGS Journal. Int J Gynecol Cancer. 2016;26:878–878.