
Abstract
Protein glycosylation is post-translational modification (PTM) which is important for pharmacokinetics and immunogenicity of recombinant glycoprotein therapeutics. As a result of variations in monosaccharide composition, glycosidic linkages and glycan branching, glycosylation introduces considerable complexity and heterogeneity to therapeutics. The host cell line used to produce the glycoprotein has a strong influence on the glycosylation because different host systems may express varying repertoire of glycosylation enzymes and transporters that contributes to specificity and heterogeneity in glycosylation profiles. In this review, we discuss the types of host cell lines currently used for recombinant therapeutic production, their glycosylation potential and the resultant impact on glycoprotein properties. In addition, we compare the reported glycosylation profiles of four recombinant glycoproteins: immunoglobulin G (IgG), coagulation factor VII (FVII), erythropoietin (EPO) and alpha-1 antitrypsin (A1AT) produced in different mammalian cells to establish the influence of mammalian host cell lines on glycosylation.
Importance of glycosylation for biologics production
Glycosylation is post-translational modification (PTM) occurring within the secretory pathways of cells, that is, endoplasmic reticulum (ER) and Golgi apparatus, where monosaccharide units such as galactose (Gal), mannose (Man), fucose (Fuc), N-acetylglucosamine (GlcNAc), N-acetylgalactosamine (GalNAc) and sialic acids are covalently attached to specific amino acids of newly synthesized proteins and lipid structures [Citation1–3]. Glycans attached to the amide nitrogen atom of asparagine (Asn) residues are termed N-linked glycans (), while glycans attached to the oxygen atom of serine (Ser) or threonine (Thr) residues are O-linked glycans (). Glycans can be attached in linear or branching chains and may be linked by α- or β-glycosidic linkages at various linkage positions such as 1➔3, 1➔6 or 2➔3. The possible variations in monosaccharide composition, glycosidic linkages and glycan branching gives rise to an extremely diverse glycan repertoire [Citation4]. Concomitantly, biologics with varying glycostructures may differ in therapeutic efficacy as glycans can significantly influence protein solubility, bioactivity, stability and immunogenicity [Citation5]. In monoclonal antibody (mAb) manufacturing, core fucosylation was shown to attenuate antibody-binding affinity via potentially masking binding moieties on N-glycans [Citation6–8]. Immunoglobulin G (IgG) with a low degree of fucosylation (21%) was found to have enhanced binding affinity to FcγIIIA Fc receptors (FcγRIIIA) up 53-fold as compared to a highly fucosylated IgG (98% fucosylated) [Citation9]. Accordingly, antibody-dependent cellular cytotoxicity (ADCC) was increased in fucose-deficient IgG [Citation9,Citation10]. Oligomannose glycostructures Man5 and Man8/9 have similarly been shown to enhance FcγRIIIA binding and ADCC activity in comparison to complex-fucosylated glycostructures and at similar levels to afucosylated complex structures [Citation11,Citation12]. One caveat, however, is that oligomannose glycostructures are rapidly cleared from circulation either via a mannose receptor or mannose-binding lectin-mediated mechanisms, thus affecting the pharmacokinetics of therapeutic antibodies [Citation12–15]. Sialylation has been shown to be important for several properties of recombinant proteins, improving solubility, biological activity, thermal stability and circulatory half-life [Citation16–18]. In vivo, sialic acids prevent the recognition of sub-terminal Gal and GalNAc residues by asialoglycoprotein receptors on hepatic cells, protecting glycoproteins from uptake and degradation [Citation19]. In most mammals, the predominant sialic acids produced are N-acetylneuraminic acid (Neu5Ac) and N-glycolylneuraminic acid (Neu5Gc). Humans do not produce Neu5Gc due to a 92-bp deleterious mutation on the CMAH gene which codes for cytidine monophospo-Neu5Ac (CMP-Neu5Ac) hydroxylase, an enzyme that converts CMP-Neu5Ac into CMP-Neu5Gc [Citation20,Citation21]. However, circulating Neu5Gc-specific antibodies have been detected in humans reflecting the presence of Neu5Gc, postulated to be derived from dietary red meat and dairy products [Citation22,Citation23]. More importantly, this demonstrates that Neu5Gc-decorated proteins have the capacity to elicit an antibody response in humans [Citation22–24]. The level of Neu5Gc in biologics is thus an essential component to assess as circulating Neu5Gc-specific antibodies can neutralize and reduce the half-life of Neu5Gc-containing glycoproteins [Citation25,Citation26]. Furthermore, prolonged treatments with Neu5Gc-containing biologics could result in the accumulation of Neu5Gc in tissues and consequently promote antibody-mediated inflammation, atherosclerosis and carcinoma progression [Citation27,Citation28].
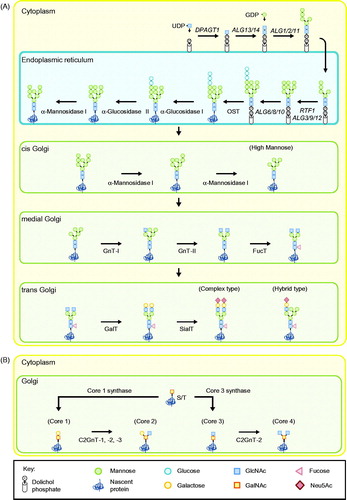
Figure 1. Glycan biosynthesis of secreted proteins in humans. (A) N-glycosylation initiates with the construction of a Glc3Man9GlcNAc2-PP-dolichol precursor glycan which is then attached onto asparagine residues of nascent proteins as part of a cotranslational modification. Properly folded proteins are then trafficked to the Golgi apparatus where a myriad of factors influence further processing of glycans to form a heterogeneous population of oligomannose, complex- or hybrid-type glycostructures. (B) O-linked glycosylation initiates in the Golgi with the attachment of N-acetylgalactosamine (GalNAc) units onto serine and threonine residues of newly translated proteins. Addition of galactose (Gal) and N-acetylglucosamine (GlcNAc) units subsequently results in the synthesis of different O-glycan core structures (core 1 to 4). Uridine diphosphate (UDP)-glucose; Guanosine diphosphate (GDP)-mannose; Dolichyl-phosphate N-acetylglucosaminephosphotransferase 1 (DPAGT1); Asparagine linked glycosylation (ALG) genes; RNA polymerase-associated protein RTF1 homolog (RTF1); Oligosaccaryl transferase (OST); N-acetylglucosaminyl Transferase I (GnT-I); N-acetylglucosaminyl Transferase II (GnT-II); Fucosyltransferase (FucT); Galactosyltransferase (GalT); Sialyltransferase (SialT); β-1,6 GlcNAc transferases (C2GnT-1, -2 or -3); N-acetylneuraminic acid (Neu5Ac).
Among several factors, protein glycosylation is influenced by host-cell type. Different host systems may express varying glycosylation enzymes and transporters, contributing to the specificity and heterogeneity in glycosylation profiles and subsequent clinical effectiveness of the therapeutic product. For example, recombinant glucocerebrosidase (GC) can potentially be produced using plant species such as the Arabidopsis thaliana and Nicotiana benthamiana, since these cells typically produce glycoproteins with oligomannose structures required for GC’s therapeutic efficacy to treat Gaucher’s disease [Citation29,Citation30]. However, glycoengineering strategies have to be employed to circumvent the attachment of plant-specific β-1,2-xylose and α-1,3-fucose [Citation31–33] to lower the risk of immunogenic reactions that may be elicited by nonhuman glycans as previously observed in some murine-derived biologics [Citation34–36]. In another example, several cases of anaphylaxis and angioedema were highlighted in clinical trial studies for cetuximab, an FDA approved mouse-human chimeric monoclonal antibody (mAb) used for treating patients with EGFR-expressing metastatic colorectal cancer [Citation37]. These side effects were ascribed to the presence of immunogenic galactose-alpha-1,3-galactose (alpha-Gal) glycans found in 30% of Cetuximab glycostructures [Citation36,Citation38,Citation39].
These studies highlight the importance of glycosylation in biologics production and correspondingly, the need to determine an appropriate host cell system that would produce a functional glycosylation profile. Glycosylation of recombinant proteins in non-mammalian systems are different from humans and thus have limited applications in the production of therapeutic glycoproteins [Citation40–47]. Nonetheless, efforts are currently underway to engineer these different expression systems for therapeutic glycoprotein production [Citation48–50]. As mammalian cells are the predominant expression systems for over 60% of recombinant protein pharmaceuticals [Citation51], we review the glycosylation potential of various mammalian host cells in the following sections. In addition, we compare reported glycosylation profiles of four recombinant glycoproteins produced in different mammalian cells to establish the influence of mammalian host cell lines on glycosylation.
Mammalian expression platforms
Mammalian cells possess the metabolic machinery to produce and secrete recombinant proteins that closely resemble or are fully compatible with humans. As such, mammalian host cells are the preferred expression systems for the manufacture of complex-type N-linked glycoproteins such as multimeric antibodies and blood clotting factors. Types of mammalian cells used for the production of approved bio-therapeutics include Chinese hamster ovary (CHO) cells, baby hamster kidney (BHK) cells, NS0 myeloma and Sp2/0 hybridoma mouse cell lines, human embryonic kidney cells 293 (HEK293) and HT-1080 human cells.
Nonhuman mammalian systems
Of the several nonhuman mammalian expression systems, CHO cells are the predominant host cell platform accounting for over 60% of currently approved bio-therapeutics [Citation52–54]. The wide use of CHO cells is related to the number of advantages they offer as host cells for production. CHO cells are well characterized with the established gene amplification methods and improved clonal selection strategies, contributing to the overall increase in protein production yield [Citation55]. In terms of culture conditions, CHO cells can be adapted to suspension cultures and in chemically defined, serum-free media which allows for large scalability and inter-batch reproducibility. CHO cells are robust, being highly tolerant for changes in culture conditions including pH, oxygen level, pressure and temperature [Citation56,Citation57]. Utilizing CHO cells for production confers safer manufacturing conditions to recombinant proteins as CHO cells have restricted susceptibility to human viruses thus reducing the risk for transmission of adventitious agents from CHO cells to humans [Citation58,Citation59]. More importantly, CHO cells are able to produce complex types of recombinant proteins with human-compatible glycosylation. CHO cells, however, do not express Gal α2,6 ST, α1,3/4 fucosyltransferase or β-1,4-N-acetylglucosaminyltransferase III (GnT-III) which are enzymes expressed in human cells () [Citation60–62]. As such, CHO-derived proteins may lack or have differing glycostructures from human-derived glycoproteins. Combined with their long history for use in manufacturing (since 1986 for the production of tissue plasminogen activator), risks of a delay for regulatory approvals are relatively lower with CHO cells as compared to novel host cell lines [Citation53].
Table 1. Glycosylation enzymes differentially expressed in different mammalian host cell lines used for commercial production compared to humans.
Other nonhuman mammalian expression systems include the BHK-21 cells, murine NS0 myeloma and Sp2/0 hybridoma cells. BHK-21 cells were derived from baby Syrian hamster (Mesocricetus auratus) kidney [Citation63] and are used for the production of coagulation factors, for example, FVII and FVIII [Citation54,Citation56,Citation64]. Murine NS0 myeloma and Sp2/0 hybridoma cells were derived from immunoglobulin-producing tumor cells [Citation65,Citation66]. These cells no longer produce their original immunoglobulins but still possess the machinery to do so. As such, NS0 and Sp2/0 cells are utilized for the production of monoclonal antibodies [Citation64,Citation67].
Despite their wide use in the production of human recombinant proteins, nonhuman mammalian cells lack or have differing machineries for human-type glycosylation. Consequently, glycosylation patterns produced in these cells can be immunogenic in humans. Certain murine cell lines, such as NS0 and Sp2/0, can potentially attach antigenic alpha-Gal onto recombinant proteins which may trigger an immunogenic response, such as anaphylactic shock in humans [Citation39,Citation68]. Most nonhuman mammalian cell lines can also attach Neu5Gc which can similarly elicit immunogenic responses [Citation26–28,Citation69,Citation70]. Due to the potential risks, nonhuman cell lines are stringently screened to identify clones that produce recombinant proteins with a desired glycan profile [Citation26].
Human cell lines for glycoprotein production
Human cell lines are a promising and emerging alternative to nonhuman host cells as the resultant recombinant protein would possess fully human PTMs, reducing the cost for downstream processing and, more importantly, circumventing the risk of immunogenic reactions from nonhuman glycans. With the capacity to produce human therapeutics that most resemble native counterparts, human host cells holds an advantage over other expression systems. Human cell lines used or developed for biopharmaceutical production include HEK293, PER.C6, CEVEC’s amniocyte production (CAP), AGE1.HN, HKB-11 and HT-1080 cells.
HEK293 cells were immortalized in the 1970 s by transforming genomic regions of an adenoviral vector into cultures of normal human embryonic kidney cells [Citation71,Citation72]. These regions are trans-complementary for adenovirus type 5 expression vectors which can replicate adenovirus to high titers in HEK293 cells. HEK293 cells are adaptable to suspension cultures in chemically-defined, serum-free media making HEK293 cells easily scalable for large-scale manufacture [Citation73]. Several derivatives of HEK293 cells, such as HEK293-T and HEK293-EBNA1, have been developed from the parental HEK293 cells for improved recombinant protein production [Citation74–76]. The highly transfectable HEK293-T cells were developed through further transformation of the simian vacuolating virus 40 (SV40) large T antigen into HEK293 cells. HEK293-T cells are notably capable of producing high titer yields of retroviral vectors expressing genes that can be used for gene therapy [Citation77]. Similarly, HEK293-EBNA1 cells are HEK293 derivatives stably transfected with the Epstein-Barr virus (EBV) nuclear antigen 1 (EBNA1). HEK293-EBNA1 cells have an improved growth rate and expression of EBV-based vectors compared to HEK293 cells [Citation78]. Notable FDA approved biologics produced in HEK293 cells include recombinant FVIII fusion protein (rFVIIIFc) (ELOCTA®) and a glucagon-like peptide 1 receptor agonist (Dulaglutide; TRULICITY®) for the treatment of Hemophilia A and Type 2 Diabetes respectively [Citation79,Citation80].
Other human cell lines immortalized through the expression of adenoviral type 5 E1 genes include PER.C6, CAP and AGE1.HN cells [Citation81]. PER.C6 cells are derived from human primary embryonic retinoblasts. These cells are reported to be capable of growing in high cell densities and can produce recombinant proteins at high yield without the amplification of the inserted gene of interest [Citation82]. CAP cells, developed from primary human amniocytes, offer similar advantages such as the capacity to grow at high cell densities in suspension cultures and the ability to stably and highly express fully glycosylated and sialylated proteins without the presence of antibiotic selection pressures [Citation83]. AGE1.HN cells, derived from the periventricular zone of a human fetal brain, are also able to grow in suspension cultures at high densities [Citation84]. Additionally, AGE1.HN cells have been engineered for apoptosis-resistance in culture media with high ammonia build-up as observed in the late stages of bioproduction [Citation84,Citation85].
Another cell line used for biologics production is the HKB-11 cell, a hybrid clone generated through polyethylene glycol fusion of HEK293-S and a modified Burkitt’s lymphoma B-cell line [Citation55,Citation86,Citation87]. HKB-11 cells are capable of producing recombinant proteins at high yields without forming aggregates as seen in human host cells [Citation87]. Interestingly, HKB-11 cells have been shown to be superior to HEK293 and BHK-21 cells for high-titer production of recombinant proteins such as human FVIII [Citation88]. Yet another cell line used for biologics production is HT-1080, a human fibrosarcoma cell line with an epithelial-like phenotype [Citation89]. By introducing DNA promoters to activate expression of endogenous genes, HT-1080 has been utilized to produce four commercial biologics, namely epoetin delta (DYNEPO®), iduronate-2-sulfatase (ELAPRASE®), agalsidase alfa (REPLAGAL®) and velaglucerase alfa (VPRIV®) [Citation90].
Similar to other expression systems, there are limitations to using human cells for production. Human cell lines have the capacity to produce sialyl-Lewisx (sLex) antennary fucosylation which localize and bind to endothelial selectins in areas of inflammation [Citation91]. Although favorable for anti-inflammatory therapeutics, undesired production of biologics with sLex antennary fucosylation could perturb the drug’s proper biodistribution and pharmacokinetics. Another constraint is that human cell lines carry a potential risk for the contamination and subsequent transmission of human pathogens such as viruses and other adventitious agents due to a lack of a species barrier that would otherwise be present in nonhuman host cells. In this aspect, it is notable that many of these human cell lines are also used for the production of viral vaccines and gene therapy vectors, underscoring the propensity of these cells to propagate human viruses [Citation92]. This will likely be of regulatory and safety considerations in therapeutic biopharmaceutical manufacturing. Nonetheless, these considerations are not insurmountable, because there are currently approved therapeutic biopharmaceutical produced using HEK293 and HT-1080 cell lines [Citation93]. Advancements in manufacturing technologies such as improvement in viral inactivation and clearance methods can also address potential contamination by adventitious pathogens [Citation94,Citation95].
Impact of mammalian host cell line on glycosylation
Different cell types naturally affect glycosylation profiles of recombinant proteins [Citation96–98]. This phenomenon is observed throughout the different classes of recombinant biologics – mAbs, vaccines, hormones, growth factors, anticoagulants, cytokines and general recombinant glycoproteins. In comparing glycosylation profiles of 12 different proteins produced in CHO-S and HEK293-EBNA1 cells, glycosylation differences were observed as evidenced from differences in molecular weight, isoelectric point (pI), glycoprotein structure and mass peak profile [Citation97]. Glycan analysis of the same biologics produced in the two different cell lines generally showed that glycoproteins expressed in HEK293 cells had more complex glycosylation profiles compared to CHO-derived proteins [Citation97,Citation99]. In contrast, CHO-derived proteins were more sialylated compared to HEK293-derived proteins [Citation97,Citation99].
To further investigate the impact of mammalian cell lines on glycosylation, we chose four glycoproteins (IgG, coagulation factor VII (FVII), erythropoietin (EPO) and recombinant alpha-1 antitrypsin (A1AT)). These four glycoproteins were chosen from different classes of recombinant biologics produced in mammalian cells and were either blockbuster drugs with large revenues or represent biologics for which more glycosylation data is available. In addition to differences in host cell line, we noted that cell cultivation and analytical methods may also contribute to observed differences in glycosylation structures: for example, cells are known to be able to incorporate Neu5Gc from serum in culture media [Citation100]; analysis of sialylated glycostructures by matrix-assisted laser desorption/ionization (MALDI) mass spectrometry (MS) is hindered by the labile nature of sialic acids which are readily lost upon ionization by MALDI [Citation101]; the negative charge on sialylated glycostructures generally have lower ionization efficiency compared to neutral glycans which results in a bias during MS quantification [Citation102]. In order to take into account glycosylation differences arising from experimental variation, we collated data on culture methods, glycan release, labeling, separation and detection techniques. As such, we review reported glycosylation profiles of the four glycoproteins across different predominant mammalian cell expression systems in the case studies discussed below.
Immunoglobulin G (IgG)
Most antibodies developed for therapeutic use are of the human IgG isotype. The fragment crystallizable (Fc) region of IgG is essential for the binding of Fc gamma receptors (FcγR) on myeloid and natural killer (NK) cells, inducing ADCC [Citation103]. The predominant glycan at position N297 of the Fc region is a biantennary complex-type structure composed of a core Man and GlcNAc residues, annotated as G0 (). Structural changes induced in this biantennary glycan core have been reported to affect FcγR binding and subsequent ADCC effector functions [Citation104]. Glycoforms with this core structure include a variable number of Gal residues annotated as G1 and G2 structures. Further, glycosylation of this biantennary structure include attachment of core-fucose and bisecting GlcNAc moieties as observed in approximately 90% and 10% of serum-derived IgG respectively [Citation105].
Figure 2. Proposed structures of major N-linked glycans on IgG produced using mammalian cells [Citation105–114]. Relative abundance of glycostructures, when determined by studies, is shown as percentage values. Relative abundance may not amount to 100% depending on the method of analysis employed by each study. Expression systems noted to produce immunogenic N-glycolylneuraminic acid (Neu5Gc) are denoted with #. Experimental methods are described in a matrix configuration [A.B.C.D.E] representing A - culture condition (“S” 10% serum; “SF” serum free); B - glycan release method (“H” hydrazinolysis; “PF” PNGase F; “T” trypsin digest); C - glycan labeling (“2AB” 2-aminobenzamide); D - separation technique (“AEC” high-performance anion-exchange chromatography; “LC” high-pressure liquid chromatography; “NLC” nano-liquid chromatography; “NP” normal-phase HPLC; “RP” reverse-phase HPLC); E - mass spectrometry ionization and detection (“ESI” electrospray ionization; “EM2” electrospray ionization coupled to tandem MS; “MAL” matrix assisted laser desorption/ionization; “−” negative ion mode; “+” positive ion mode). Methods not specified are denoted as “0”.
![Figure 2. Proposed structures of major N-linked glycans on IgG produced using mammalian cells [Citation105–114]. Relative abundance of glycostructures, when determined by studies, is shown as percentage values. Relative abundance may not amount to 100% depending on the method of analysis employed by each study. Expression systems noted to produce immunogenic N-glycolylneuraminic acid (Neu5Gc) are denoted with #. Experimental methods are described in a matrix configuration [A.B.C.D.E] representing A - culture condition (“S” 10% serum; “SF” serum free); B - glycan release method (“H” hydrazinolysis; “PF” PNGase F; “T” trypsin digest); C - glycan labeling (“2AB” 2-aminobenzamide); D - separation technique (“AEC” high-performance anion-exchange chromatography; “LC” high-pressure liquid chromatography; “NLC” nano-liquid chromatography; “NP” normal-phase HPLC; “RP” reverse-phase HPLC); E - mass spectrometry ionization and detection (“ESI” electrospray ionization; “EM2” electrospray ionization coupled to tandem MS; “MAL” matrix assisted laser desorption/ionization; “−” negative ion mode; “+” positive ion mode). Methods not specified are denoted as “0”.](/cms/asset/035d2a50-4182-4cbe-9d98-b23e564fe2cc/ibty_a_1416577_f0002_c.jpg)
The major glycoform expressed in plasma-, CHO-K1-, HEK293-derived IgG are core-fucosylated G1 structures [Citation105–111]. Studies using high-performance liquid chromatography (HPLC) and MS analysis of IgG glycostructures derived from human plasma, CHO and HEK293 cells showed that a majority were core-fucosylated with approximately half of glycostructures containing Gal residues [Citation106,Citation109–111]. On the other hand, Hills et al. reported that IgG derived from NS0 mouse cell lines has only 21% or about 0.3 mols galactose per complex N-linked glycan, although the glycans were also predominantly core-fucosylated biantennary complex-type structures [Citation112]. In the same study, about 29% of the glycostructures identified in NS0-derived IgG were high-mannose type glycans [Citation112], while Mimura et al. reported that high-mannose N-glycans detected in CHO-K1, J558L and HEK293 cells were relatively lower with 3.5%, 5.5% and 1.1% respectively [Citation110].
Terminal sialylation was detected at low amounts of under 5% on IgG derived from human plasma, CHO-K1, J558L and HEK293 cells [Citation105,Citation106,Citation108–110,Citation112,Citation113]. Due to low abundance, sialylated glycoforms of CHO-derived IgG was not clearly distinguishable through HPLC chromatograms and was thus not depicted during the two studies that utilized only liquid chromatography for glycan profiling [Citation108,Citation110]. In contrast, Montesino et al. reported that 11.8% of total N-glycans detected on NS0-derived IgG were sialylated [Citation114], relatively more abundant than IgG derived from human plasma, CHO-K1, J558L and HEK293 cells. We noted that an absence of sialic acid in NS0-derived IgG was reported by Sheeley et al. and Hills et al. [Citation111,Citation112], but this may be explained by the analytical methods employed: Sheeley et al. focused only on analyzing expected biantennary structures, and Hills et al. proposed carbohydrate structures primarily based on HPLC chromatograms which could have omitted minor glycan structures.
Coagulation factor VII (FVII)
Activated FVII is a glycoprotein used to treat hemophilia A/B patients, stimulating the coagulation cascade to alleviate prolonged and frequent bleeding symptoms. More importantly, activated FVII is used as a bypassing agent for hemophilia patients who have developed antibodies against prophylaxis treatments with recombinant FVIII or FIX [Citation115,Citation116]. Post-translational modification of FVII is highly complex which includes γ-carboxylation and β-hydroxylation in addition to N- and O-linked glycosylation [Citation117,Citation118]. To ensure proper expression of the glycan profile, recombinant FVII is produced in mammalian cells.
N-glycosylation of FVII occurs primarily on the light chain at N145 and the heavy chain at N322 (). Characterization of N-glycans by reverse-phase HPLC and high pH anion exchange chromatography-pulse amperometric detection (HPAEC-PAD) showed that plasma-derived FVII were predominantly complex-type bi- and tri-antennary structures without core-fucosylation [Citation119,Citation120]. The primary N-linked glycostructure on CHO- and BHK-derived FVII was reported to be complex-type biantennary di-sialylated core-fucosylated structures [Citation119–121], while HEK293-derived FVII was characterized to have the most heterogeneous N-linked glycans of about 20 different glycostructures, the main glycostructure being an agalacto-type biantennary core-fucosylated glycan with variable (0 to 3) Lex fucosylation on its antennae [Citation120]. Comparing the glycosylation profile across different cell lines, Bohm et al. showed that plasma-derived FVII contained the highest degree of terminal sialylation, followed by CHO-, BHK- and HEK293-derived FVII at 4.06, 3.14, 2.96 and 0.57 sialic acids per molecule of FVII, respectively [Citation120]. Liquid Chromatography (LC)-MS analysis of O-linked glycans showed that plasma-, CHO- and HEK293-derived FVII had a 55–70% xylose content on the S52 O-glycosylation site, while BHK-derived FVII had lower xylose content at 21% on the same site [Citation120].
Figure 3. Proposed structures of major N-linked glycans on FVII produced using mammalian cells [Citation119–121]. Relative abundance of glycostructures, when determined by studies, is shown as percentage values. Relative abundance may not amount to 100% depending on the method of analysis employed by each study. Expression systems noted to produce immunogenic N-glycolylneuraminic acid (Neu5Gc) are denoted with #. Experimental methods are described in a matrix configuration [A.B.C.D.E] representing A - culture condition (“S” 2–10% serum; “SF” serum free); B - glycan release method (“AN” endoproteinase AspN; “H” hydrazinolysis; “PF” PNGase F; “T” trypsin digest); C - glycan labeling (“2AB” 2-aminobenzamide; “APTS” 8-aminopyrene-1,3,6-trisulfonate); D - separation technique (“AEC” high-performance anion exchange chromatography; “RP” reverse-phase HPLC; “SEC” size exclusion chromatography); E - mass spectrometry ionization and detection (“ESI” electrospray ionization; “MAL” matrix-assisted laser desorption/ionization; “−” negative-ion mode; “+” positive-ion mode). Methods not specified are denoted as “0”.
![Figure 3. Proposed structures of major N-linked glycans on FVII produced using mammalian cells [Citation119–121]. Relative abundance of glycostructures, when determined by studies, is shown as percentage values. Relative abundance may not amount to 100% depending on the method of analysis employed by each study. Expression systems noted to produce immunogenic N-glycolylneuraminic acid (Neu5Gc) are denoted with #. Experimental methods are described in a matrix configuration [A.B.C.D.E] representing A - culture condition (“S” 2–10% serum; “SF” serum free); B - glycan release method (“AN” endoproteinase AspN; “H” hydrazinolysis; “PF” PNGase F; “T” trypsin digest); C - glycan labeling (“2AB” 2-aminobenzamide; “APTS” 8-aminopyrene-1,3,6-trisulfonate); D - separation technique (“AEC” high-performance anion exchange chromatography; “RP” reverse-phase HPLC; “SEC” size exclusion chromatography); E - mass spectrometry ionization and detection (“ESI” electrospray ionization; “MAL” matrix-assisted laser desorption/ionization; “−” negative-ion mode; “+” positive-ion mode). Methods not specified are denoted as “0”.](/cms/asset/579d9b70-6322-4fa4-a0c6-a28cc8079d5c/ibty_a_1416577_f0003_c.jpg)
Erythropoietin (EPO)
EPO is a hematopoietic hormone that regulates the proliferation and differentiation of erythroid precursor cells [Citation122]. As such, recombinant EPO has been employed for the treatment of anemia resulting from conditions such as chronic renal failure and AIDS [Citation123,Citation124]. Native EPO is N-linked glycosylated at sites N24, N38 and N83 and is primarily decorated with tetra-antennary complex-type glycostructures () [Citation125,Citation126]. Similarly, glycan profiles of recombinant EPO expressed in BHK-21, CHO and HT-1080 cells were predominantly tetra-antennary accounting for a relative abundance of 60–80% of total glycostructures observed [Citation127–130]. One study by Park et al., however, reports that the primary glycan structure observed in recombinant EPO derived from CHO cells was a bi-antennary complex-type glycan [Citation131]. We note that disparities for this study may be due to determining a relative abundance of glycostructures from the area of MS peaks instead of quantitative HPLC peaks as analyzed in other studies.
Figure 4. Proposed structures of major N-linked glycans on EPO produced using mammalian cells [Citation126–131]. Relative abundance of glycostructures, when determined by studies, is shown as percentage values. Relative abundance may not amount to 100% depending on the method of analysis employed by each study. Studies that analyzed glycoprotein sialylation separately are denoted with *. Expression systems noted to produce immunogenic N-glycolylneuraminic acid (Neu5Gc) are denoted with #. Experimental methods are described in a matrix configuration [A.B.C.D.E] representing A – culture condition (“S” 10% serum; “SF” for serum free); B – glycan release method (“H” hydrazinolysis; “PF” PNGase F); C – glycan labeling (“2AB” 2-aminobenzamide); D – separation technique (“AEC” high-performance anion-exchange chromatography; “LEC” lectin affinity chromatography; “RP” reverse-phase HPLC; “SPE” solid-phase extraction; “WAX” weak anion exchange chromatography); E - mass spectrometry ionization and detection (“ESI” electrospray ionization; “FAB” fast atom bombardment; “MAL” matrix assisted laser desorption/ionization; “−“ negative-ion mode; “+” positive-ion mode). Methods not specified are denoted as “0”.
![Figure 4. Proposed structures of major N-linked glycans on EPO produced using mammalian cells [Citation126–131]. Relative abundance of glycostructures, when determined by studies, is shown as percentage values. Relative abundance may not amount to 100% depending on the method of analysis employed by each study. Studies that analyzed glycoprotein sialylation separately are denoted with *. Expression systems noted to produce immunogenic N-glycolylneuraminic acid (Neu5Gc) are denoted with #. Experimental methods are described in a matrix configuration [A.B.C.D.E] representing A – culture condition (“S” 10% serum; “SF” for serum free); B – glycan release method (“H” hydrazinolysis; “PF” PNGase F); C – glycan labeling (“2AB” 2-aminobenzamide); D – separation technique (“AEC” high-performance anion-exchange chromatography; “LEC” lectin affinity chromatography; “RP” reverse-phase HPLC; “SPE” solid-phase extraction; “WAX” weak anion exchange chromatography); E - mass spectrometry ionization and detection (“ESI” electrospray ionization; “FAB” fast atom bombardment; “MAL” matrix assisted laser desorption/ionization; “−“ negative-ion mode; “+” positive-ion mode). Methods not specified are denoted as “0”.](/cms/asset/9569ab5d-f6f6-497b-8213-fc74043d51ee/ibty_a_1416577_f0004_c.jpg)
Analysis of EPO fucosylation in urinary, BHK, CHO and HT-1080 showed predominant α(1,6) core-fucosylation [Citation127–129,Citation131]. Interestingly, Shahrokh et al. detected the minute presence of a sialyl Lex epitope in HT-1080-derived EPO, not identified previously in other studies [Citation130]. The majority of reported EPO glycostructures are acidic, capped mainly with terminal sialic acids. For example, the study carried out by Sasaki et al. reported that virtually all glycans detected were sialylated [Citation129]. In another study, Llop et al. compared the number of sialic acid residues per EPO protein: IEF analysis of glycoprotein’s pI showed the presence of more acidic bands with an estimated average of 12.02 sialic acid residues per glycoprotein in HT-1080-derived EPO compared to CHO-derived analogs with an estimated average of 11.53 sialic acid residues per glycoprotein molecule [Citation128]. A slightly larger portion of tetra-charged structures were found in CHO-derived EPO as compared to HT-1080-derived EPO: Weak anion-exchange (WAX) HPLC profiling of charge-state glycans showed that recombinant EPO derived from CHO contained 47.85% tetra-charged, 36.85% tri-charged, 12.63% di-charged, 0.76% mono-charged and 1.9% neutral structures, while recombinant EPO derived from HT-1080 contained 44.28% tetra-charged, 37.69% tri-charged, 16.81% di-charged and 1.2% neutral structures [Citation128]. In comparison, Skibeli et al. reported that tetra-charged moieties were not detected in EPO isolated from sera of anemic patients [Citation132]. In our review, we noted one study that reported undetectable levels of sialylation in CHO-derived EPO [Citation131]. While the authors did not specifically address this disparity, we postulate that it may be attributable to the specific CHO cell line used by the study.
Structural analysis showed less antenna variability and a moderately lower number of N-acetyllactosaminyl (LacNAc) repeats in HT-1080 EPO compared to CHO-derived analogs () [Citation128,Citation130]: Shahrokh et al. reported that LacNAc repeats accounted for approximately 30% of total glycans in HT-1080-derived EPO and 32–38% of total glycans in CHO-derived EPO [Citation130], while Sasaki et al. reported that 56.8% of total glycans in CHO-derived EPO contains LacNAc repeats [Citation122]. Analysis of reported structures showed that LacNAc repeats was more abundant in BHK-derived EPO (26.8% total glycans) and HT-1080-derived EPO (30% total glycans) than was observed in native urinary EPO (7.5% total glycans) [Citation122].
Lastly, Llop et al. observed that CHO-derived EPO contained approximately 1.3% of immunogenic Neu5Gc which was devoid in recombinant EPO derived from HT-1080 (128). Traces of Neu5Gc detected in CHO-derived EPO was consistent across commercial erythropoiesis stimulating agents NEORECORMON® (epoetin beta), EXPREX® (epoetin alfa) and ARANESP® (darbepoetin alfa) which contained 1.1%, 1.4% and 1.1% Neu5Gc [Citation130].
Alpha-1 antitrypsin (A1AT)
A1AT is a serine protease inhibitor with three main N-linked glycosylation sites at positions N46, N83 and N247 [Citation133]. The predominant glycans exhibited in native A1AT and are typically di-sialylated biantennary complex-type structures () [Citation134,Citation135]. In a study by Lee et al., N-linked glycan analysis of plasma-derived A1AT showed that the most abundant glycan structure detected was a di-sialylated biantennary glycan at 77.1% [Citation136]. The predominant glycan found in CHO-derived recombinant A1AT was also a di-sialylated complex-type biantennary structure but at a lower percentage of 47.9% [Citation136]. Other studies in human cell lines AGE1.HN, HEK293, HEK293T and PER.C6 showed similar findings where the predominant glycan observed in recombinant A1AT was a biantennary complex-type structure [Citation137–140].
Figure 5. Proposed structures of major N-linked glycans on A1AT produced using mammalian cells [Citation135–140]. Relative abundance of glycostructures, when determined by studies, is shown as percentage values. Relative abundance may not amount to 100% depending on the method of analysis employed by each study. Studies that analyzed glycoprotein sialylation separately are denoted with *. Expression systems noted to produce immunogenic N-glycolylneuraminic acid (Neu5Gc) are denoted with #. Experimental methods are described in a matrix configuration [A.B.C.D.E] representing A – culture condition (“S” 10% serum; “SF” serum free); B – glycan release method (“PF” PNGase F); C – glycan labeling (“2AA” 2-aminobenzoic acid; “2AB” 2-aminobenzamide; “2PA” 2-aminopyridine); D – separation technique (“AEC” high-performance anion exchange chromatography; “NP” normal-phase HPLC; “RP” reverse-phase HPLC); E – mass spectrometry ionization and detection (“ESI” electrospray ionization; “EM2” electrospray ionization coupled to tandem MS; “M2” tandem MS; “MAL” matrix-assisted laser desorption/ionization; “−“ negative-ion mode; “+” positive-ion mode). Methods not specified are denoted as “0”.
![Figure 5. Proposed structures of major N-linked glycans on A1AT produced using mammalian cells [Citation135–140]. Relative abundance of glycostructures, when determined by studies, is shown as percentage values. Relative abundance may not amount to 100% depending on the method of analysis employed by each study. Studies that analyzed glycoprotein sialylation separately are denoted with *. Expression systems noted to produce immunogenic N-glycolylneuraminic acid (Neu5Gc) are denoted with #. Experimental methods are described in a matrix configuration [A.B.C.D.E] representing A – culture condition (“S” 10% serum; “SF” serum free); B – glycan release method (“PF” PNGase F); C – glycan labeling (“2AA” 2-aminobenzoic acid; “2AB” 2-aminobenzamide; “2PA” 2-aminopyridine); D – separation technique (“AEC” high-performance anion exchange chromatography; “NP” normal-phase HPLC; “RP” reverse-phase HPLC); E – mass spectrometry ionization and detection (“ESI” electrospray ionization; “EM2” electrospray ionization coupled to tandem MS; “M2” tandem MS; “MAL” matrix-assisted laser desorption/ionization; “−“ negative-ion mode; “+” positive-ion mode). Methods not specified are denoted as “0”.](/cms/asset/9d373e7f-91de-4411-b01f-e4c78a05b540/ibty_a_1416577_f0005_c.jpg)
Fucose was not detected in the predominant glycan in plasma-derived A1AT unlike in CHO- and human cell-derived A1AT which exhibited varying degrees of fucosylation. Total fucosylation in plasma-derived A1AT was minimal, present only in 10.2% of A1AT glycostructures as reported by Lee et al. [Citation136], while recombinant A1AT produced in CHO and human cell lines predominantly contained core α(1,6) fucose and to a certain degree, Lex fucose [Citation136–138,Citation140]. Interestingly, we observed that Lex fucosylation was expressed the most in AGE1.HN-derived A1AT at about 48.5% compared to plasma-derived A1AT (<10%) and A1AT derived from other cell lines [Citation135,Citation136,Citation138–140]. Amongst the cell lines assessed, the degree of sialylation in PER.C6-derived A1AT (96.7–99% sialylated) was most similar to plasma-derived A1AT (99.2% sialylated) [Citation136,Citation140]. The degree of sialylation in recombinant A1AT expressed in CHO, AGE1.HN and HEK293 cells were moderately lower at about 90% of total glycostructures [Citation136–138]. The level of sialylation in HEK293-derived A1AT was not depicted in one study by Lusch et al. as samples were desialylated prior to MS analysis, although a supplementary analysis showed presence of sialic acid in about 90% of total glycan structures [Citation139].
Notably, CHO-derived A1AT was found to contain both Neu5Ac (98.7%) and traces of nonhuman Neu5Gc at 1.3% of total sialic acids [Citation136]. Sialic acids found in CHO-derived A1AT were exclusively α(2,3)-linked in contrast to the native and human cell line-derived A1AT which contained both α(2,3)-linked and α(2,6)-linked sialic acids [Citation136].
Implication of selecting a suitable host cell line
Glycosylation plays an important role in the pharmacokinetics and immunogenicity of recombinant biotherapeutics. From the case studies presented above, it can be observed that protein glycosylation is significantly dependent on the host-cell type. This should form part of the considerations for the choice of a host cell line for the manufacture of recombinant biotherapeutics, in addition to production yield and cost, scalability, and regulatory acceptance. Given the role of glycans, this consideration will contribute to the functional similarity or enhancement of the recombinant product when compared to the native protein.
Due to a large degree of familiarity, CHO cells have been the predominant choice for the production of many recombinant proteins. This choice is augmented by several glycoengineering strategies to humanize CHO cells and similarly, other nonhuman cell lines to produce recombinant proteins with human-compatible glycosylation [Citation141–145]. An alternative strategy would be to utilize human host cells thus circumventing the risk for synthesizing antigenic glycans. This measure is especially important for the production of therapeutics as these recombinant proteins are administered to nonhealthy individuals, sometimes at high doses and over long periods which may increase the incidence of an immunogenic response. With the importance of glycosylation on the physical and functional properties of recombinant proteins, continued efforts in glycobiology will contribute to more informed decisions on biotherapeutic manufacturing, product quality and efficacy.
Disclosure statement
No potential conflict of interest was reported by the authors.
Additional information
Funding
References
- Apweiler R, Hermjakob H, Sharon N. On the frequency of protein glycosylation, as deduced from analysis of the SWISS-PROT database. Biochim Biophys Acta. 1999;1473:4–8.
- Yan A, Lennarz WJ. Unraveling the mechanism of protein N-glycosylation. J Biol Chem. 2005;280:3121–3124.
- Wong CH. Protein glycosylation: new challenges and opportunities. J Org Chem. 2005;70:4219–4225.
- Brooks SA. Appropriate glycosylation of recombinant proteins for human use: implications of choice of expression system. Mol Biotechnol. 2004;28:241–255.
- Walsh G. Biopharmaceutical benchmarks 2010. Nat Biotechnol. 2010;28:917–924.
- Ferrara C, Stuart F, Sondermann P, et al. The carbohydrate at FcgammaRIIIa Asn-162. An element required for high affinity binding to non-fucosylated IgG glycoforms. J Biol Chem. 2006;281:5032–5036.
- Matsumiya S, Yamaguchi Y, Saito J, et al. Structural comparison of fucosylated and nonfucosylated Fc fragments of human immunoglobulin G1. J Mol Biol. [Research Support, Non-U.S. Gov't]. 2007;368:767–779.
- Iida S, Misaka H, Inoue M, et al. Nonfucosylated therapeutic IgG1 antibody can evade the inhibitory effect of serum immunoglobulin G on antibody-dependent cellular cytotoxicity through its high binding to FcgammaRIIIa. Clin Cancer Res. 2006;12:2879–2887.
- Shields RL, Lai J, Keck R, et al. Lack of fucose on human IgG1 N-linked oligosaccharide improves binding to human Fcgamma RIII and antibody-dependent cellular toxicity. J Biol Chem. 2002;277:26733–26740.
- Shinkawa T, Nakamura K, Yamane N, et al. The absence of fucose but not the presence of galactose or bisecting N-acetylglucosamine of human IgG1 complex-type oligosaccharides shows the critical role of enhancing antibody-dependent cellular cytotoxicity. J Biol Chem. 2003;278:3466–3473.
- Kanda Y, Yamada T, Mori K, et al. Comparison of biological activity among nonfucosylated therapeutic IgG1 antibodies with three different N-linked Fc oligosaccharides: the high-mannose, hybrid, and complex types. Glycobiology. 2007;17:104–118.
- Yu M, Brown D, Reed C, et al. Production, characterization, and pharmacokinetic properties of antibodies with N-linked mannose-5 glycans. MAbs. 2012;4:475–487.
- Goetze AM, Liu YD, Zhang Z, et al. High-mannose glycans on the Fc region of therapeutic IgG antibodies increase serum clearance in humans. Glycobiology. 2011;21:949–959.
- Alessandri L, Ouellette D, Acquah A, et al. Increased serum clearance of oligomannose species present on a human IgG1 molecule. Mabs. 2012;4:509–520.
- Arnold JN, Wallis R, Willis AC, et al. Interaction of mannan binding lectin with alpha2 macroglobulin via exposed oligomannose glycans: a conserved feature of the thiol ester protein family? J Biol Chem. 2006;281:6955–6963.
- Sinclair AM, Elliott S. Glycoengineering: the effect of glycosylation on the properties of therapeutic proteins. J Pharm Sci. 2005;94:1626–1635.
- Goldwasser E, Kung CK, Eliason J. On the mechanism of erythropoietin-induced differentiation. 13. The role of sialic acid in erythropoietin action. J Biol Chem. 1974;249:4202–4206.
- Narhi LO, Arakawa T, Aoki K, et al. Asn to Lys mutations at three sites which are N-glycosylated in the mammalian protein decrease the aggregation of Escherichia coli-derived erythropoietin. Protein Eng. 2001;14:135–140.
- Chiu MH, Tamura T, Wadhwa MS, et al. In vivo targeting function of N-linked oligosaccharides with terminating galactose and N-acetylgalactosamine residues. J Biol Chem. 1994;269:16195–16202.
- Schlenzka W, Shaw L, Kelm S, et al. CMP-N-acetylneuraminic acid hydroxylase: the first cytosolic Rieske iron-sulphur protein to be described in Eukarya. FEBS Lett. 1996;385:197–200.
- Chou HH, Takematsu H, Diaz S, et al. A mutation in human CMP-sialic acid hydroxylase occurred after the Homo-Pan divergence. Proc Natl Acad Sci USA. 1998;95:11751–11756.
- Zhu A, Hurst R. Anti-N-glycolylneuraminic acid antibodies identified in healthy human serum. Xenotransplantation. 2002;9:376–381.
- Tangvoranuntakul P, Gagneux P, Diaz S, et al. Human uptake and incorporation of an immunogenic nonhuman dietary sialic acid. Proc Natl Acad Sci USA. 2003;100:12045–12050.
- Samraj AN, Pearce OM, Laubli H, et al. A red meat-derived glycan promotes inflammation and cancer progression. Proc Natl Acad Sci USA. 2015;112:542–547.
- Nguyen DH, Tangvoranuntakul P, Varki A. Effects of natural human antibodies against a nonhuman sialic acid that metabolically incorporates into activated and malignant immune cells. J Immunol. 2005;175:228–236.
- Ghaderi D, Taylor RE, Padler-Karavani V, et al. Implications of the presence of N-glycolylneuraminic acid in recombinant therapeutic glycoproteins. Nat Biotechnol. 2010;28:863–867.
- Hedlund M, Padler-Karavani V, Varki NM, et al. Evidence for a human-specific mechanism for diet and antibody-mediated inflammation in carcinoma progression. Proc Natl Acad Sci USA. 2008;105:18936–18941.
- Pham T, Gregg CJ, Karp F, et al. Evidence for a novel human-specific xeno-auto-antibody response against vascular endothelium. Blood. 2009;114:5225–5235.
- He X, Galpin JD, Tropak MB, et al. Production of active human glucocerebrosidase in seeds of Arabidopsis thaliana complex-glycan-deficient (cgl) plants. Glycobiology. 2012;22:492–503.
- Limkul J, Iizuka S, Sato Y, et al. The production of human glucocerebrosidase in glyco-engineered Nicotiana benthamiana plants. Plant Biotechnol J. 2016;14:1682–1694.
- Strasser R, Stadlmann J, Schahs M, et al. Generation of glyco-engineered Nicotiana benthamiana for the production of monoclonal antibodies with a homogeneous human-like N-glycan structure. Plant Biotechnol J. 2008;6:392–402.
- Mercx S, Smargiasso N, Chaumont F, et al. Inactivation of the β(1,2)-xylosyltransferase and the α(1,3)-fucosyltransferase genes in Nicotiana tabacum BY-2 Cells by a multiplex CRISPR/Cas9 strategy results in glycoproteins without plant-specific glycans. Front Plant Sci. 2017;8:403.
- Hanania U, Ariel T, Tekoah Y, et al. Establishment of a tobacco BY2 cell line devoid of plant-specific xylose and fucose as a platform for the production of biotherapeutic proteins. Plant Biotechnol J. 2017;15:1120–1129.
- Curtis BR, Swyers J, Divgi A, et al. Thrombocytopenia after second exposure to abciximab is caused by antibodies that recognize abciximab-coated platelets. Blood. 2002;99:2054–2059.
- Suntharalingam G, Perry MR, Ward S, et al. Cytokine storm in a phase 1 trial of the anti-CD28 monoclonal antibody TGN1412. N Engl J Med. 2006;355:1018–1028.
- O'Neil BH, Allen R, Spigel DR, et al. High incidence of cetuximab-related infusion reactions in Tennessee and North Carolina and the association with atopic history. J Clin Oncol. 2007;25:3644–3648.
- Mendelsohn J, Baselga J. Epidermal growth factor receptor targeting in cancer. Semin Oncol. 2006;33:369–385.
- Qian J, Liu T, Yang L, et al. Structural characterization of N-linked oligosaccharides on monoclonal antibody cetuximab by the combination of orthogonal matrix-assisted laser desorption/ionization hybrid quadrupole-quadrupole time-of-flight tandem mass spectrometry and sequential enzymatic digestion. Anal Biochem. 2007;364:8–18.
- Chung CH, Mirakhur B, Chan E, et al. Cetuximab-induced anaphylaxis and IgE specific for galactose-alpha-1,3-galactose. N Engl J Med. 2008;358:1109–1117.
- Peters J, Stoger E. Transgenic crops for the production of recombinant vaccines and anti-microbial antibodies. Hum Vaccin. 2011;7:367–374.
- Graumann K, Premstaller A. Manufacturing of recombinant therapeutic proteins in microbial systems. Biotechnol J. 2006;1:164–186.
- Nothaft H, Szymanski CM. Bacterial protein N-glycosylation: new perspectives and applications. J Biol Chem. 2013;288:6912–6920.
- Bosch D, Castilho A, Loos A, et al. N-glycosylation of plant-produced recombinant proteins. Curr Pharm Des. 2013;19:5503–5512.
- Webster DE, Thomas MC. Post-translational modification of plant-made foreign proteins; glycosylation and beyond. Biotechnol Adv. 2012;30:410–418.
- Tanner W, Lehle L. Protein glycosylation in yeast. Biochim Biophys Acta. 1987;906:81–99.
- Gomord V, Fitchette AC, Menu-Bouaouiche L, et al. Plant-specific glycosylation patterns in the context of therapeutic protein production. Plant. Biotechnol J. 2010;8:564–587.
- Shi X, Jarvis DL. Protein N-glycosylation in the baculovirus-insect cell system. Curr Drug Targets. 2007;8:1116–1125.
- Anyaogu DC, Mortensen UH. Manipulating the glycosylation pathway in bacterial and lower eukaryotes for production of therapeutic proteins. Curr Opin Biotechnol. 2015;36:122–128.
- Naegeli A, Aebi M. Current Approaches to Engineering N-Linked Protein Glycosylation in Bacteria. Methods Mol Biol. 2015;1321:3–16.
- Hamilton SR, Zha D. Progress in yeast glycosylation engineering. Methods Mol Biol. 2015;1321:73–90.
- Wurm FM. Production of recombinant protein therapeutics in cultivated mammalian cells. Nat Biotechnol. 2004;22:1393–1398.
- Hacker DL, De Jesus M, Wurm FM. 25 years of recombinant proteins from reactor-grown cells - where do we go from here? Biotechnol Adv. 2009;27:1023–1027.
- Kim JY, Kim YG, Lee GM. CHO cells in biotechnology for production of recombinant proteins: current state and further potential. Appl Microbiol Biotechnol. 2012;93:917–930.
- Butler M, Spearman M. The choice of mammalian cell host and possibilities for glycosylation engineering. Curr Opin Biotechnol. 2014;30:107–112.
- Durocher Y, Butler M. Expression systems for therapeutic glycoprotein production. Curr Opin Biotechnol. 2009;20:700–707.
- Ghaderi D, Zhang M, Hurtado-Ziola N, et al. Production platforms for biotherapeutic glycoproteins. Occurrence, impact, and challenges of non-human sialylation. Biotechnol Genet Eng Rev. 2012;28:147–175.
- Lai T, Yang Y, Ng SK. Advances in mammalian cell line development technologies for recombinant protein production. Pharmaceuticals (Basel). 2013;6:579–603.
- Berting A, Farcet MR, Kreil TR. Virus susceptibility of Chinese hamster ovary (CHO) cells and detection of viral contaminations by adventitious agent testing. Biotechnol Bioeng. 2010;106:598–607.
- Xu X, Nagarajan H, Lewis NE, et al. The genomic sequence of the Chinese hamster ovary (CHO)-K1 cell line. Nat Biotechnol. 2011;29:735–741.
- Campbell C, Stanley P. A dominant mutation to ricin resistance in Chinese hamster ovary cells induces UDP-GlcNAc:glycopeptide beta-4-N-acetylglucosaminyltransferase III activity. J Biol Chem. 1984;259:13370–13378.
- Stanley P, Sundaram S, Tang J, et al. Molecular analysis of three gain-of-function CHO mutants that add the bisecting GlcNAc to N-glycans. Glycobiology. 2005;15:43–53.
- Sallustio S, Stanley P. Novel genetic instability associated with a developmentally regulated glycosyltransferase locus in Chinese hamster ovary cells. Somat Cell Mol Genet. 1989;15:387–400.
- Macpherson I, Stoker M. Polyoma transformation of hamster cell clones–an investigation of genetic factors affecting cell competence. Virology. 1962;16:147–151.
- Barnes LM, Bentley CM, Dickson AJ. Advances in animal cell recombinant protein production: GS-NS0 expression system. Cytotechnology. 2000;32:109–123.
- Potter M, Boyce CR. Induction of plasma-cell neoplasms in strain BALB/c mice with mineral oil and mineral oil adjuvants. Nature. 1962;193:1086–1087.
- Galfre G, Milstein C. Preparation of monoclonal antibodies: strategies and procedures. Meth Enzymol. 1981;73:3–46.
- Kohler G, Milstein C. Continuous cultures of fused cells secreting antibody of predefined specificity. Nature. 1975;256:495–497.
- Galili U. Immune response, accommodation, and tolerance to transplantation carbohydrate antigens. Transplantation. 2004;78:1093–1098.
- Varki A. Glycan-based interactions involving vertebrate sialic-acid-recognizing proteins. Nature. 2007;446:1023–1029.
- Muchmore EA, Diaz S, Varki A. A structural difference between the cell surfaces of humans and the great apes. Am J Phys Anthropol. 1998;107:187–198.
- Graham FL, Smiley J, Russell WC, et al. Characteristics of a human cell line transformed by DNA from human adenovirus type 5. J Gen Virol. 1977;36:59–74.
- Louis N, Evelegh C, Graham FL. Cloning and sequencing of the cellular-viral junctions from the human adenovirus type 5 transformed 293 cell line. Virology. 1997;233:423–429.
- Henry O, Jolicoeur M, Kamen A. Unraveling the metabolism of HEK-293 cells using lactate isotopomer analysis. Bioprocess Biosyst Eng. 2011;34:263–273.
- Yamaguchi K, Itoh K, Ohnishi N, et al. Engineered long terminal repeats of retroviral vectors enhance transgene expression in hepatocytes in vitro and in vivo. Mol Ther. 2003;8:796–803.
- Berkner KL. Expression of recombinant vitamin K-dependent proteins in mammalian cells: factors IX and VII. Meth Enzymol. 1993;222:450–477.
- Vink T, Oudshoorn-Dickmann M, Roza M, et al. A simple, robust and highly efficient transient expression system for producing antibodies. Methods. 2014;65:5–10.
- Pear WS, Nolan GP, Scott ML, et al. Production of high-titer helper-free retroviruses by transient transfection. Proc Natl Acad Sci USA. 1993;90:8392–8396.
- Schlaeger EJ, Christensen K. Transient gene expression in mammalian cells grown in serum-free suspension culture. Cytotechnology. 1999;30:71–83.
- McCue J, Kshirsagar R, Selvitelli K, et al. Manufacturing process used to produce long-acting recombinant factor VIII Fc fusion protein. Biologicals. 2015;43:213–219.
- Glaesner W, Vick AM, Millican R, et al. Engineering and characterization of the long-acting glucagon-like peptide-1 analogue LY2189265, an Fc fusion protein. Diabetes Metab Res Rev. 2010;26:287–296.
- Havenga MJ, Holterman L, Melis I, et al. Serum-free transient protein production system based on adenoviral vector and PER.C6 technology: high yield and preserved bioactivity. Biotechnol Bioeng. 2008;100:273–283.
- Jones D, Kroos N, Anema R, et al. High-level expression of recombinant IgG in the human cell line per.c6. Biotechnol Prog. 2003;19:163–168.
- Schiedner G, Hertel S, Bialek C, et al. Efficient and reproducible generation of high-expressing, stable human cell lines without need for antibiotic selection. BMC Biotechnol. 2008;8:13.
- Niklas J, Schrader E, Sandig V, et al. Quantitative characterization of metabolism and metabolic shifts during growth of the new human cell line AGE1.HN using time resolved metabolic flux analysis. Bioprocess Biosyst Eng. 2011;34:533–545.
- Priesnitz C, Niklas J, Rose T, et al. Metabolic flux rearrangement in the amino acid metabolism reduces ammonia stress in the alpha1-antitrypsin producing human AGE1.HN cell line. Metab Eng. 2012;14:128–137.
- Cho MS, Yee H, Brown C, et al. Versatile expression system for rapid and stable production of recombinant proteins. Biotechnol Prog. 2003;19:229–232.
- Picanco-Castro V, Biaggio RT, Cova DT, et al. Production of recombinant therapeutic proteins in human cells: current achievements and future perspectives. Protein. 2013;20:1373–1381.
- Mei B, Chen Y, Chen J, et al. Expression of human coagulation factor VIII in a human hybrid cell line, HKB11. Mol Biotechnol. 2006;34:165–178.
- Rasheed S, Nelson-Rees WA, Toth EM, et al. Characterization of a newly derived human sarcoma cell line (HT-1080). Cancer. 1974;33:1027–1033.
- Moran N. Shire's replacement enzymes validate gene activation. Nat Biotechnol. 2010;28:1139–1140.
- Mulligan MS, Warner RL, Rittershaus CW, et al. Endothelial targeting and enhanced antiinflammatory effects of complement inhibitors possessing sialyl Lewisx moieties. J Immunol. 1999;162:4952–4959.
- Rodrigues AF, Soares HR, Guerreiro MR, et al. Viral vaccines and their manufacturing cell substrates: New trends and designs in modern vaccinology. Biotechnol J. 2015;10:1329–1344.
- Dumont J, Euwart D, Mei B, et al. Human cell lines for biopharmaceutical manufacturing: history, status, and future perspectives. Crit Rev Biotechnol. 2016;36:1110–1122.
- Tsao YS, Condon R, Schaefer E, et al. Development and improvement of a serum-free suspension process for the production of recombinant adenoviral vectors using HEK293 cells. Cytotechnology. 2001;37:189–198.
- Ye GJ, Scotti MM, Thomas DL, et al. Herpes simplex virus clearance during purification of a recombinant adeno-associated virus serotype 1 vector. Hum Gene Ther Clin Dev. 2014;25:212–217.
- Zeck A, Pohlentz G, Schlothauer T, et al. Cell type-specific and site directed N-glycosylation pattern of FcgammaRIIIa. J Proteome Res. 2011;10:3031–3039.
- Croset A, Delafosse L, Gaudry JP, et al. Differences in the glycosylation of recombinant proteins expressed in HEK and CHO cells. J Biotechnol. 2012;161:336–348.
- Go EP, Liao HX, Alam SM, et al. Characterization of host-cell line specific glycosylation profiles of early transmitted/founder HIV-1 gp120 envelope proteins. J Proteome Res. 2013;12:1223–1234.
- Uchida Y, Tsukada Y, Sugimori T. Enzymatic properties of neuraminidases from Arthrobacter ureafaciens. J Biochem. 1979;86:1573–1585.
- Bardor M, Nguyen DH, Diaz S, et al. Mechanism of uptake and incorporation of the non-human sialic acid N-glycolylneuraminic acid into human cells. J Biol Chem. 2005;280:4228–4237.
- Nie H, Li Y, Sun XL. Recent advances in sialic acid-focused glycomics. J Proteomics. 2012;75:3098–3112.
- Nishikaze T. Sensitive and structure-informative N-glycosylation analysis by MALDI-MS; ionization, fragmentation, and derivatization. Mass Spectrom (Tokyo). 2017;6:A0060.
- Vidarsson G, Dekkers G, Rispens T. IgG subclasses and allotypes: from structure to effector functions. Front Immunol. 2014;5:520.
- Subedi GP, Hanson QM, Barb AW. Restricted motion of the conserved immunoglobulin G1 N-glycan is essential for efficient FcgammaRIIIa binding. Structure. 2014;22:1478–1488.
- Bakovic MP, Selman MH, Hoffmann M, et al. High-throughput IgG Fc N-glycosylation profiling by mass spectrometry of glycopeptides. J Proteome Res. 2013;12:821–831.
- Dekkers G, Plomp R, Koeleman CA, et al. Multi-level glyco-engineering techniques to generate IgG with defined Fc-glycans. Sci Rep. 2016;6:36964.
- Zhang Y, Go EP, Desaire H. Maximizing coverage of glycosylation heterogeneity in MALDI-MS analysis of glycoproteins with up to 27 glycosylation sites. Anal Chem. 2008;80:3144–3158.
- van Berkel PH, Gerritsen J, Perdok G, et al. N-linked glycosylation is an important parameter for optimal selection of cell lines producing biopharmaceutical human IgG. Biotechnol Prog. 2009;25:244–251.
- Chung CY, Wang Q, Yang S, et al. Integrated genome and protein editing swaps alpha-2,6 sialylation for alpha-2,3 sialic acid on recombinant antibodies from CHO. Biotechnol J. 2017;12:1600502.
- Mimura Y, Kelly RM, Unwin L, et al. Enhanced sialylation of a human chimeric IgG1 variant produced in human and rodent cell lines. J Immunol Methods. 2016;428:30–36.
- Sheeley DM, Merrill BM, Taylor LC. Characterization of monoclonal antibody glycosylation: comparison of expression systems and identification of terminal alpha-linked galactose. Anal Biochem. 1997;247:102–110.
- Hills AE, Patel A, Boyd P, et al. Metabolic control of recombinant monoclonal antibody N-glycosylation in GS-NS0 cells. Biotechnol Bioeng. 2001;75:239–251.
- Vestrheim AC, Moen A, Egge-Jacobsen W, et al. Different glycosylation pattern of human IgG1 and IgG3 antibodies isolated from transiently as well as permanently transfected cell lines. Scand J Immunol. 2013;77:419–428.
- Montesino R, Calvo L, Vallin A, et al. Structural characterization of N-linked oligosaccharides on monoclonal antibody Nimotuzumab through process development. Biologicals. 2012;40:288–298.
- Scharrer I. Recombinant factor VIIa for patients with inhibitors to factor VIII or IX or factor VII deficiency. Haemophilia. 1999;5:253–259.
- Ng HJ, Lee LH. Recombinant activated clotting factor VII (rFVIIa) in the treatment of surgical and spontaneous bleeding episodes in hemophilic patients. Vasc Health Risk Manag. 2006;2:433–440.
- Jurlander B, Thim L, Klausen NK, et al. Recombinant activated factor VII (rFVIIa): characterization, manufacturing, and clinical development. Semin Thromb Hemost. 2001;27:373–384.
- Hansson K, Stenflo J. Post-translational modifications in proteins involved in blood coagulation. J Thromb Haemost. 2005;3:2633–2648.
- Fenaille F, Groseil C, Ramon C, et al. Mass spectrometric characterization of N- and O-glycans of plasma-derived coagulation factor VII. Glycoconj J. 2008;25:827–842.
- Bohm E, Seyfried BK, Dockal M, Graninger M, Hasslacher M, Neurath M, Konetschny C, Matthiessen P, Mitterer A, Scheiflinger F. Differences in N-glycosylation of recombinant human coagulation factor VII derived from BHK, CHO, and HEK293 cells. BMC Biotechnol. 2015;15:87.
- Klausen NK, Bayne S, Palm L. Analysis of the site-specific asparagine-linked glycosylation of recombinant human coagulation factor VIIa by glycosidase digestions, liquid chromatography, and mass spectrometry. Mol Biotechnol. 1998;9:195–204.
- Jacobson LO, Goldwasser E, Fried W, et al. Role of the kidney in erythropoiesis. Nature. 1957;179:633–634.
- Adamson JW, Eschbach JW. Treatment of the anemia of chronic renal failure with recombinant human erythropoietin. Annu Rev Med. 1990;41:349–360.
- Meidani M, Rezaei F, Maracy MR, et al. Prevalence, severity, and related factors of anemia in HIV/AIDS patients. J Res Med Sci. 2012;17:138–142.
- Tsuda E, Goto M, Murakami A, et al. Comparative structural study of N-linked oligosaccharides of urinary and recombinant erythropoietins. Biochemistry. 1988;27:5646–5654.
- Takeuchi M, Takasaki S, Miyazaki H, et al. Comparative study of the asparagine-linked sugar chains of human erythropoietins purified from urine and the culture medium of recombinant Chinese hamster ovary cells. J Biol Chem. 1988;263:3657–3663.
- Nimtz M, Martin W, Wray V, et al. Structures of sialylated oligosaccharides of human erythropoietin expressed in recombinant BHK-21 cells. Eur J Biochem. 1993;213:39–56.
- Llop E, Gutierrez-Gallego R, Segura J, et al. Structural analysis of the glycosylation of gene-activated erythropoietin (epoetin delta, Dynepo). Anal Biochem. 2008;383:243–254.
- Sasaki H, Bothner B, Dell A, et al. Carbohydrate structure of erythropoietin expressed in Chinese hamster ovary cells by a human erythropoietin cDNA. J Biol Chem. 1987;262:12059–12076.
- Shahrokh Z, Royle L, Saldova R, et al. Erythropoietin produced in a human cell line (Dynepo) has significant differences in glycosylation compared with erythropoietins produced in CHO cell lines. Mol Pharmaceutics. 2011;8:286–296.
- Park JH, Wang Z, Jeong HJ, et al. Enhancement of recombinant human EPO production and glycosylation in serum-free suspension culture of CHO cells through expression and supplementation of 30Kc19. Appl Microbiol Biotechnol. 2012;96:671–683.
- Skibeli V, Nissen-Lie G, Torjesen P. Sugar profiling proves that human serum erythropoietin differs from recombinant human erythropoietin. Blood. 2001;98:3626–3634.
- Carrell RW, Jeppsson JO, Vaughan L, et al. Human alpha 1-antitrypsin: carbohydrate attachment and sequence homology. FEBS Lett. 1981;135:301–303.
- Mega T, Lujan E, Yoshida A. Studies on the oligosaccharide chains of human alpha 1-protease inhibitor. II. Structure of oligosaccharides. J Biol Chem. 1980;255:4057–4061.
- Kolarich D, Turecek PL, Weber A, et al. Biochemical, molecular characterization, and glycoproteomic analyses of alpha(1)-proteinase inhibitor products used for replacement therapy. Transfusion. 2006;46:1959–1977.
- Lee KJ, Lee SM, Gil JY, et al. N-glycan analysis of human alpha1-antitrypsin produced in Chinese hamster ovary cells. Glycoconj J. 2013;30:537–547.
- Blanchard V, Liu X, Eigel S, et al. N-glycosylation and biological activity of recombinant human alpha1-antitrypsin expressed in a novel human neuronal cell line. Biotechnol Bioeng. 2011;108:2118–2128.
- Rosenlocher J, Sandig G, Kannicht C, et al. Recombinant glycoproteins: the impact of cell lines and culture conditions on the generation of protein species. J Proteomics. 2016;134:85–92.
- Lusch A, Kaup M, Marx U, et al. of alpha 1-antitrypsin neoglycoproteins: the impact of additional N-glycosylation sites on serum half-life. Mol Pharmaceutics. 2013;10:2616–2629.
- Wang Z, Hilder TL, van der Drift K, et al. Structural characterization of recombinant alpha-1-antitrypsin expressed in a human cell line. Anal Biochem. 2013;437:20–28.
- Yang Z, Wang S, Halim A, et al. Engineered CHO cells for production of diverse, homogeneous glycoproteins. Nat Biotechnol. 2015;33:842–844.
- Meuris L, Santens F, Elson G, et al. GlycoDelete engineering of mammalian cells simplifies N-glycosylation of recombinant proteins. Nat Biotechnol. 2014;32:485–489.
- Jacobs PP, Geysens S, Vervecken W, et al. Engineering complex-type N-glycosylation in Pichia pastoris using GlycoSwitch technology. Nat Protoc. 2009;4:58–70.
- Kallolimath S, Castilho A, Strasser R, et al. Engineering of complex protein sialylation in plants. Proc Natl Acad Sci U S A. 2016;113:9498–9503.
- Mabashi-Asazuma H, Kuo CW, Khoo KH, Jarvis DL. Modifying an Insect Cell N-Glycan Processing Pathway Using CRISPR-Cas Technology. ACS Chem Biol. 2015;10:2199–2208.
- Narasimhan S. Control of glycoprotein synthesis. UDP-GlcNAc:glycopeptide beta 4-N-acetylglucosaminyltransferase III, an enzyme in hen oviduct which adds GlcNAc in beta 1-4 linkage to the beta-linked mannose of the trimannosyl core of N-glycosyl oligosaccharides. J Biol Chem. 1982;257:10235–10242.
- Sburlati AR, Umana P, Prati EG, et al. Synthesis of bisected glycoforms of recombinant IFN-beta by overexpression of beta-1,4-N-acetylglucosaminyltransferase III in Chinese hamster ovary cells. Biotechnol Prog. 1998;14:189–192.
- Ihara H, Ikeda Y, Koyota S, et al. A catalytically inactive beta 1,4-N-acetylglucosaminyltransferase III (GnT-III) behaves as a dominant negative GnT-III inhibitor. Eur J Biochem. 2002;269:193–201.
- Kukowska-Latallo JF, Larsen RD, Nair RP, et al. A cloned human cDNA determines expression of a mouse stage-specific embryonic antigen and the Lewis blood group alpha(1,3/1,4)fucosyltransferase. Genes Dev. 1990;4:1288–1303.
- Orntoft TF, Vestergaard EM, Holmes E, et al. Influence of Lewis alpha1-3/4-L-fucosyltransferase (FUT3) gene mutations on enzyme activity, erythrocyte phenotyping, and circulating tumor marker sialyl-Lewis a levels. J Biol Chem. 1996;271:32260–32268.
- Zhang A, Potvin B, Zaiman A, et al. The gain-of-function Chinese hamster ovary mutant LEC11B expresses one of two Chinese hamster FUT6 genes due to the loss of a negative regulatory factor. J Biol Chem. 1999;274:10439–10450.
- Galili U, Swanson K. Gene sequences suggest inactivation of alpha-1,3-galactosyltransferase in catarrhines after the divergence of apes from monkeys. Proc Natl Acad Sci USA. 1991;88:7401–7404.
- Lanteri M, Giordanengo V, Vidal F, et al. A complete alpha1,3-galactosyltransferase gene is present in the human genome and partially transcribed. Glycobiology. 2002;12:785–792.
- Lee EU, Roth J, Paulson JC. Alteration of terminal glycosylation sequences on N-linked oligosaccharides of Chinese hamster ovary cells by expression of beta-galactoside alpha 2,6-sialyltransferase. J Biol Chem. 1989;264:13848–13855.
- Colley KJ, Lee EU, Adler B, et al. Conversion of a Golgi apparatus sialyltransferase to a secretory protein by replacement of the NH2-terminal signal anchor with a signal peptide. J Biol Chem. 1989;264:17619–17622.
- Hard K, Mekking A, Damm JB, et al. Isolation and structure determination of the intact sialylated N-linked carbohydrate chains of recombinant human follitropin expressed in Chinese hamster ovary cells. Eur J Biochem. 1990;193:263–271.
- Schlenke P, Grabenhorst E, Nimtz M, et al. Construction and characterization of stably transfected BHK-21 cells with human-type sialylation characteristic. Cytotechnology. 1999;30:17–25.
- Muchmore EA, Milewski M, Varki A, et al. Biosynthesis of N-glycolyneuraminic acid. The primary site of hydroxylation of N-acetylneuraminic acid is the cytosolic sugar nucleotide pool. J Biol Chem. 1989;264:20216–20223.
- Irie A, Koyama S, Kozutsumi Y, et al. The molecular basis for the absence of N-glycolylneuraminic acid in humans. J Biol Chem. 1998;273:15866–15871.