
Abstract
Novel therapeutics are needed for patients with relapsed or refractory diffuse large B-cell lymphoma (R/R DLBCL). Everolimus is an mTOR pathway inhibitor with synergistic anti-tumor activity when combined with histone deacetylase inhibitors, such as panobinostat, in preclinical lymphoma models. In this Phase II study, we evaluated overall response rate to single and combination everolimus and panobinostat in R/R DLBCL. Fifteen patients were enrolled to single-agent and 18 to combination. One patient responded to everolimus, while none responded to panobinostat. Though 25% of patients responded to combination therapy, responses were not durable with significant toxicity. We demonstrated minimal single-agent activity and prohibitive toxicity with combination therapy.
Introduction
Diffuse large B-cell lymphomas (DLBCL) constitute approximately 1/3 of adult non-Hodgkin lymphomas (NHL) (Citation1). Despite recent therapeutic advances in the rituximab era, nearly half of patients responding to initial chemoimmunotherapy will relapse (Citation2). The mainstay of treatment in such settings is chemoimmunotherapy followed by autologous stem cell transplant with the potential for long-term remission and cure for some (Citation3). Prognosis is extremely poor, however, for those who are ineligible or refractory to such treatments, as continued chemoimmunotherapy regimens result in a low response rate and overall survival of less than a year (Citation4). Immunotherapies such as monoclonal antibodies and CAR-T have advanced the field for some with improved durable responses, though the overall impact on long-term outcomes is less clear (Citation5). Thus, there remains an urgent, unmet clinical need for safe and efficacious alternatives to conventional therapy and newer immunotherapies in this space. Novel targeted agents may complement some of the initial improved outcomes seen with recent advances in immunotherapies.
Aberrant signaling of the phosphoinositide 3-kinase (PI3K)/protein kinase B (AKT)/mammalian target of rapamycin (mTOR) signaling pathway has been implicated in malignant cellular proliferation and survival, and several therapeutics targeting this pathway have been developed for lymphoma (Citation6). Everolimus (RAD001) is a novel oral derivative of rapamycin that inhibits mTOR (mTORi) and has demonstrated significant preclinical anti-tumor activity when tested in lymphoma cell lines (Citation7,Citation8). Phase II clinical trials of everolimus in relapsed aggressive NHL have demonstrated moderate clinical efficacy with a tolerable toxicity profile; in one study, approximately a third of DLBCL patients experienced partial responses to single-agent therapy (Citation9,Citation10).
Synergistic efficacy is seen when everolimus is combined with other antineoplastic agents, including histone deacetylase inhibitors (HDACi) (Citation8,Citation11). Mutated, aberrant chromatin remodeling is an oncogenic driver of lymphomagenesis; HDACi restore normal gene expression by reversing aberrant acetylation (Citation12). Panobinostat (LBH589) is an HDACi with demonstrated preclinical activity in multiple tumor cell lines (Citation13). Preclinical studies have demonstrated antiproliferative and pro-apoptotic effects from panobinostat and everolimus combination therapy in Hodgkin lymphoma (Citation14,Citation15). Phase I and II trials of single-agent and combination therapy panobinostat in Hodgkin lymphoma and NHL have demonstrated moderate clinical activity (Citation16–20).
Based on the preclinical synergy in B-cell lymphoma lines, combined with their demonstrated clinical efficacy and tolerability as single-agents (Citation8,Citation11,Citation14,Citation15), we designed a study to evaluate the clinical benefit of the combination of the mTORi, everolimus, and the HDACi, panobinostat, in patients with relapsed or refractory (R/R) DLBCL.
Methods
Study design
This was a single-center, phase II, prospective, non-randomized, open-label clinical trial of the HDACi, panobinostat, and the mTORi, everolimus, in patients with R/R DLBCL. Eligible patients had an Eastern Cooperative Group (ECOG) performance status of 2 or less, liver function <2.5× upper limit of normal (unless involved by lymphoma, then <5× upper limit of normal), creatinine clearance ≥30 mL/min, and hematologic parameters defined by an absolute neutrophil count (ANC) ≥1.0 × 109/L and platelets ≥75 × 109/L, unless due to bone marrow involvement by lymphoma. Previous exposure to everolimus or other medications primarily targeting the mTOR pathway, previous exposure to panobinostat or other HDACi, diabetes, steroid use >20 mg/day prednisone or equivalent, known infection with HIV, hepatitis B, or HCV, with additional viral testing at screening only for those with known risk factors, and use of concurrent QT-prolonging medications excluded enrollment. This study was approved by the Institutional Review Board (IRB) at Duke and was registered on www.clinicaltrials.gov, identifier NCT00978432. Data were collected, analyzed, and interpreted by the authors. All authors reviewed the manuscript.
We initially enrolled patients into a lead-in phase of sequential single-agent HDACi and single-agent mTORi (Part 1) which was to be followed by combination therapy (Part 2) in adult patients with R/R DLBCL. With this design, after providing informed consent, patients were non-randomly assigned to either begin receiving panobinostat or everolimus single-agent, alternating the assigned starting drug with every third enrolled patient. Patients were evaluated for response after every 4th cycle using a modified version of the Cheson revised response criteria for malignant lymphoma, and response was coded as progressive disease (PD), stable disease (SD), partial response (PR), or complete response (CR) (Citation21). Based on response assessments, patients either continued receiving initial single-agent drugs, changed to the alternate drug, or proceeded to combination therapy. However, an unplanned interim analysis after the first 15 patients were treated on the study demonstrated that none of the patients were able to move from single to doublet therapy; furthermore, there were few responses to either panobinostat or everolimus single-agent therapy. Therefore, the study was amended to remove the single-agent lead-in. Instead, all subsequently enrolled patients began with combination therapy ().
Figure 1. Planned accrual design. (a) Schema for sequential single-agent lead-in to doublet therapy. (b) Following a protocol amendment, sequential lead-in was eliminated and all patients were enrolled to doublet therapy upfront.
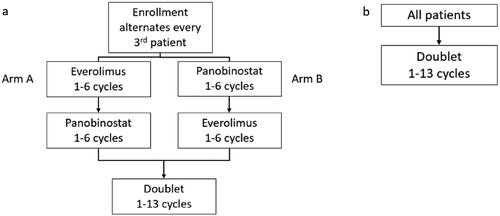
Initial trial design Part 1, Arm A: Everolimus followed by panobinostat
In the original design, patients were assigned to Arm A and non-randomized enrollment continued as discussed above. Patients received oral everolimus 10 mg daily during a 28-day cycle. Patients with a CR after 4 cycles could continue to receive everolimus until progression or up to 6 cycles, followed by a drug-free interval until disease progression at which time they would be started on panobinostat. For patients with ≤ PR after 4 cycles, study drug was switched to single-agent panobinostat following a 1- to 6-week washout period, at the providers’ discretion, unless there was rapid progression suggesting therapy should not be withheld.
Once switched, patients received oral panobinostat 40 mg 3 times a week at least 2 days apart during a 28-day cycle. Patients with a CR after 4 cycles could continue to receive panobinostat until progression or up to 6 cycles, followed by a drug-free interval until disease progression at which time they would be started on the doublet of panobinostat plus everolimus. Patients with ≤ PR after 4 cycles of panobinostat were switched to doublet using the dosages described in Part 2 below.
Initial trial design Part 1, Arm B: Panobinostat followed by everolimus
Patients who were assigned to Arm B started with panobinostat followed by single-agent everolimus with the intent to ultimately receive combination therapy. The drug dosages and general design were the same as described in Arm A above; the only difference was the order in which the single-agent drugs were administered.
Part 2: Doublet therapy of panobinostat plus everolimus
Following a protocol amendment as discussed above, patients began doublet therapy following enrollment (). Panobinostat was started at a dose of 20 mg orally three times a week at least 2 days apart and everolimus was dosed as 10 mg orally daily. Each cycle was 28 days. Due to toxicity from the doublet, the starting dose was subsequently decreased to panobinostat 15 mg 3 days a week and everolimus 7.5 mg daily for the 28-day cycle. Responses were assessed following cycle 2 and every 4th cycle thereafter. Patients with SD or better could continue to receive up to 13 cycles of doublet therapy. Patients with PD halted treatment.
All patients were followed until alternative therapy was begun, until disease progression, death, or for every 3 months up to 2 years.
End points
Co-primary objectives for the original study design included estimating the overall response rate (ORR) of single and multi-agent epigenetic targets and estimating the association of observed responses to everolimus and panobinostat with the responses predicted by molecular signatures developed in our preclinical models; however, insufficient response rates precluded planned correlative analyses of target suppression and molecular signature associations.
Patients were considered evaluable for response and toxicity assessment if they had received any study drug. Toxicity was graded using the CTCAE version 4.0 (http://ctep.info.nih.gov). Subjects were followed for toxicity from the first dose of study drug until 4 weeks after drug discontinuation.
Statistical analysis
Patients were evaluated in an intent-to-treat analysis. The ORR was calculated using descriptive statistics. The association between response rate and treatment was tested using the Fisher’s exact test. SAS version 9.0 (Cary, NC) was used for statistical analysis.
Results
Patients
Fifty patients signed informed consent between October 2009 and November 2014 at Duke University. The patient population represented a heavily pre-treated cohort with a median number of prior therapies between 2 and 4, and most patients were refractory to their most recent line of therapy (). Information on high-risk cytogenetics was not available for 28 (85%) of patients, given this testing was not routine clinical practice during the time of enrollment. As such, this information is not reported in baseline characteristics. The patients were disproportionally male and white. Of these 50 patients, 33 were enrolled and received study drug ().
Figure 2. Enrollment. IF: informed consent; PI: principal investigator.
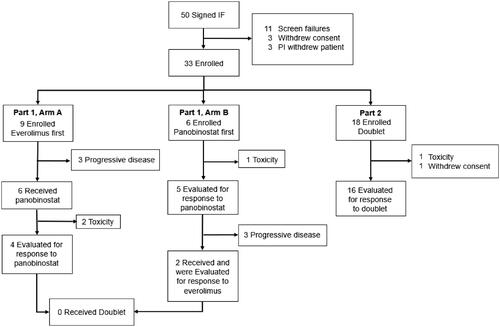
Table 1. Baseline patient characteristics.
Part 1: Single-agent everolimus or panobinostat
Nine patients were enrolled to Part 1, Arm A and thus began with single-agent everolimus. One patient achieved a CR following 6 cycles of everolimus and progressed 3 months after stopping everolimus. No responses to everolimus were seen in the other 8 patients. Out of the nine patients, three were taken off study by investigator decision due to PD while on everolimus, and six went on to receive sequential single-agent panobinostat. Of the 6 patients who received sequential panobinostat, 2 were removed early before response evaluations due to toxicity (Grade 3 diarrhea and nausea), and no responses to panobinostat were seen in the 4 remaining patients. No patients proceeded to the doublet therapy, due to a combination of toxicity or patient or investigator decision to pursue alternate therapy, including comfort care (, ).
Table 2. Best response by treatment arm and drug.
Six patients were enrolled to Part 1, Arm B and therefore started with single-agent panobinostat. One patient was removed from study before response assessment due to toxicity from panobinostat (Grade 3 thrombocytopenia). None of the 5 remaining evaluable patients had a response to panobinostat. Of these 5 patients, 3 were removed from protocol due to patient/investigator decision for PD, and 2 went on to receive sequential single-agent everolimus, neither of whom responded to the second study drug. No patients proceeded to doublet therapy due to investigator decision to pursue alternative therapy considering rapidly progressing disease (, ).
Part 2: Doublet therapy
Following the amendment, 18 patients were enrolled to Part 2, the upfront doublet of panobinostat plus everolimus. Two patients were not included in response evaluations, as 1 was removed due to early toxicity from Grade 3 colitis requiring hospitalization, and 1 withdrew consent prior to response evaluation.
The ORR for the 16 remaining patients was 25.0% (95% CI: 0.0727, 0.5238), all of which were PRs (); however, the benefit was short lived with all responders coming off study drugs before 6 months. Of the four total patients who had a PR, one patient experienced a PR following cycle 2 but progressed after cycle 4, and three patients experienced a PR following cycle 4 but all of them discontinued treatment due to toxicity either immediately following cycle 4 or cycle 5 ().
Safety and toxicity
All patients experienced an adverse event of at least grade 1, the most common being hematologic and gastrointestinal including fatigue, anorexia, nausea, vomiting, and diarrhea (). Grade 4 thrombocytopenia was common, especially in patients receiving panobinostat. Greater toxicity was noted with doublet therapy than with sequential single-agent therapy, with all 18 patients experiencing at least one grade 3 or 4 adverse events and half experiencing an SAE (). Due to the high number of toxicities observed with doublet therapy, after the first seven patients were treated, a protocol amendment decreased the starting dose of everolimus from 10 mg orally daily to 7.5 mg orally daily, and panobinostat from 20 mg orally 3 times a week to 15 mg orally 3 times a week. Despite this, all 11 patients receiving decreased doses still experienced a grade 3/4 AE ().
Table 3. Adverse events.
The most frequent Grade 3 or 4 AEs occurred with doublet therapy, and included hematologic toxicity, especially thrombocytopenia (15 patients), which was likely attributable to panobinostat. Other significant AEs included fatigue (12 patients), hypoxia confirmed or suspected secondary to pneumonitis (3 patients), cardiac (2 patients), colitis (1 patient), gastrointestinal (2 patients), and infection (2 patients) (). Ultimately, 3 patients in Part 1 Arm A, 1 patient in Part 1 Arm B, and 6 patients in Part 2 were removed from protocol primarily due to toxicity concerns.
Discussion
This was intended as a feasibility study of an mTORi, everolimus, and an HDACi, panobinostat, given as single-agents to be immediately followed as doublet therapy for patients with R/R DLBCL. Our trial design incorporated sequential and doublet therapy to provide patients with highly refractory disease with as many novel agents within the same trial design as possible, and to increase the efficacy of study drugs by targeting separate, complementary pathways vital to lymphomagenesis. Furthermore, given demonstrated synergistic preclinical activity of combination therapy (Citation14,Citation15), we hypothesized that patients who did not respond to single-agent therapy could still benefit from doublet therapy. In our small study, we found minimal single-agent activity of either agent, and responses were not durable. While doublet therapy had greater response, suggesting that combination therapy was more clinically effective as demonstrated from preclinical models, the toxicity rates were prohibitive for use in clinical practice.
In our trial, the combination of everolimus plus panobinostat demonstrated modest efficacy in R/R DLBCL with an ORR of 25%. However, most patients experienced Grade 3 or 4 adverse events including thrombocytopenia in 83% and fatigue in 55% of patients. A subsequent protocol amendment of dose reduction for both agents did not improve tolerability. Our results are consistent with a phase I study of combination panobinostat and everolimus in R/R lymphoma,; while the ORR was 43%, there were no responders among 7 patients enrolled with R/R DLBCL (Citation14). Like our study, thrombocytopenia severely limited treatment tolerability (Citation14).
Unlike the findings in our study, single-agent panobinostat has demonstrated some efficacy in R/R DLBCL. Zaja and colleagues evaluated single-agent clinical activity of panobinostat administered at 40 mg three times a week in a cohort of 35 patients with R/R DLBCL, and reported an ORR of 17.1% with a CR of 11.4%, which was durable in a few patients (Citation20). However, similar to our study, thrombocytopenia was severely limiting and resulted in dose reductions or suspensions for 19 (54%) patients, ultimately limiting clinical use (Citation20). In one of the largest trials of panobinostat in R/R DLBCL to date, Assouline and colleagues reported a 28% ORR in 40 patients receiving panobinostat 30 mg three times a week with or without rituximab, and demonstrated no added benefit to the addition of an anti-CD20 monoclonal antibody (Citation16). Thrombocytopenia necessitated dose interruptions or reductions in over half of the patients enrolled (Citation16). Correlative biomarkers in this study identified distinct genomic variations in responders; patients with MEF2B mutations (15% of patients enrolled) had 3.67 (1.46–9.19) likelihood ratio for response versus 8.1 (1.94–33.62) hazard ratio for progression in patients harboring a CD79A/B mutation (Citation16). Genomic variation with enrichment for aggressive pathology in our highly refractory patient population may account for differences in ORR noted between our two trials. In a third study, Barnes et al reported an ORR of 11% to combination therapy with panobinostat and rituximab in a heavily refractory patient cohort like ours, with a duration of response under 2 months and thrombocytopenia limiting use (Citation17). Our study did not demonstrate any responses to single-agent panobinostat. However, only 12 patients were exposed to the single agent, and it is possible our small sample size lacked power to demonstrate a very low response rate such as the 11% ORR reported by Barnes and colleagues.
Single-agent activity for everolimus in DLBCL was previously demonstrated by Witzig and colleagues, in which 14 of 47 (30%) R/R DLBCL patients reported PRs with a tolerable toxicity profile, with an average time to response of 2.0 months for responders (Citation10). Comparatively, most patients reported SD on single-agent everolimus in our trial, and those who progressed did so rapidly within 1–2 cycles. An impressive ORR of 38% in a cohort of 24 heavily refractory R/R DLBCL patients to combination everolimus and rituximab was previously reported by Barnes et al, with most responders exhibiting PR by 2 cycles of therapy, suggesting anti-CD20 directed therapy may synergize more effectively than an HDACi (Citation22). Our data, combined with these previously published results, suggest that there is a modest clinical benefit at best from single-agent or combination mTORi and HDACi in R/R DLBCL.
The intent of this novel trial design of sequential single-agents followed by the doublet was twofold. First, it would enable us to evaluate the synergistic effect of the doublet, by observing whether we would see higher response rates with the doublet than we saw with the single-agents in the same patient population. Second, if effective, the design could be an efficient way to evaluate multiple different drugs quickly within one trial rather than three different trials, thereby providing patients with R/R disease access to several different potentially effective therapeutic options. Unfortunately, we demonstrated that in a group of heavily pretreated patients with R/R DLBCL, this was not an effective trial design as patients were not able to proceed through all intended iterations of drugs. This study design may be more suitable in less aggressive malignancies.
There are several limitations to our study. It is a single-center study and lacks a control arm, which introduces bias. Our small sample size limits statistical power and as such, most of our responses are presented descriptively. Our protocol underwent several amendments throughout trial conduct due to excessive toxicity and low response rates, resulting in patients observed under multiple administration schedules and dosages, which affects the quality of our statistical analysis. Due to rapid PD, many patients could not complete an adequate washout period which may confound response and toxicity assessments. Lastly, multiple patients withdrew prior to response evaluation, resulting in a lower number of evaluable patients.
Conclusion
Our study contributes to a growing body of clinical literature regarding the use of mTORi and HDACi in NHL. While we have demonstrated minimal clinical efficacy in R/R DLBCL, other investigators have reported modest clinical efficacy with improved responses in patients with targeted mutations (Citation6,Citation10,Citation14,Citation16,Citation17,Citation20,Citation22–24). R/R patients with DLBCL suffer an aggressive lymphoma with few effective treatment options, and as such, larger, well-designed clinical trials will be needed to establish any possible role for mTORi or HDACi. Future investigators are cautioned regarding toxicity; ultimately, these agents may be better suited to the treatment of indolent lymphoma, in which lower doses and longer treatment periods may result in better tolerability and response.
Declaration of interest
Dr. Rizzieri is on the Advisory Board for Novartis.
Additional information
Funding
References
- Swerdlow SH, Campo E, Harris NL, Jaffe ES, Pileri SA, Stein H. WHO classification of tumours of haematopoietic and lymphoid tissues, fourth edition. Lyon, France: IARC; 2008.
- Coiffier B, Thieblemont C, Van Den Neste E, Lepeu G, Plantier I, Castaigne S, et al. Long-term outcome of patients in the LNH-98.5 trial, the first randomized study comparing rituximab-CHOP to standard CHOP chemotherapy in DLBCL patients: a study by the Groupe d'Etudes des Lymphomes de l'Adulte. Blood. 2010;116(12):2040–2045. doi:https://doi.org/10.1182/blood-2010-03-276246.
- Aksentijevich I, Jones RJ, Ambinder RF, Garrett-Mayer E, Flinn IW. Clinical outcome following autologous and allogeneic blood and marrow transplantation for relapsed diffuse large-cell non-Hodgkin’s lymphoma. Biol Blood Marrow Transplant. 2006;12(9):965–972. doi:https://doi.org/10.1016/j.bbmt.2006.05.018.
- Crump M, Neelapu SS, Farooq U, Van Den Neste E, Kuruvilla J, Westin J, et al. Outcomes in refractory diffuse large B-cell lymphoma: results from the international SCHOLAR-1 study. Blood. 2017;130(16):1800–1808. doi:https://doi.org/10.1182/blood-2017-03-769620.
- Neelapu SS, Locke FL, Bartlett NL, Lekakis LJ, Reagan PM, Miklos DB, Jacobson CA, et al. A comparison of 2-year outcomes in ZUMA-1 (axicabtagene ciloleucel [axi-cel]) and SCHOLAR-1 in patients (Pts) with refractory large B cell lymphoma (LBCL). Biol Blood Marrow Transplant. 2020;26:S1–S412.
- Calimeri T, Ferreri AJM. m-TOR inhibitors and their potential role in haematological malignancies. Br J Haematol. 2017;177(5):684–702. doi:https://doi.org/10.1111/bjh.14529.
- Wanner K, Hipp S, Oelsner M, Ringshausen I, Bogner C, Peschel C, et al. Mammalian target of rapamycin inhibition induces cell cycle arrest in diffuse large B cell lymphoma (DLBCL) cells and sensitises DLBCL cells to rituximab. Br J Haematol. 2006;134(5):475–484. doi:https://doi.org/10.1111/j.1365-2141.2006.06210.x.
- Haritunians T, Mori A, O'Kelly J, Luong QT, Giles FJ, Koeffler HP. Antiproliferative activity of RAD001 (everolimus) as a single agent and combined with other agents in mantle cell lymphoma. Leukemia. 2007;21(2):333–339. doi:https://doi.org/10.1038/sj.leu.2404471.
- Yee KWL, Zeng Z, Konopleva M, Verstovsek S, Ravandi F, Ferrajoli A, et al. Phase I/II study of the mammalian target of rapamycin inhibitor everolimus (RAD001) in patients with relapsed or refractory hematologic malignancies. Clin Cancer Res. 2006;12(17):5165–5173. [Database] doi:https://doi.org/10.1158/1078-0432.CCR-06-0764.
- Witzig TE, Reeder CB, LaPlant BR, Gupta M, Johnston PB, Micallef IN, et al. A phase II trial of the oral mTOR inhibitor everolimus in relapsed aggressive lymphoma. Leukemia. 2011;25(2):341–347. [Database] doi:https://doi.org/10.1038/leu.2010.226.
- Yazbeck VY, Buglio D, Georgakis GV, Li Y, Iwado E, Romaguera JE, et al. Temsirolimus downregulates p21 without altering cyclin D1 expression and induces autophagy and synergizes with vorinostat in mantle cell lymphoma. Exp Hematol. 2008;36(4):443–450. doi:https://doi.org/10.1016/j.exphem.2007.12.008.
- Richon VM, O'Brien JP. Histone deacetylase inhibitors: a new class of potential therapeutic agents for cancer treatment. Clin Cancer Res. 2002;8(3):662–664.
- Beckers T, Burkhardt C, Wieland H, Gimmnich P, Ciossek T, Maier T, et al. Distinct pharmacological properties of second generation HDAC inhibitors with the benzamide or hydroxamate head group. Int J Cancer. 2007;121(5):1138–1148. doi:https://doi.org/10.1002/ijc.22751.
- Oki Y, Buglio D, Fanale M, Fayad L, Copeland A, Romaguera J, et al. Phase I study of panobinostat plus everolimus in patients with relapsed or refractory lymphoma. Clin Cancer Res. 2013;19(24):6882–6890. doi:https://doi.org/10.1158/1078-0432.CCR-13-1906.
- Lemoine M, Derenzini E, Buglio D, Medeiros LJ, Davis RE, Zhang J, et al. The pan-deacetylase inhibitor panobinostat induces cell death and synergizes with everolimus in Hodgkin lymphoma cell lines. Blood. 2012;119(17):4017–4025. doi:https://doi.org/10.1182/blood-2011-01-331421.
- Assouline SE, Nielsen TH, Yu S, Alcaide M, Chong L, MacDonald D, et al. Phase 2 study of panobinostat with or without rituximab in relapsed diffuse large B-cell lymphoma. Blood. 2016;128(2):185–194. doi:https://doi.org/10.1182/blood-2016-02-699520.
- Barnes JA, Redd R, Fisher DC, Hochberg EP, Takvorian T, Neuberg D, et al. Panobinostat in combination with rituximab in heavily pretreated diffuse large B-cell lymphoma: results of a phase II study. Hematol Oncol. 2018;36(4):633–637. doi:https://doi.org/10.1002/hon.2515.
- DeAngelo DJ, Spencer A, Bhalla KN, Prince HM, Fischer T, Kindler T, et al. Phase Ia/II, two-arm, open-label, dose-escalation study of oral panobinostat administered via two dosing schedules in patients with advanced hematologic malignancies. Leukemia. 2013;27(8):1628–1636. doi:https://doi.org/10.1038/leu.2013.38.
- Maly JJ, Christian BA, Zhu X, Wei L, Sexton JL, Jaglowski SM, et al. A phase I/II trial of panobinostat in combination with lenalidomide in patients with relapsed or refractory Hodgkin lymphoma. Clin Lymphoma Myeloma Leuk. 2017;17(6):347–353. doi:https://doi.org/10.1016/j.clml.2017.05.008.
- Zaja F, Salvi F, Rossi M, Sabattini E, Evangelista A, Ciccone G, et al. Single-agent panobinostat for relapsed/refractory diffuse large B-cell lymphoma: clinical outcome and correlation with genomic data. A phase 2 study of the Fondazione Italiana Linfomi. Leuk Lymphoma. 2018;59(12):2904–2910. doi:https://doi.org/10.1080/10428194.2018.1452208.
- Cheson BD, Pfistner B, Juweid ME, Gascoyne RD, Specht L, Horning SJ, International Harmonization Project on Lymphoma, et al. Revised response criteria for malignant lymphoma. J Clin Oncol. 2007;25(5):579–586. doi:https://doi.org/10.1200/JCO.2006.09.2403.
- Barnes JA, Jacobsen E, Feng Y, Freedman A, Hochberg EP, LaCasce AS, et al. Everolimus in combination with rituximab induces complete responses in heavily pretreated diffuse large B-cell lymphoma. Haematologica. 2013;98(4):615–619. doi:https://doi.org/10.3324/haematol.2012.075184.
- Apuri S, Sokol L. An overview of investigational histone deacetylase inhibitors (HDACis) for the treatment of non-Hodgkin’s lymphoma. Expert Opin Investig Drugs. 2016;25(6):687–696. doi:https://doi.org/10.1517/13543784.2016.1164140.
- Batlevi CL, Crump M, Andreadis C, Rizzieri D, Assouline SE, Fox S, et al. A phase 2 study of mocetinostat, a histone deacetylase inhibitor, in relapsed or refractory lymphoma. Br J Haematol. 2017;178(3):434–441. doi:https://doi.org/10.1111/bjh.14698.