
Abstract
Healthcare systems worldwide are struggling to find ways to fund the cost of innovative treatments such as gene therapies, regenerative medicine, and monoclonal antibodies (mAbs). As the world’s best known mAbs are close to facing patent expirations, the biosimilars market is poised to grow with the hope of bringing prices down for cancer treatment and autoimmune disorders, however, this has yet to be realized. The development costs of biosimilars are significantly higher than their generic equivalents due to therapeutic equivalence trials and higher manufacturing costs. It is imperative that academics and relevant companies understand the costs and stages associated with biologics processing. This article brings these costs to the forefront with a focus on biosimilars being developed for Rheumatoid Arthritis (RA). mAbs have remarkably changed the treatment landscape, establishing their superior efficacy over traditional small chemicals. Five blockbuster TNFα mAbs, considered as first line biologics against RA, are either at the end of their patent life or have already expired and manufacturers are seeking to capture a significant portion of that market. Although in principle, market-share should be available, withstanding that the challenges regarding the compliance and regulations are being resolved, particularly with regards to variation in the glycosylation patterns and challenges associated with manufacturing. Glycan variants can significantly affect the quality attributes requiring characterization throughout production. Successful penetration of biologics can drive down prices and this will be a welcome change for patients and the healthcare providers. Herein we review the biologic TNFα inhibitors, which are on the market, in development, and the challenges being faced by biosimilar manufacturers.
Introduction
As a result of substantial growth during the past decade, the biopharmaceutical industry is at the forefront of clinical care, with monoclonal antibodies (mAbs) generating the highest revenue. Therapeutic glycoproteins account for more than two-thirds of recombinant therapeutic proteins on the current market, whilst also seeing the number of approved therapies increasing each year. In future years, patent protections will soon expire (), and subsequently initiate a new wave of development of biosimilars and biobetters [Citation1]. The global biosimilars market is predicted to reach to $35 billion by 2020 [Citation2] while the mAb market is expected to exceed $65 billion by 2023. There are currently 214 approved mAbs with almost around 1000 under development for several indications in various phases of development (). This does look like a daunting number but most of them are withdrawn at phase 1 human clinical trials and only 23% of those, which reach phase 2 trials, make it to the market [Citation3]. The gene and cellular therapeutic approaches are rapidly increasing for phase 1 development. Nevertheless, market access and reimbursement remains the biggest challenge for these while mAbs commercialization routes are relatively well established [Citation4].
Figure 1. Patent expirations for major biologics. Biosimilars are under development for the biologics (originators) which are going off patents. The exclusivity period differs in the US and EU for the same molecules, hence represented separately but color coded to make it easier for the comparison. Brand names, scientific names and the company holding the patent are provided for each.
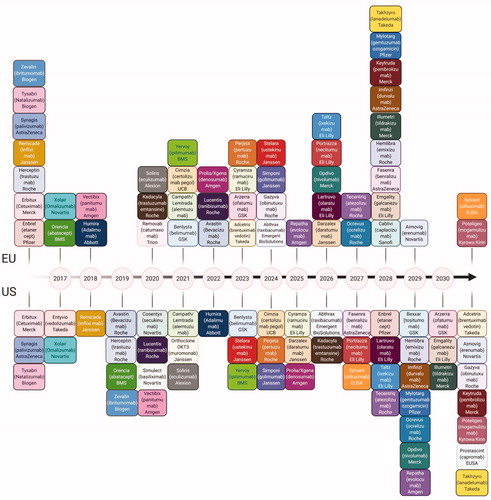
Figure 2. Market Trends in Biopharmaceuticals. (A) Top selling biopharmaceuticals from 2018 globally. (B) As patents for top selling biologics are expiring, an increasing amount of biosimilars and biobetters are reaching developmental milestones. (C) The number of biotherapeutics in phase 1 by category demonstrating the developmental superiority of mAbs in the current biopharmaceutical market. (D) Number of compounds in development according to the therapeutic category.
Nevertheless, the administration of biosimilars in an ageing population is resulting in healthcare payer systems, public and private, to struggle particularly in a post COVID-19 world. There is an urgent need to reduce the drug development and manufacturing costs. In the United States healthcare expenditure was $3.3 trillion in 2016 where $329 billion was spent on prescription drugs [Citation5]. Any form of financial plastering and insurance plans may help in the short term. However, longer-term solutions involve investing in the prevention and lowering of actual drug expenditure. The largest amount incurred by the developed nations is demonstrated with cancer, arthritis, and neurodegenerative disorders, where the treatment of choice for these diseases is primarily expensive biologics. Price competition between off-patent substances has been the driving force to maintain prices in check for “generics” but not so much for “biosimilars.” Therefore, this review focuses on the regulatory and manufacturing challenges faced by biosimilars, and introduces readers to the various stages and the costs involved in developing a biosimilar. It also provides details of the requisite tools and techniques required for the characterization of the biosimilars and gives a detailed account of the biosimilars being developed against anti TNFα.
Over the last two centuries, patents have been the incentive to drive innovation in the pharmaceutical sector. It has allowed the pharmaceutical companies to charge high prices and make enough profits during the exclusivity period that allowed them to keep funding the cost of clinical trials and research for newer targets and better drugs. For buyers, patients and the healthcare systems, the respite used to come at the end of the exclusivity period, when competing manufacturers could sell the same drug, usually at a fraction of the price. For small molecules (chemicals with a fixed molecular formula such as antihistamines, aspirin, codeine etc.), products from competing manufacturers as compared to the innovator are known as “generic” while for biologicals the term used is “biosimilar.” Such price control has been achieved for small molecules because generic manufacturers can produce these drugs at very low prices without enduring extensive clinical trials (). Phase 1 is the first stage of the clinical trials, where approximately 15 − 30 healthy human volunteers are subjected to the drug at different dosages to determine its risk profile. Phase 2 involves up to 200 patients at the optimized dose and finally, phase 3 enrolls up to three thousand patients expanding on effectiveness and drug safety. These clinical trials are the most expensive element of new drug development and any reduction of the phases or curtailing of the patient numbers can result in significant cost savings for the manufacturers. The biosimilar manufacturers cannot avoid clinical trials but aim for an abbreviated pathway while for generic small molecule manufacturers, the requirement is to prove that their chemical is identical to the reference product which can be easily confirmed through mass spectrometry and relatively simple pharmacokinetic studies [Citation6].
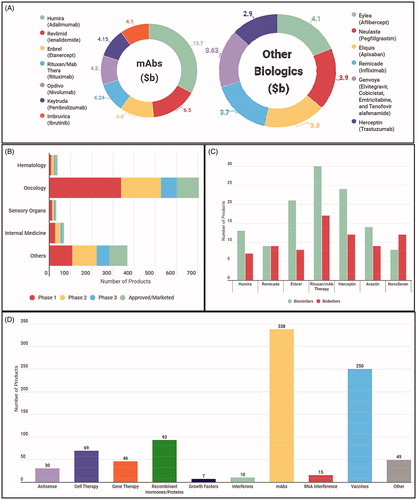
Figure 3. Comparison of Originator and Biosimilar Development Pathways. (A) Development pathway of new originator molecules. This is the likely pathway to be followed by the biobetters as well. (B) Abbreviated pathway followed for the biosimilars. (C) Physicochemical characteristics that require monitoring and characterization, some of them for, the critical quality attributes. (D) Process related impurities that require removal from the final batches. (E) Other factors to consider that may cause little variations hence, extremely important while determining similarity.
The first and foremost challenge for biosimilar manufacturers is due to the inherent nature of biomolecules, which have complex structures that may result in batch-to-batch variations, even in the originator molecules. A fact which is recognized by Biologics Price Competition and the Innovation Act of 2009 in these words: “A ‘biosimilar’ product is a biological product that is ‘highly similar’ to the reference product notwithstanding minor differences in clinically inactive components and with respect to which ‘there are no clinically meaningful differences between the biological product and the reference product in terms of the safety, purity, and potency of the product.’ FDA advises that they “will likely take a two-step approach in reviewing applications that seek to establish that generic biologics are ‘biosimilar’ to a licensed ‘reference’ biological product. First, they will review ‘analytic’ data showing how similar [company] compounds are to an FDA-approved innovator version. Second, the agency will then determine on a case-by-case basis how much animal and clinical data is required for approval [Citation7].” A biosimilar manufacturer can and does show that the primary structure, namely the amino acid sequence of the produced protein is similar to the originator, but it is almost impossible to demonstrate that the folded higher order tertiary or quaternary structure (if multimeric) is similar [Citation6]. The characterization techniques which can be used to probe three dimensional folded structures in a research environment, such as nuclear magnetic resonance and x ray crystallography are not yet robust enough for industrial use. Therefore, a cohort of biophysical parameters are used as a proxy to characterize the structure of the proteins (). Throughout the manufacturing process, the higher order protein structure, should be maintained and if the information could not be reliably obtained then a relevant biological activity assay can lead the way. The World Health Organization (WHO) has published a set of guidelines for the “evaluation of mAbs as similar therapeutic proteins” with the objective to have coherent global approaches [Citation8]. Indeed, variation in the structure may or may not change the clinical significance, however, these protein biologics still need to be closely monitored throughout development [Citation9] The objective, therefore, is to demonstrate that biosimilar should be “highly similar” to the reference product with “no clinically meaningful difference” whilst maintaining that the products are safe, efficient and are of appropriate quality. Although the production process for biosimilars attempts to follow the process pathway of the original drug, including characterization, pre-clinical and clinical testing, the emphasis within these pathways differs () [Citation10]. The development of the originators centers around the clinical trials to evaluate safety and efficacy, as with all new chemical or molecular entities (NCE/NME) (). Whereas, extensive biologic characterization and comparative analysis acts as a focal point for biosimilars, because the therapeutic benefit has already been identified [Citation11]. Therefore, when highly similar, the requirement to re-assess the biosimilars clinical benefit in completion is by-passed as it relies on the originators previous clinical trials [Citation12]. Hence, abbreviating the clinical pathway for biosimilars () and bypassing phase II is justified, as there would be no further knowledge obtained on the drug other than what is already understood for the originator [Citation13,Citation14]. This is a significant saving, both in terms of reducing the costs of expensive clinical trials, but also savings realized from producing smaller biopharmaceutical batches for testing.
The first and foremost challenge for biosimilar manufacturers is due to the inherent nature of biomolecules, which have complex structures that may result in batch-to-batch variations, even in the originator molecules. A fact which is recognized by Biologics Price Competition and the Innovation Act of 2009 in these words: “A ‘biosimilar’ product is a biological product that is ‘highly similar’ to the reference product notwithstanding minor differences in clinically inactive components and with respect to which ‘there are no clinically meaningful differences between the biological product and the reference product in terms of the safety, purity, and potency of the product.’ FDA advises that they “will likely take a two-step approach in reviewing applications that seek to establish that generic biologics are ‘biosimilar’ to a licensed ‘reference’ biological product. First, they will review ‘analytic’ data showing how similar [company] compounds are to an FDA-approved innovator version. Second, the agency will then determine on a case-by-case basis how much animal and clinical data is required for approval [Citation7].” A biosimilar manufacturer can and does show that the primary structure, namely the amino acid sequence of the produced protein is similar to the originator, but it is almost impossible to demonstrate that the folded higher order tertiary or quaternary structure (if multimeric) is similar [Citation6]. The characterization techniques which can be used to probe three dimensional folded structures in a research environment, such as nuclear magnetic resonance and x ray crystallography are not yet robust enough for industrial use. Therefore, a cohort of biophysical parameters are used as a proxy to characterize the structure of the proteins (). Throughout the manufacturing process, the higher order protein structure, should be maintained and if the information could not be reliably obtained then a relevant biological activity assay can lead the way. The World Health Organization (WHO) has published a set of guidelines for the “evaluation of mAbs as similar therapeutic proteins” with the objective to have coherent global approaches [Citation8]. Indeed, variation in the structure may or may not change the clinical significance, however, these protein biologics still need to be closely monitored throughout development [Citation9] The objective, therefore, is to demonstrate that biosimilar should be “highly similar” to the reference product with “no clinically meaningful difference” whilst maintaining that the products are safe, efficient and are of appropriate quality. Although the production process for biosimilars attempts to follow the process pathway of the original drug, including characterization, pre-clinical and clinical testing, the emphasis within these pathways differs () [Citation10]. The development of the originators centers around the clinical trials to evaluate safety and efficacy, as with all new chemical or molecular entities (NCE/NME) (). Whereas, extensive biologic characterization and comparative analysis acts as a focal point for biosimilars, because the therapeutic benefit has already been identified [Citation11]. Therefore, when highly similar, the requirement to re-assess the biosimilars clinical benefit in completion is by-passed as it relies on the originators previous clinical trials [Citation12]. Hence, abbreviating the clinical pathway for biosimilars () and bypassing phase II is justified, as there would be no further knowledge obtained on the drug other than what is already understood for the originator [Citation13,Citation14]. This is a significant saving, both in terms of reducing the costs of expensive clinical trials, but also savings realized from producing smaller biopharmaceutical batches for testing.
Apart from challenges associated with structural confirmation and characterization, manufacturing biologics is further complicated by changes in scale up, process-related impurities and post translational modification of the proteins (PTMs). lists the PTMs where the most significant and the foremost reason for batch to batch variability is the control of the glycosylation process, which changes with the expression system employed, the fermentation and culture conditions and the choice of the growth medium and a myriad of external factors. Sometimes the variation can be seen in the proteins produced by the same cell, a phenomenon known as isoform microheterogeneity. The variation in glycoforms can significantly alter important therapeutic attributes and ultimately the protein quality. Particularly in terms of antibodies, glycosylation can impact stability, solubility and secretion as well as biological activity and antigenicity, hence, determining the overall functional ability of the antibody. Therefore, glycoforms require characterization throughout the production process incurring costs and time [Citation15] and are discussed in detail later in this article. In addition to glycosylation, deamidation and oxidation patterns must be highly similar within testing [Citation16]. All these variations may vary from batch-to-batch, especially when there is more than one originator manufacturer [Citation17]. Therefore, the processes of biosimilar synthesis must be modulated to decrease the variation and the manufacture of a product as similar as possible to the originator in order to reduce efficacy or toxicity concerns. Quantitative mass spectrometry can detect these small changes and assess the extent of the modifications occurring. Alternatively, they can be analyzed using a combination of HPLC, and peptide mapping [Citation18].
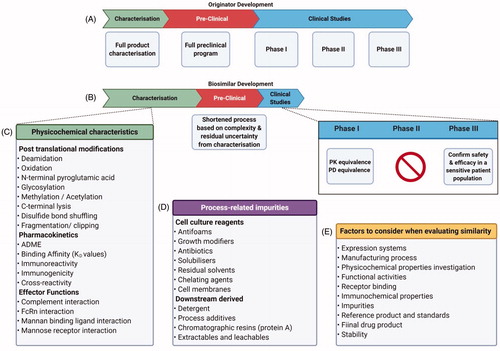
The objective of this review is not the research processes and costs associated with finding an appropriate druggable target, but to elaborate out the chemistry, manufacturing and control (CMC) activities required to develop a robust, reliable and reproducible process to make it safe to administer biologics. The key stages involved in the production of biologics includes upstream process development, downstream process development, formulation development, scale up and bulk drug substance manufacturing and final drug product manufacturing ( and ). Alongside these activities, suitable analytical methods are developed for complete characterization of biologicals at every stage. These methods inform the quality control release specifications for the product and are increasingly developed to determine the manufacturability of the molecules earlier in the process. This preliminary characterization allows manufacturers to select the subset of the physicochemical characteristics () which are relevant to inform Critical Quality Attributes (CQAs). CQAs only entail the parameters that have the direct impact on the efficacy or safety of the product and have clinical significance. CQAs are divided into three tiers according to their clinical relevance. Tier 1 is an equivalence test to assess the analytical similarity between the most clinically relevant attributes of the originator and the biosimilar factor. Tier 2 is for its mild/moderate attributes and is known as the quality range. CQAs in Tier 3 have the lowest risk ranking, where raw data and graphical comparisons are used which are acceptable because of the minimal impacts on the clinical outcomes [Citation19]. Biosimilar manufacturers try to match the originator’s CQA as closely as possible. They have to continue refining until the process is robust enough to keep the manufactured biosimilar within the decided range of CQAs. One of the major bottlenecks for biosimilar manufacturers and CDMOs is to obtain enough quality reference material and Bio-CMC data to formulate the CQAs which were followed by the originator. To obtain this essentially, a biosimilar manufacturer has to conduct their own experiments. During the development of SB2, a biosimilar for infliximab, Samsung Bioepis Co. had to characterize 80 lots of US and EU marketed infliximab from different batches, in order to give them enough breadth to develop a consistent target product profile, which could be followed for further development. During this development, they used 60 different assays to quantify various CQAs [Citation20]. The same company characterized 60 batches (30 from the EU and 30 from the US) of originator etanercept to develop a biosimilar named SB4 [Citation21]. It is noteworthy that there is regional conditionality to the bioequivalence trials. If a manufacturer is intending to sell in a particular area, it needs to source the originator from that area. For example, to assume a biosimilar manufacturer based in the United States of America has developed biosimilar matching of a referenced product in America. After approval they will be able to market in the States but would not be able to sell in Europe. To circumvent the problem, they would need to perform comparability studies from an originator sourced from Europe. These comparability studies are known as bridging studies or trials [Citation6].
Figure 4. CMC activities over an 18-month period for the development of monoclonal antibodies Analytical Methods Development is carried almost throughout the 10 months while upstream process development leads to downstream process development with some important overlap. Formulation development and drug manufacturability become a very important consideration early on, the details of individual steps are entailed in (Created with Biorender.com).
Table 1. Stages and timescales involved in the production of a monoclonal antibody biosimilars.
In pharmaceutical processes, early failure is always better than failing late! There is no doubt that most of the drugs that fail the clinical trials for lack proving efficacy or being too immunogenic, hence a space and race for biobetters exists. However, it can be a nightmare for a company to have their drug on a clinical hold due to a production error. Kozlowski and Swann [Citation22] have made a very interesting contribution in this regard. They looked at 50 failed submissions from various mAb developers and found that the most common hold reasons were fairly rectifiable. For example, a lack of data on the clearance of endogenous virus, appropriate specifications of critical tests, lack of basic molecular characterization (including glycosylation) and only a handful were held due to stability issues or lack of data on the critical drug components.
Historically, the antibody discovery and development of a lead candidate into a drug product was compartmentalized into different departments within pharmaceutical companies and CDMOs. However, this has started to change and CQAs that determine the manufacturability of proteins are being considered at a very early stage. Given the heavy costs associated with the biosimilar development, the definitive goal is to reduce costs and increase efficiency throughout development, whilst providing superior biopharmaceuticals. These efficiencies are sought throughout the multiple stages of production, including clonal selection, cell culture, protein purification and final formulation. Most of the companies therefore, prefer to develop their drug on an established platform for reliability while improving their time to market it. The first step in this process is to have a well characterized cell line. For most Contract Development and Manufacturing Organization (CDMOs) Chinese Hamster Ovary (CHO) cell line is the most popular for its ability to grow in suspension cultures leading to a high cell density, high specific yield and its stability to changes in both pH and oxygen. Lonza has a Potelligent® CHOK1VS® developed through their GS gene expression system® which is capable of generating fucose free antibodies which enhances ADCC functions in the body, Fujifilm Diosynth Biotechnologies has developed SaturnTM mAb solutions which uses their Apollo XTM expression system based on CHO-DG44 derived host cell lines containing an efficient secretory mechanism, Catalent has GPEx® cell line development technology which uses a retrovector technology boosting 100% efficient transduction and combines it with CHOZN® a glutamine synthase (GS) knock out CHO which allows them to produce high titers. Companies have to consider the final yield of the proteins, just a decade ago the concentrations produced were 0.5–1 g/L while now most of the companies boast upto 10 g/L. Nevertheless, final applications may dictate which cell line you select. For example, Potelligent® CHOK1VS® has a considerably lower yield just above 2 g/L but if you are designing a molecule which has to enhance antibody dependent cell mediated cytotoxicity (ADCC) then it is a much better choice. There have been many attempts on producing whole mAbs in cell lines other than CHO, but none of them has had the commercial success CHO has seen. The goal is to overcome some of CHO’s limitations such as the production of α-gal and N-glycocylneuraminic acid, which could lead to increase immunogenicity [Citation23]. An admirable collaboration has been formed recently between Prof. Chris Love’s group at MIT and leading pharmaceutical companies including Biogen, Pfizer, Genentech and Sanofi to invest in looking at alternatives to CHO [Citation24] where they are considering reviving research into the yeast species, Pichia pastoris. It will be an interesting space to observe in the coming years because recent gains made in synthetic biology will helping all the strains to overcome their problems. In addition to cell line engineering, novel ways are being developed to better screen and produce protein fragments. Iontas, a small company based in Cambridge UK, is pioneering the use of mammalian display technology which not only helps with antibody discovery and optimization, but also aids with manufacturability early in the process. Therapeutic antibodies must be able to withstand a wide variety of stresses during manufacture including changes in pH, temperature, ionic strength, mechanical strain and sheer forces, exposure to air-water interfaces and high protein concentrations. During development certain challenges which are sequence related can be addressed by removal or modifying sites which are liable to deamidation or glycosylation. However, the biggest challenge in mAb production is to optimize pharmacokinetic profile while formulating at high concentrations because of the increase in nonspecific self-association of proteins. This is a growing problem exacerbated by the fact that companies are selecting sub-cutaneous administration as a favored route to deliver biologics. This requires concentrated antibody formulations (>50 mg/mL) with the aim to deliver not more than 2 ml in a single injection. Dobson et al. have demonstrated a good strategy to eliminate the amino acids responsible for self-association using Hydrogen/Deuterium Mass Spectrometry (HDX-MS) and cross linking MS (XL-MS) combined with the negative stain EM and in silico aggregation prediction tools [Citation25]. They showed rescue of a mAb against nerve growth factor (MEDI1912), which had good biophysical characteristics but was being marred by self-association leading to precipitation, increased viscosity and short half-life in vivo. Mammalian display allows the selection of all clones with good biophysical characteristics applying to an aggregation screen early on.
It is confounding that the drug development costs are always discussed from the stand point of new drugs, but it is becoming increasingly important to look at the costs associated with biosimilars development on a stand-alone basis or in a combination with small molecules to be marketed as combination therapy. It is imperative to understand the manufacturing and process development costs to appreciate the pressure faced by a biosimilar manufacturer. The costs associated with drug development have been debated in numerous research and commercial publications from average costs ranging from $1.3 billion to as high as $2.8 billion [Citation26]. This is over the period of 10–15 years and the majority of the costs are incurred during extensive clinical trials. On the other hand, having abbreviated pathways decreases the cost of developing a biosimilar to US$200 million and may take upto 5 years to complete. Farid et al. [Citation27] has recently published a very interesting paper where they have concluded that process development and manufacturing costs for biopharmaceuticals to be upto 13–17% of the overall drug development costs. Modeling on the basis of a 12% clinical success rate, they have concluded that a pharmaceutical company needs to allocate $60 million toward manufacturing pre-clinical to phase 2 materials and $70 million for phase 3 regulatory review material preparation. Obviously, projections such as these are dependent upon the number of compounds being developed, their likely success, quantity of the materials being prepared etc. Even by conservative estimates, it would take around 18 months and £7 million to manufacture enough product i.e. a clinical candidate to be taken first into human clinical trials ( and ). The rest of the clinical trials expenditure is on top of it. Several companies are simultaneously competing for the same target, therefore, the time spent developing the drug becomes imperative. Coming second to the market after originator (or becoming a first biologics) results in a significant market share and for some developers that is the only hope to obtain a return on their investment. Hence, CDMOs take pride in their faster times to the clinic. Pantheon, now part of Thermo Fisher Scientific and has a Quick to ClinicTM program for first in human biologic clinical trials boasting only 14 months to develop the drug. and list the details of the steps entailed from start to finish with likely timescales to completionFootnote1. The table is curated from the perspectives of the development of monoclonal antibodies, for other biologics that include the use of human cell lines, and developers are required to follow ICH Q5D. One needs to provide the detailed cultivation history alongside the ethnic and geographical origin of the cell line. The need of increased investment also gives an opportunity for biosimilar manufacturers to improve upon the innovator molecule. They do not need to invest in finding and approving new molecules but instead can improve existing characteristics of the innovator molecule and such drugs are known as “biobetters.” To demonstrate the increased efficacy and to get exclusivity, often biobetters have to follow the regulatory pathway closer to the originator and a phase 2 trial may not be exempt. One can argue that Humira (adalimumab) was actually a biobetter for Remicade (infliximab) because it essentially reduced the immunogenicity by humanizing the antibody [Citation28]. Nevertheless, some researchers believe that biobetters is a term more suited to a molecule with minor improvements such as one which would change the pharmacokinetics/pharmacodynamic properties of the same molecule. The most common modification is the addition of polyethylene glycol (PEG) to the Fc region of the mAb. This prolongs renal clearance thus giving additional time for the mAb to stay within the blood, and hence a better chance to bind to its targets. PEG addition also stops antibodies from crossing placenta thus making them safe for pregnant women. Further modifications include modifying a particular mAb with a known toxic agent to produce a drug conjugate, with an example being modification of trastuzumab to ado-trastuzumab emantisine to further make the mAb effective by adding a microtubule assembly inhibitor [Citation29]. Another important modification is to modify a mAb with a radioisotope or a comparative detection label for use in tumor identification.
Currently, there are numerous biosimilars approved for the European market with many more awaiting approvals, forming the largest growth segment of the global pharmaceutical market. Adding to this eagerness is the patent fall, which is about to effect some leading biopharmaceuticals, particularly monoclonal antibodies, as shown in and . Their leading sales figure has pushed competitors to produce biosimilars and biobetters for the molecules with the largest market share. This is not uncommon, as similar trends can be seen with other leading biologics, particularly with insulin and erythropoietin (). lists the number in development for most sold biological products. Traditionally, highly regulated processes have had nationwide regulatory bodies such as the European Medicines Agency (EMA), United States (US) Food and Drug Administration (FDA) and Chinese NMPA (National Medical Products Administration) for assessing biosimilars as appropriate therapies specifying numerous practices. The biomanufacturers must adhere to ensure that a biosimilar is highly analogous to the reference drug and to ultimately conserve the fidelity and features of the associated glycan structures; ICH Q5E details the evaluation and comparability exercises for biological products before and after changes in the manufacturing process including considerations for quality control. The document does not prescribe to any particular analytical, non-clinical or clinical strategy but give advice on the quality aspects. However, the guidelines do put manufacturing comparability in the context of process stages extremely well, “The demonstration of comparability does not necessarily mean that the quality attributes of the pre-change and post-change product are identical, but that they are highly similar and that the existing knowledge is sufficiently predictive to ensure that any differences in quality attributes have no adverse impact upon safety or efficacy of the drug product. A determination of comparability can be based on a combination of analytical testing, biological assays, and, in some cases, nonclinical and clinical data. If a manufacturer can provide assurance of comparability through analytical studies alone, nonclinical or clinical studies with the post-change product are not warranted [Citation30].” The analytical methods used may require modification to suit the change particularly if the manufacturing process gives rise to any undesirable changes. For example, if changes give rise to a different impurity profile of host cell proteins, the manufacturer should be able to confirm that the tests and procedures used to quantitate and to remove the impurities are still valid. The comparability exercise should include a side-by-side analytical profile comparison of the pre- and post-change products demonstrating consistency with regards to the selected characterization test and critical quality attributes. Any process related impurities are dealt with in accordance with ICHQ6B. These tests and procedures should be validated in accordance with ICH guidelines particularly Q2A andQ2B and for stability testing withQ5C ().
Table 2. ICH guidance documents covering the testing & characterization of biological products.
To meet the aims of the guidelines, the drug product must be characterized throughout product development particularly keeping a stringent check on critical processing perimeters. These perimeters include specifications for raw, source and starting materials, reagents, extent of bioburden and viral safety testing of the post-change cell banks. Quantifying removal of adventitious agents and product/process related impurities such as residual host cell DNA and proteins and keeping the purity in check also forms part of these critical parameters. All the changes expected to occur during the manufacturing process which could impact final drug quality (identity, potency, safety and efficacy) require testing pre- and post-process. Moreover, stability is extremely important because slight modifications in the process can lead to stability issues in future. For example, the presence of trace amounts of a protease might only be detected by product degradation that occurs over an extended period of time. For stability testing, drugs are exposed to accelerated degradation studies up to 50 °C and exposed to light where degradation products are studied and quantified. Similar degradation products are also studied after hydrolysis and oxidation. ICH Q5C provides detailed guideline on the stability testing.
Rheumatoid arthritis
Rheumatoid arthritis (RA), the 2nd most common type of arthritis, behind osteoarthritis, and is an autoimmune disease effecting approximately 0.5–1% of the population across the globe [Citation31,Citation32]. This equates to approximately 78 million people in the world and around 400,000 people in the UK. Despite the broad spectrum of the available drugs, the disease is not controlled in almost 30% of the patients, which equates to 23–25 million patients looking for alternative treatments [Citation33,Citation34]. Pharmaceutical companies recognize this, therefore, there is a competition to obtain the market share from the biologics going off patents. shows the number of biosimilars and biobetters in the running for various biologics currently on the market.
Table 3. Selected examples of top selling biologics compared to other leading biologics demonstrating the impact mAbs on global market share (2018).
RA is a leading cause of work disability; two-thirds of the RA patients lose 39 workdays on average each year, accumulating costs of £1.8 billion in terms of their productivity annually in the UK [Citation35,Citation36]. The average annual costs associated with RA treatment is high, and patients in the US are expected to pay from $1300 to $3000 per treatment, annually accumulating up to $30,000 [Citation37]. Whilst in the UK, the estimated costs for the National Health Service (NHS) each year for RA is approximately £560 million [Citation38]. As for privatized healthcare and Medicare in the US, annual costs are predicted in a recent study to accrue to $306 million and $600 million respectively [Citation39]. Hence, RA demonstrates a significant burden on healthcare systems worldwide [Citation40–42]. Unfortunately, if the disease is treated with less effective small molecules such as Aspirin, Ibuprofen, Methotrexate, Prednisone etc. the cost per patient varies from $100 to $300, whereas, if advanced cases are treated with more effective biologicals such as Enbrel, Remicade, Humira, Rituxan etc. and the cost per patient is $15,000–$24,250 [Citation43]. This 80-fold increase is too steep for all healthcare systems, hence, there is a need to understand RA better and to decreases the price of biosimilars.
RA was first described by Dr. Augustin Jacob Landre-Beauvais in 1800 A.D. as a systemic autoimmune disorder primarily affecting the metacarpophalangeal (MCP) and proximal interphalangeal (PIP) joints, which leads to the hallmark features of the condition. This hand involvement typically occurs early in the disease progression [Citation44–46]. In autoimmune disorders, immune cells and antibodies recognize the body’s own proteins, cells or other agents as potential antigens. This autoimmunity may start years before the onset of active RA. The period before the development of clinical manifestation of the symptoms is referred to as pre-articular disease and is marked with the formation of autoantibodies that may be a rheumatoid factor (RF), anti-cyclic citrullinated peptidal (ACCP) antibodies or anti-collagen antibodies. It may initiate due to some genetic factors or environmental factors such as smoking, stress, lifestyle changes or maybe a combination of both. Some biomechanical, neurological or traumatic factors (not well identified), may cause the transition from pre-articular to articular disease [Citation47].
Immune complexes of rheumatoid factors and/or anti-citrulinated protein antibodies when present in the synovium provoke an inflammatory response by the infiltration of leukocytes in the synovial compartments. This is initiated by the expression of adhesion molecules (e.g. selectins, integrins, and other microglobulins) in the synovial microvessels or chemokines produced by the synovial cells. Macrophage like synovial cells are activated by these complexes either by surface receptors or by antigen-presenting cells through phagocytosis. Fibroblasts like synoviocytes also activate the T-cells to release TNFα, interleukins and other cytokines. This further leads to the stimulation of macrophages and thus a cascade starts resulting in an uncontrolled autoimmune response. The role of B-cells extends beyond antigen presentation to the fabrication of antibodies (autoantibodies in rheumatoid arthritis) and the release of cytokines. Cell surface proteins like immunoglobulins and differentiation antigens (CD20 and CD22) are also expressed on B-cells [Citation48]. B-cell produced autoantibodies fabricate larger immune complexes which further activate the complement system and Fc receptor and thus provokes the production of cytokines [Citation48]. The increased production of these cytokines activates more T and B-cells and macrophages. Macrophages themselves produce many important cytokines such as interleukins (IL) and tumor necrosis factor (TNFα) and thus are involved in osteoclastogenesis [Citation32,Citation48].
Another source of inflammatory cytokines is fibroblast-like synoviocytes present in the synovial membrane [Citation32]. Activated synoviocytes grow up as pannus and release collagenases, MMPs, proteases and reactive oxygen intermediates (ROIs). IL-1, IL-6, and TNFα are also released in excess amounts in synovial fluid which summons the destructive catabolism of chondrocytes and stimulates osteoclasts thereby, initiating bone erosion.
As a result of this continuous immune response, synovial membrane becomes hyperplastic with two distinct layers or zones. The upper layer comprises macrophages and fibroblast-like synoviocytes while the underlying layer contains the cellular infiltrate of CD4+ T cells, CD8+ T cells, B cells, plasma cells, natural killer cells, macrophages, and fibroblasts. The numbers of macrophages and synoviocytes are fairly high in RA as compared to normal joints. Dendritic cells are also higher numbers in both the synovium and synovial fluid highlighting the involvement of T cells in the RA pathogenesis. As both the dendritic cells and synoviocytes are efficient in antigen presentation, their presence highlights the role of perivascular infiltrates in T cell activation and disease progression. CD4+ T cells are found abundantly in these infiltrates while the CD8+ T cells and plasma cells are also seen [Citation49]. The inflamed synovium overruns the cartilage as pannus and accelerates cartilage destruction through chondrocytes, osteoclasts, and MMPS. Some aggregation of T cells and B cells is also seen in the bone marrow which destroys the bone from inside [Citation47,Citation50]. The story does not end here. The cartilage and bone damage produces a lush source of neo-antigens to further call upon the autoimmune response. Rheumatoid joints so contain a remarkably distinct pathophysiological environment characterized by hypoxia, biochemical stress and angiogenesis [Citation51–53].
The therapeutic options for rheumatoid arthritis have increased tremendously over the last three decades. There is no cure for the disease -documented due to a myriad of complex mechanisms involved in the pathogenesis. However, many therapies have been entered in this space to manage the symptoms and progression of the disease, enhancing the quality of life and reducing morbidity. These include conventional synthetic disease modifying antirheumatic drugs (csDMARDs), glucocorticoids, biological disease modifying antirheumatic drugs (bDMARDs) and targeted disease modifying antirheumatic drugs (tsDMARDs). The patient responses to these available therapies vary greatly. Rheumatoid arthritis is an incurable disease and treatment targets aim at sustained remission and low disease activity. Methotrexate, a csDMARD, is recommended as the first medication to patients on the diagnosis of rheumatoid arthritis due to its safety, efficacy, mode of administration and low cost. A large proportion of patients remain unresponsive or intolerant to methotrexate. In that case Leflunomide or Sulfasalazine can be recommended as the first therapy. Glucocorticoids can be combined with these csDMARDS when starting treatment but these should be tapered as soon as it is clinically reasonable [Citation54]. If remission and low disease activity is not achieved in the first 6 months with csDMARDS, then a bDMARD or a tsDMARD is added. According to the EULAR recommendations of 2016, preference is given to the bDMARDs over the tsDMARDS. Similarly, the TNF inhibitors have been given preference over other biologic drugs as long term registary data is available for the TNF inhibitors as compared to other biologics [Citation55]. The usual practice has been to use a bDMARD in case of a poor response to csDMARDs. However, EULAR recommendations of 2019 have not given any such preference or vice versa due to the evidence of similar safety and efficacy of tsDMARDs in recent years [Citation54]. TNF inhibitors as a combination therapy with methotrexate have shown radiographic improvement as compared to methotrexate alone resulting in sustained remission and low disease activity in a larger proportion of patients. The patients with active disease treated with etanercept and methotrexate combination therapy for one year achieve lower disease activity and clinical remission as compared to the ones receiving methotrexate monotherapy [Citation56]. In case of failure of one TNF- inhibitor the treatment is switched to another TNF inhibitor or a bDMARD with a different mode of action such as interleukin-6 inhibitor, interleukin-1inhibitor, co-stimulation inhibitor or anti-b-cell therapies are employed. There are some studies showing the efficacy of a TNF inhibitor after the failure of one TNF inhibitor [Citation57]. However, the biologic with a different mode of action is better than using another TNF inhibitor [Citation58–60]. The combination therapies with two or more biologic drugs are prohibited due the intolerable rate of adverse events.
Mostly the biologic drugs are recommended as combination therapy with methotrexate. Among bDMARDs only Tocilizumab has shown some efficacy as monotherapy compared to methotrexate [Citation61]. Monotherapy of TNF inhibitors or other biologics has not been superior to the methotrexate monotherapy [Citation62–64]. However, it can be used for patients having very low disease activity. In actual practice almost one-third of the RA patients use biologic drugs as monotherapy [Citation65]. Anakinra IL-1 receptor antagonist has also been found effective in causing radiographic improvements in the patients but its efficacy is inferior to TNF inhibitors [Citation66]. Abatacept has been found to be efficacious in patients with a poor response to methotrexate and TNF inhibitors [Citation67]. Rituximab, Anakinra and abatacept show improvements in disease symptoms but these have a higher risk of serious infections [Citation68,Citation69]. In case of failure of bDMARDs or tsDMARDs another biologic or target synthetic drug can be used of the same class or a different class. Switching to JAK inhibitors after the failure of TNFα inhibitors has the same effects as switching from JAK inhibitors to TNFα inhibitors [Citation70]. The tsDMARDs have the advantage of being orally administered. These have comparable efficacy to that of biologics causing low disease activity and disease remission. Both the biologic drugs and kinase inhibitors have an increased rate of adverse effects when compared to the conventional drug. The most common adverse effects of these drugs are opportunistic infections and cardiovascular disorders [Citation71]. However, the risk of malignancies is not much prevalent in biologic drugs [Citation72,Citation73]. The opportunistic infections are more common in case of JAK inhibitors especially the herpes zoster infection [Citation71]. The events of thrombosis have occurred due to JAK inhibitors [Citation74,Citation75]. The FDA approved JAK inhibitors contain black box warnings with them [Citation76–78]. So, extreme care should be taken to select the therapeutic drugs for RA patients keeping in mind the disease activity level, patient health factors, comorbidities and risk factors. As RA is an incurable disease, so the therapeutic option that causes remission or at least slows disease activity should be continued. In spite the availability of treatment with different modes of action, 20–30% of the patients remain unresponsive to all the available therapeutics [Citation54,Citation79].
TNFα inhibitors
TNFα inhibitors were the first biologics approved by Food and Drug Administration (FDA) for the treatment of RA [Citation80,Citation81]. Although hyperactivated immune cells are the major factor for the development of RA, cytokines also participate in this process [Citation82,Citation83]. Cytokines are the intracellular messengers of the immune system, controlling many phases of immunity and inflammation, from development and differentiation of cells to the suppression of responses [Citation84–86]. A distinct cytokine pattern of interleukin-1, 4, 6, 8, 13, 15 and TNFα is seen in RA [Citation87]. The analysis of synovial fluid and synovium shows remarkable amounts of IL-1 and TNF, IFNα, granulocyte-macrophage colony-stimulating factor (GM-CSF), macrophage colony-stimulating factor (M-CSF) and leukocyte inhibitory factor (LIF) [Citation87,Citation88]. The central role of these cytokines in the rheumatoid pathogenesis has been confirmed by studies involving the blockage or inhibition of these cytokines. The therapeutic reduction of the disease progression is also marked by the decrease of inflammatory cytokines in the synovial fluid such as IL-1 and TNFα [Citation82,Citation89,Citation90]. Cytokines are produced by a variety of cells in the synovium including macrophages, mast cells, FLS, CD4+T cells and B cells [Citation84,Citation85]. Cytokines also may initiate a positive feedback loop to produce further cytokines which in turn may activate different cell types [Citation85]. Among different cytokines found in the synovial fluid, TNFα originating from macrophages, neutrophils, mast cells, T lymphocytes and B lymphocytes, is the major culprit that trigger the inflammatory process, and is found in large quantities in the synovial fluid [Citation88,Citation91–93]. It lies upstream of other pro-inflammatory cytokines and initiates the cascade of activation and secretion that results in synovial hyperplasia. Its production is instigated by all the possibly lethal provocations such as heat, x-rays, UV light or any type of immunological or chemical irritants. Cytokines are produced immediately on stimulation and can be detected in mouse serum within 30 min. Analysis of synovium in arthritic joints shows its presence at the interface between cartilage and pannus. It initiates leukocyte infiltration, upsurges adhesion molecules, stimulates the production of IL-1, IL-6, IL-8, prostaglandin E2 (PGE2), matrix metalloproteinases (MMPs) and GM-CSF, accelerates cartilage destruction, activates osteoclasts and causes bone resorption. It also interferes with the synthesis of cartilage matrix (proteoglycans) [Citation94,Citation95].
The pathological roles of TNFα has been confirmed by in vitro studies and clinical investigations in patients treated with TNFα inhibitors [Citation96,Citation97]. Decreased concentrations of GM-CSF, IL-1, IL-6 and IL-8 have been observed by in vitro studies involving the neutralization of TNFα. IL-1, 8 and TNFα functions in a synchronized manner [Citation98]. The IL-1 produced as a result of TNFα stimulation in turn stimulates the production of TNFα. The TNFα inhibitors have been proven to downregulate other pro-inflammatory cytokines and degrading enzymes such as MMPs, inhibit the expression of adhesion molecules, reducing synovial inflammation and joint destruction [Citation91,Citation93,Citation99–103]. A brief summary of the roles of TNFα are presented in and .
Figure 5. Downstream signaling of TNF alpha. TNF alpha has impact on cartilage matrix, endothelial cells, synoviocytes, macrophages/monocytes and prostanoids; playing a key role the pathogenesis of rheumatoid arthritis (Created with Biorender.com).
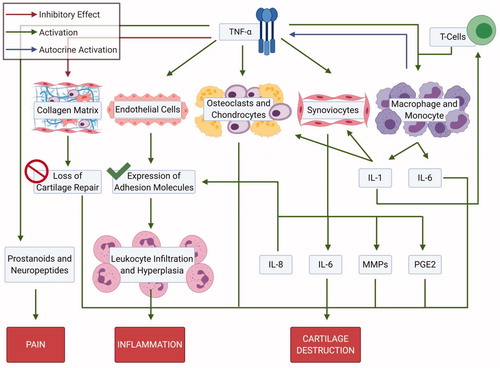
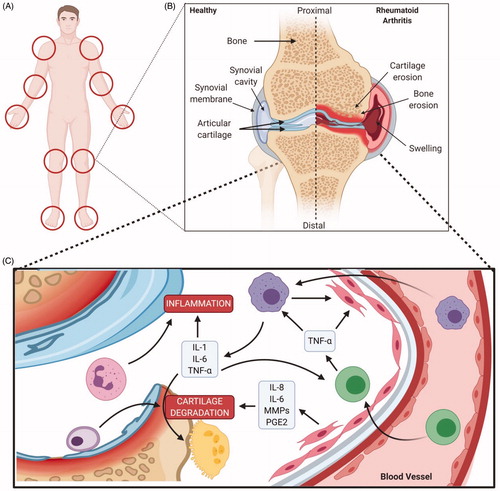
Figure 6. Pathophysiology of rheumatoid arthritis. (A) Locations of impacted areas in Rheumatoid Arthritis. (B) A healthy synovial joint and the impact of RA. In a healthy joint (left), there is no swelling in the synovial joint capsule, cartilage and bone are intact. Whereas, in RA (right), there is swelling, cartilage and bone erosion. (C) Mechanism of RA within the joint capsule. TNF alpha impacts synoviocytes, macrophages/monocytes and osteoclasts; inititating RA pathogenesis. Synoviocytes line the capsule and T-cells infiltrate the synovial membrane, initiating inflammation via TNF alpha. Joint degradation occurs via recruitment of macrophages and secretion of inflammatory cytokines. Bone erosion occurs by osteoclasts and inhibition of collagen secretion by synoviocytes (Created with Biorender.com).
Two receptors of TNFα; p55 and p75, are found on the surface of many cells including FLS. In healthy joints, the expression of TNFα is regulated by the soluble receptors (sTNFR) which can bind to superfluous TNFα, inhibiting its access to cell surface receptors and thus providing a setback to its inflammatory roles. The detection of sTNFR and the understanding of their regulatory role has opened new avenues for anti-TNFα therapies soon after TNFα inhibitors became the first biologics approved by FDA for the treatment of RA [Citation104]. Anti-TNF therapies have been proven to be efficacious through clinical investigations with remarkable markdown of the disease symptoms and radiographic improvements [Citation105–107]. The two approaches for inhibiting TNFα are based on monoclonal antibodies against TNFα and fusion proteins acting as soluble TNF receptors (sTNFR). Both of these approaches have been designed to inhibit TNFα, curbing its proficiency to interact with cell surface receptors to initiate the inflammatory response. represents FDA approved TNFα inhibitors in use.
Table 4. FDA approved Anti-TNFα biologics and ts-DMARDS.
Infliximab
Infliximab (Remicade), the first introduced TNFα inhibitor, is a monoclonal chimeric antibody fabricated by coupling human IgG1 κ with murine Fv regions of anti-Human TNFα antibody and shows high neutralizing capabilities against TNFα () [Citation108]. Initially approved by FDA in 1998 for Crohn’s disease, infliximab received FDA approval for RA in 2004 and for ulcerative colitis in 2005. Infliximab does not cure the disease but manages its symptoms. It works by binding to TNFα and hinders its approach to cell surface receptors, thus nullifying its effects [Citation109]. Infliximab reduces the production of IL-1 and 6, controls the expression of adhesion molecules, moderates leukocyte migration and reduces cartilage and bone resorption by reducing the production of degrading enzymes. It is administered intravenously every 4–8 weeks and the standard recommended dose is 5 mg kg−1 of patient body weight (). However, Infliximab has been shown to exhibit low drug survival [Citation110]. Several studies have shown higher rates for Infliximab drug discontinuation than any other TNFα inhibitor [Citation110]. This can be attributed to the infusion reactions with enhanced immunogenicity due to the presence of the murine elements in this drug. It is not associated with immunogenicity found with subcutaneous mode of administration. Other complications associated with use of Infliximab include: headaches, nausea, abdominal pain, heartburns, reactivation of hepatitis, development of psoriasis, optic neuritis, higher risk of cognitive heart failure, lymphomas, lupus like syndrome and bone marrow suppression along with its interactions with warfarin and theophylline drugs [Citation20]. These adverse events and lower efficacy has resulted in higher drug withdrawal for Infliximab relative to Adalimumab and Etanercept [Citation20]. In spite of all this, Infliximab (Remicade) has remained the bestselling drug of Janssen.
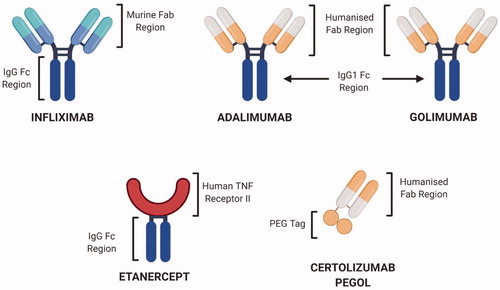
Figure 7. Structural representation of selected FDA-approved anti-TNFα biologics. Infliximab, Adalimumab and Golimumab are complete antibodies while Etanercept is a fusion protein whereas Certolizumab pegol has a humanized Fab region, hence devoid of effector functions (Created with Biorender.com).
Adalimumab
To avoid any possibilities of an immune reaction against the mouse proteins, drug development was diverted toward the generation of fully humanized antibodies and resulted in the introduction of Adalimumab (D2E7) to the therapeutic arsenal [Citation111]. Adalimumab, under the trade name Humira, is prepared by phage display technology using murine anti-TNFα monoclonal antibody as a template and replacing murine parts with human regions [Citation112]. This development resulted in Sir Gregory Winter (University of Cambridge) being awarded the 2018 Nobel Prize in Chemistry. Humira was approved by the FDA in December 2002 for RA treatment. Its response rate is comparable to that of methotrexate and gives twofold efficacy when used as a combination therapy [Citation113]. In 2014, IMS health announced it as the bestselling drug on the current market (). It is also specific for TNFα and blocks its ability to interact with surface receptors p55 and p75. In in vitro models, Humira disrupts the cells presenting TNFα surface receptors. Reduced concentrations of IL-1and 6 and MMPs have been revealed through clinical investigations which, in turn, prevents cartilage destruction [Citation114]. It is administered subcutaneously on alternate weeks (). Adverse effects of Humira include: upper respiratory tract infection, nausea, abdominal pain, sinusitis, intensified tuberculosis infection along with the common harmful effects of TNFα inhibitors. There is some evidence for the development of anti-adalimumab antibodies leading to a poor response [Citation115].
Certolizumab pegol
Certolizumab pegol (Cimzia®), approved by FDA in May 2009, is a humanized PEGylated (polyethylene-glycol) monoclonal antibody, which is unglycosylated due to its bacterial origin and lack of Fc region which contains the most probability of being glycosylated. It is fabricated by combining the Fab fragment of monoclonal humanized antibody with PEG [Citation116]. PEG increases the bioavailability by decreasing the rate of blood clearance and effecting pharmacokinetics–with the half-life found to be approximately 14 days [Citation117–119] (). Due to the absence of the Fc fragment (), it is unable to interact with the Fc receptor and stimulate complement-dependent cytotoxicity (CDC) or antibody-dependent cytotoxicity (ADCC), a prevalent risk associated with the use of Infliximab and Adalimumab. It interacts with both the soluble and transmembrane TNFα, downregulating its ability to provoke an inflammatory response through both receptors [Citation120]. Its efficacy and ability to produce a quick and persistent response, leading to reduced joint damage and increased physical functionality, has been established through clinical investigations [Citation121,Citation122]. Studies on mouse models have shown its inflammation-dependent, enhanced, and persistent penetration in the tissues as compared to other TNFα inhibitors. It has also shown that it is incapable of persuading neutrophil degradation and apoptosis in lymphocytes and monocytes of human blood origin [Citation120,Citation123]. It produces appreciable results when used alone with MTX or other DMARDS, when DMARDS alone fails to produce a satisfactory response. In 2013 Cimzia® received FDA approval for Psoriasis Arthritis in adults [Citation124]. Common side effects of Cimzia® include: upper respiratory tract infection, urinary tract infections, stomach pain, constipation, and diarrhea in some cases. Its adverse effects include malignancies and heart failure [Citation125].
Golimumab
Golimumab, a humanized anti-TNFα monoclonal antibody (IgG) is effective against both the transmembrane and soluble TNFα, was approved by FDA in 2009 for active RA (). It is specific for TNFα and is inactive against TNFβ. Golimumab is also used against psoriatic arthritis and ankylosing spondylitis [Citation126–128]. Its long-term efficacy and capability to produce radiographic improvements have been proven with clinical trials [Citation129]. Golimumab reduces the concentration of IL-1, 6, MMP-3, VEGF, CRP and ICAM-1. It can be used as an alternative therapy when other TNFα inhibitors and disease-modifying antirheumatic drugs (DMARDs) are not effective, and provides enhanced efficacy in combination with methotrexate [Citation100,Citation130]. Additionally, the immunogenicity and adverse effects are reduced in combination with methotrexate, although it can activate preexisting psoriasis and fungal infection. However, it does not counterpoise the enhanced efficacy of combination therapy [Citation131]. It is administered subcutaneously once a month which makes it preferable over other TNFα inhibitors, which show rather complex dosing regimens. It is characterized with reduced immunogenicity and fewer adverse effects [Citation129]. Only 2.1–6.3% of patients have been shown to develop antibodies against Golimumab [Citation132]. Another study regarding the immunogenicity of Golimumab has reported the development of antinuclear antibodies (ANA) at the rate of 12.2% in patients treated with Golimumab in combination with methotrexate, while 14.9% of the placebo developed such antibody concentrations. Also, 6.3% of the patients have been reported to develop anti-drug antibodies [Citation133,Citation134]. Furthermore, the adverse effects of these inhibitors are not as pronounced as is shown with other medications. It has lesser frequency and severity of injection site reactions including erythema. There are fewer incidences of developing tuberculosis or activation of the existing dormant tuberculosis, while, some opportunistic infections have been observed, including upper respiratory tract reactions and nasopharyngitis [Citation135]. The risk factors associated with other anti-TNFα therapies such as lymphomas and cognitive heart failure have also been reported in patients receiving Golimumab.
Etanercept
Another approach to impede TNFα is based on engineered fusion proteins mimicking soluble TNFα receptors [Citation136]. The fusion proteins have ligand binding sites of TNF RI (p55) and II (p75) linked with an immunoglobin like molecule. Etanercept under the trade name of Enbrel was the first fusion protein approved by FDA for RA in 1998. This approval was extended to Juvenile RA in 1999, to Psoriatic Arthritis in 2002, to Ankylosing Spondylitis (AS) in 2003 and Plaque Psoriasis in 2004. It is a dimeric entity consisting of two p75 (TNFR2) receptors bound to a human immunoglobulin IgG1 through its Fc fragment [Citation137]. It shows three orders of magnitude higher affinity for TNFα as compared to the monomeric entities. Its role is to mimic soluble TNFα receptors; however, due to its longer half-life in the blood when compared to the natural sTNFR, it is more effective and supplements their function in regulating the inflammatory roles of TNFα [Citation138]. Adverse effects of Etanercept include worsening of tuberculosis, sepsis, hepatitis B and other opportunistic infections. Recently, one case has been reported to develop Pasteurella multocida bacteremia in response to Etanercept [Citation139]. Strongyloides hyper-infection cases have also been registered and a black box warning was placed on the Etanercept by the FDA in 2008. shows a comparison of different FDA-approved TNFα inhibitors.
Table 5. TNF inhibitor biosimilars approved worldwide [Citation81,Citation140–144].
Neovius et al. have found a 38% drug survival for Infliximab, 50% for Adalimumab and 55% for Etanercept after five years of follow up in patients using these drugs as their first line anti-TNFα therapy [Citation145]. The reduced drug survival may be attributed to a lower response, reduced efficacy, disease remission and adverse events (especially infusion reactions in the case for Infliximab). Development of antidrug antibodies also contributes to disease remission. The results of clinical investigation shows that reduced efficacy and disease remission are the major cause of increasing the rate of drug withdrawal in the last decade as been compared to adverse events [Citation145].
Challenges with glycosylation and their characterization
The major goal for any biosimilar manufacturer is to make it interchangeable with the original reference product [Citation16,Citation146]. The market share of biosimilars is expected to grow to $61.47 billion by 2025 with a compound average growth rate (CAGR) of 34.2% [Citation147]. Many biosimilars of biologic TNFα inhibitors are currently in development [Citation140,Citation148–150]. Biosimilar development has an added advantage that patents of many anti-TNFα biologics are either close to expiration or have already expired (). Almost all the approved anti-TNFα biologics will expire in the US and EU by 2030.
A biosimilar can only be used for the indications approved for the reference drug; however, it is not necessary for a biosimilar to meet with all those indications. It may be approved for some of the indications of the reference drug and not for others. Hence, it is necessary to determine those indications before exchanging it for the reference drug. Furthermore, the exchanging or switching to the biosimilar on and off can reduce the safety issues of using the biologic alone for a long time. Biosimilars, to be approved by FDA, should have same mode of action, identical administration routes, comparable strength and similar dosing regimens as that of the reference product [Citation151]. Due to the high complexity of the antibodies, the generation of exact replicas of their structure is a hard task and no batch of any biologic is exactly identical to the original approved one. Small changes in the manufacturing processes can alter their function as discussed in a previous section of this review [Citation152,Citation153] One of the most difficult challenges is with glycosylation, which is a ubiquitous post-translational modification (PTM) and can affect important critical attributes during manufacturing, particularly the pharmacokinetic and pharmacodynamic properties. The safety, quality and efficacy of the final drug can be affected with the addition of glycans to different consensus sites. These oligosaccharides play a significant role in the activation of the effector functions of the immune system such as Antibody dependent cellular toxicity (ADCC) and complement dependent cytotoxicity (CDC). If the effector response is crucial to antibody’s activity, then the glycosylation pattern plays a critical role. However, if a mAb is being developed where its therapeutic activity is entirely based on blocking a receptor or any other protein from binding to the target then glycosylation is not important from functional aspects. However, from a manufacturing perspective heterogeneity should be kept under 5% to comply with FDA regulations. It should be noted that the microheterogeneity can significantly affect: the solubility, stability, immunogenicity and receptor binding [Citation154].
These glycans have complex biantennary structures where three mannose residues form the core structure with two N-acetylglucosamines (GlnNAc) bound toward the proximal end (toward the protein) with central mannose through β1 − 4 linkages. Another bisecting N-acetylglucosamine (GlcNAc) toward the proximal end may or may not be present (), which may lead to the enhancement of ADCC [Citation156]. Another important residue is fucose which is attached to the most proximal GlcNAc through the α1 − 6 linkage. It is not always present but it can lead to a decreased ADCC but has no effect on CDC. The oligosaccharides may contain a terminal galactose giving confirmations in case of one G1F, and in two G2F and G0 in case of the absence of galactose. Higher galactose content results in increased CDC activity () [Citation155]. Galactose residues can attach themselves to sialic acid residues through a α2 − 6 linkage. Increases in sialic acid residues can lead to a longer serum half-life. Furthermore, desialylation of intravenous IgG can abrogate the anti-inflammatory properties of the biological therapeutics [Citation158]. High-mannose glycans have been reported to critically affect the ADCC properties and the PK/PD, increasing serum clearance [Citation159].
Figure 8. Glycan variation shown in biotherapeutics. Variation occurring through development can be accounted to distinctive glycan machinery present in the chosen expression system. Adapted from several sources [Citation154–157].
![Figure 8. Glycan variation shown in biotherapeutics. Variation occurring through development can be accounted to distinctive glycan machinery present in the chosen expression system. Adapted from several sources [Citation154–157].](/cms/asset/8261e4ac-fa19-4b74-bd13-c1825b922b86/ibty_a_1830746_f0008_c.jpg)
Advances are being made to understand how to control glycosylation during the fermentation process and how rapid detection of glycans can be made to reduce heterogeneity within glycoproteins. It is a complex task to control glycosylation as within the typical structure of the mAbs, and varying covalent structures can occur. Thus, it is important to understand and have the ability to manipulate the chosen cell culture. N-linked glycosylation occurs in the endoplasmic reticulum and Golgi apparatus, with the addition of a great number of precursor enzymes [Citation159]. Irregularity of these structures is influenced by a number of factors: host cell type, nutrients and supplementation, pH, temperature and by-product accumulation [Citation160]. Furthermore, each cell line possesses its own distinctive glycosylation machinery which impacts the final glycosylation patterns [Citation161]. Specific glycan-processing enzymes are accountable for the events of glycosylation, such as trimming and glycan-epitope alterations. The resulting affect is a diverse library of possible glycans. The extent and type of glycosylation can be controlled by cell engineering and by manipulating the feed. All the CDMOs take exceptional care to make sure that they can control the glycoform. Pharmaceutical companies have had historical challenges with glycan engineering. One such example is when Merck acquired GlycoFi, a Darmouth spin off for $400 million in 2006. The technology was based on selective glycosylation in yeast. Merck could not capitalize this in a timely fashion. However, CDMOs now have much more promising tools at their disposal. Lonza has in-licensed BioWa’s Potelligent® technology where antibodies are produced with a reduced fucose content leading to higher ADCC. Lonza’s cell line has FUT-8 gene knocked out guaranteeing fucose free antibodies. demonstrates variation in common glycan structures.
Antibody glycosylation is the major cause of batch to batch variability, therefore regulatory authorities require complete characterization of the identity and extent of glycosylation. It is important to determine the percent occupancy of each glycosylation site. Glycans can be analyzed from an intact glycoprotein, using methods including HPLC, matrix-assisted laser desorption ionization (MALDI) MS and CE-MS. MALDI-MS offers the ability for the analysis of small intact proteins, however this has decreased mass accuracy [Citation159]. To increase this sensitivity, mAbs may either be cleaved with proteases into the respective heavy and light chains or be chemically reduced. A common analytical tool used to identify glycoprotein sialyation is capillary-electrophoresis CE-MS, although it is often conserved for quality testing and is useful in site-specific glycan heterogeneity at the glycopeptide level. As a result, other charged-based electrophoresis methods could be utilized for intact analysis such as ion-exchange chromatography (IEC) [Citation162]. Another approach is to release the glycans from the glycopeptide with an amidase, such as PNGase F, and subsequently label them with a fluorescent tag to facilitate their quantification. These labeled N-glycans can then be separated using HILIC which reportedly offers greater resolution over HPLC methods [Citation159]. The analysis of glycans often takes advantage of multiple complementary approaches to correctly identify structures [Citation155,Citation163]. lists various analytical techniques that are used to determine critical quality attributes to determine the glycosylation. A small deviation in any of the steps involved in manufacturing process such as fermentation, pharmaceutical preparation, purification or a slight change in pH, buffer, stabilizer, bulking agent, solubility and mode of molecular aggregation may cause an unpredictable change in the resulting product. It necessitates extensive analytical, preclinical and clinical studies to ensure its comparable efficacy, safety and immunogenicity (). Hence, biosimilars have to pass additional measures or biocompatibility analyses to prove its equivalence or non-inferiority to the reference product in order to obtain regulatory approval. A number of biosimilars are being used in different treatment schemes. shows a brief description of the anti-TNFα biosimilars approved worldwide [Citation140] ().
Figure 9. Glycan analysis. Flowchart depicting sample preparation for glycan analysis, which typically involve lengthy denaturation and labeling steps to acquire sample for analysis.
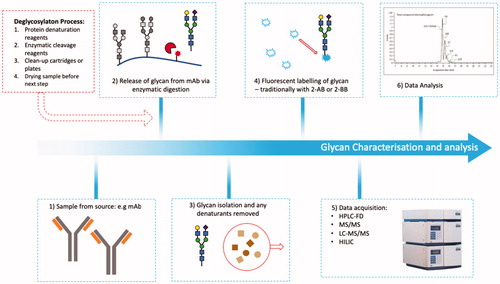
Table 6. Analytical procedures for the analysis of mAb critical quality attributes.
Infliximab biosimilars
On 5th April, 2016 the FDA granted approval for Inflectra (CT-P13), a biosimilar to reference drug Infliximab (INX) for all its recommended indications [Citation169]. It is the second FDA-approved biosimilar and the first monoclonal antibody biosimilar on the portfolio of FDA-approved drugs. CT-P13 under the brand names of Remsima (by Celltrion) and Inflectra (by Hospira) have been approved by EMEA in 2013 for Crohn’s Disease and Ulcerative Colitis. It possesses the same amino acid sequence as that of INX and is engineered in equivalent cell lines. It has comparable pharmacokinetic and pharmacodynamics, with binding affinities to hTNF, Fcγ and FcRn [Citation170]. It also shows the high specificity of infliximab to hTNFα and does not bind to TNFβ [Citation171]. Comparability is also observed in terms of CDC and ADCC. Two double-blind, multicenter trials named as PLAETAS and PLANETRA were designed to compare the efficacy, safety and pharmacokinetics of CT-P13, biosimilar for infliximab in AS. In PLAETAS equal doses (5 mg kg−1) of the reference and biosimilar were given to the patients. Both drugs produced similar pharmacokinetics and immunogenicity. The assessment by spondyloarthritis International Society, ASAS 20, at week 30 was found to be 70.5% for CT-P13 and 72.4% for Infliximab while 51.8% and 47.4% ASAS 40 was achieved for both, respectively. 3.9% of the patients in CT-P13 arm and 4.9% in infliximab arm developed infusion reactions [Citation172]. PLANETRA, a phase III trial was designed to compare the clinical outcomes of CT-P13 and Infliximab in RA patients with the active disease unresponsive to methotrexate. Equal doses (3 mg kg−1 every two weeks) of both the biosimilar and reference along with methotrexate (12.5–25 mg per week) as combination therapy were given to the patients. Both drugs produced comparable efficacy, safety, pharmacokinetics and immunogenicity. The efficacy of these biosimilars can be measured on a standard criteria developed by American College of Rheumatology (ACR). The criteria was to measure improvement in tender or swollen joints through patient assessment, physician assessment, pain scale, disability/functional questionnaire, and acute phase reactant. ACR20 is ≥ 20% improvement, ACR50 is ≥ 50% improvement while ACR70 is ≥70% improvement [Citation173]. An ACR 20 response of 60.9% and 58.6% was produced for CT-P13 and infliximab respectively at week 30 among the intention-to-treat population. Anti-drug antibodies at week 14 were found to be 25.4% and 25.8% respectively in Infliximab and CT-P13 arm while 48.2% and 48.4% respectively at week 30 [Citation174]. Pharmacokinetic factors such as the concentration of serum C-reactive protein (CRP), anticyclic citrullinated peptide, erythrocyte sedimentation rate was also comparable. 6.6% and 8.3% of the patients in both the biosimilar and reference arms respectively developed infusion reactions while the occurrence of severe adverse events was 14% and 10%, respectively. This inequality of adverse events was linked with the tuberculosis and pneumonia cases in the CT-P13 treated patients. Investigators of this trial have related these diseases to the preexisting conditions or the patients’ personal risk factors [Citation174].
A similar randomized, double-blind and multi-centered clinical trial (phase III) was also designed in Japan to compare the effects of CT-P13 and Infliximab in the RA patients with an equal dose (3 mg kg−1) of the concerned drugs in both arms along with 6–16 mg per week of the methotrexate as a combination therapy. Comparable pharmacokinetics, ACR20 and ACR5O and adverse events were observed at week 14, 30 and 54 [Citation175]. Based on the favorable results of clinical trials, the British Society of Gastroenterology (BSG) in February 2016 recommended the switching of stable patients using Infliximab to the biosimilar with same dosing regimen [Citation176]. EMEA has approved a second Remicade biosimilar “Flixabi” in May 2016, clearing it for commercialization [Citation144]. Many pharmaceutical companies have joined the biosimilars development race and several infliximab biosimilars are in clinical development. The results of their clinical trials are promising with favorable primary and secondary end points.
Etanercept biosimilars
Benepali (SB4) entered the European market in 2016 as the first subcutaneous anti-TNFα biosimilar. [Citation176]. It has been approved by the EU in 16th January 2016 as a biosimilar for Enbrel (etanercept) against the indications of RA, psoriatic arthritis, plaque psoriasis and axial spondyloarthritis. Clinical trials have demonstrated Benepali to have comparable efficacy, immunogenicity and safety profile to that of Enbrel [Citation177]. The clinical trials have shown equivalent efficacy in terms of ACR 20 response and the safety profile [Citation177]. ACR 20 response of 78.1% in the Benapali arm has been achieved as compared to 80.3% in the Enbrel arm at week 24. While 81.8% in Benepali as compared to 81.5% in Enbrel arm at week 52. ACR 50 and ACR 70 was 46.6% and 25.5% respectively for SB4 against 42.3% and 22.6% in the Enbrel arm, respectively. Adverse events were developed in 55.2% patients in SB4 as compared to 58.2% in the Enbrel group. Serious adverse events were reported in 13 patients in each group. 5% patients of SB4 patients discontinued treatment due to adverse events as compared to the 6.4% of the Enbrel group. 0.3% and 1.3% respectively in SB4 and Enbrel arm developed serious infections. Malignancies were developed in 1% of patients in SB4 and 0.3% in Enbrel arm while the injection site reactions were observed in 3.7% in SB4 as compared to 17.2% of the Enbrel arm. The antidrug antibodies were developed in 0.7% and 13.1% patients of the SB4 and Enbrel group, respectively. However, after week 12, these antidrug antibodies (ADAs) disappeared. This clinical trial has demonstrated the equivalent clinical efficacy of the biosimilar to the reference drug. [Citation172,Citation178]
One biosimilar of Enbrel, Brenzys, has already been approved in South Korea in September 2015 for the treatment of psoriasis, axial spondyloarthritis and rheumatoid arthritis and psoriatic arthritis. Many Etanercept biosimilars are in development [Citation81,Citation179].
Adalimumab biosimilars
Many Humira biosimilars are also in development. ABP501 is a biosimilar registered by Amgen in November 2015 for FDA approval. Clinical trials III have been conducted to prove its equivalence efficacy, safety and immunogenicity in psoriasis and RA FDA accepted Amgen’s license application for ABP-501 on September 23rd, 2016. In December 2015 Amgen has also submitted the license application to EMEA for the marketing authorization of ABP501 in the EU. In the phase III clinical trials organized to compare the biosimilar to the reference Humira in RA patients, ACR 20 response was achieved in 74.6% and 72.4% in ABP-501 and adalimumab arm respectively at week 24. ACR 50 and 70 was found to be 49.25% and 26.0% respectively for ABP-501, and 52.05% and 22.9% respectively for adalimumab. Adverse events were reported in 50% of the patients in the ABP-501 arm and 55% in the adalimumab arm with serious adverse events to be 3.8% and 5.0% respectively at week 24. This data was presented by Amgen in the Association for Rheumatology Health Professionals (ARHP) and American College of Rheumatology (ACR) annual meeting in November 2015 [Citation180]. Since then ABP501 has been proven to work equally for patients with moderate-to-sever plaque psoriasis [Citation181]. Markus et al. [Citation182] has recently reviewed the Totality of Evidence (TOE) comparing ABP501 and can serve as a good example for other biosimilar manufacturers looking for guidance on building an evidence file. Many other Humira biosimilar candidates are in late stages of clinical development () with highly equivalent end points achieved by initial clinical trials [Citation183] ().
Table 7. TNF inhibitor biosimilars in development with respective developmental stages.
Intended copies or biomimics
Intended copies are the copy versions of biologics which are marketed without undergoing stringent regulatory processes. These are not available in highly regulated markets such as US, Europe and Australia but are available in countries with relaxed regulations such as China, Colombia and India. Shanghai CP Goujian Pharmaceutical Co has launched an Enbrel biomimic under the trade name of Yisaipu in China and Etanar in Colombia. One intended copy of Enbrel has also been launched in Mexico by Probiomed biopharmaceutical under the brand name of Infinitam based on only one study to compare its pharmacokinetics and pharmacodynamics to the reference product [Citation179].
India has been most welcoming to biosimilars and has granted approval to the intended copies as early as of 2000, while the first biosimilar was approved by EMEA in 2006 and by USA in 2015. Currently, the Indian biopharmaceutical market has more than 20 approved biomimics [Citation187]. The reason for this high uptake in India is that their laws allow to only obtain patents on manufacturing processes and not on the actual products. This allows them to boldly reverse the engineered drugs developed by international companies. In September 2014, Ranbaxy Laboratories (India) launched a Remicade biosimilar with the trade name Infimab (BOW015). The results from the clinical trials of BOW015 show its bioequivalence to the reference product Remicade. Reditux, an intended copy of rituximab, has been available in India since 2007. It was launched without any head to head trial with rituximab and efficacy had to be determined through a retrospective audit [Citation188]. Kikuzubam, yet another copy of rituximab had to be withdrawn from South America due to its side effects and toxicity [Citation6].
The first ever approved copy of Humira developed by Zydus Cadila (India) also lies on the portfolio of the Indian drug market which was approved by the Drug Controller General of India (DCGI) in December 2014 under the brand name Exemptia (ZRC3197) for RA, psoriasis arthritis, AS and juvenile inflammatory arthritis. Zydus Cadila has conducted clinical trials to compare the pharmacokinetics and pharmacodynamics of this copy version with the reference drug and outcomes were promising [Citation189]. However, it has not been approved yet by any other regulatory body and further clinical studies are awaiting to grant it the status of biosimilar. In January 2016, Department of Biotechnology (DBT) India granted marketing approval to Adfrar, a copy version of Humira, developed by Torrent Pharmaceuticals India [Citation179]. represents the intended copies of the mAbs allowed to be marketed worldwide.
Table 8. Intended copies launched worldwide.
Although Exemptia, and Infimab showed comparable efficacy, safety and immunogenicity to their respective reference products, the adverse effects are much more prevalent in the cases of the intended copies. There is a need for harmonization for the approval of copy versions. Every country should follow a standard set of regulations and guidelines in order to obtain approval for a drug [Citation81,Citation141]. Despite advances, biosimilars have failed to penetrate into the drug market and health systems of the poorer countries and it does not look feasible until the manufacturing costs and approval costs decrease. The patent holding companies are endeavoring to gain patent extension as the TNF inhibitors have been the best-selling drugs. A new patent has been issued to Enbrel that was due to expire in 2012 but now it is patent protected till 2028 [Citation81]. Humira was the second best selling drug of 2015 with annual worldwide sales of $14.95 bn, whilst Enbrel was the 4th and Remicade the 5th, with annual sales of $9.47bn and $8.2bn, respectively [Citation191]. It will be a financial setback for the patent holding companies if they fail to compete with the emerging biosimilars against their products. Pay for delay, where the originator patent holding companies pay the biosimilar developers to delay the launch of the biosimilar, and hinders the introduction of the biosimilars into the drug market. Along with the competition from the reference drugs, the TNFα biosimilars also have to compete with other biologic inhibitors such as Rituximab, the CD20 inhibitor and Anakinra IL-1 inhibitor, which shows parallel efficacy and safety profiles [Citation192]. Furthermore, the availability of the orally administered kinase inhibitors in rheumatology and other inflammatory diseases pose challenges to the biosimilar development [Citation193,Citation194]. Many kinase inhibitors have succeeded to penetrate in rheumatology with a reliable pharmacokinetics, pharmacodynamics and safety profile along with relatively less prices as compared to biologic therapies [Citation147,Citation192]. Fostamatinib, Tofacitinib (kinase inhibitors) entered clinical practice with a significant patient response and disease remission. All the anti-TNF therapies show reduced efficacy alone and are only useful in combination with some DMARDs or nonsteroidal anti-inflammatory drugs (NSAIDS) [Citation63,Citation64,Citation126,Citation127,Citation195]. Antidrug antibodies have also been observed in many cases. [Citation115,Citation134]. Some enhanced efficacy is observed in combination therapy with methotrexate [Citation113,Citation131]. However, almost 30% of the patients do not respond to anti-TNFα therapies [Citation33]. These factors along with the associated adverse events lead to the discontinuation of the TNFα inhibitors. Although the availability of other therapeutic possibilities contribute a major share in it, global data shows a stable discontinuation rate of anti-TNFα therapies over the last fifteen years [Citation145].
Conclusion
Healthcare systems are under extreme pressure from the rising costs of biologics to the newer personalized therapies which are currently being approved [Citation196]. Voxelotor, sickle hemoglobin (HbS) polymerization inhibitor costs $1,25,000 per patient. Sickle Cell Anemia is most common in Africa, where no one can afford it. The newer therapies such as Sprinraza® (nusinersen) a promising treatment for Spinal Muscular Atrophy (SMA) costs $750,000 for the first inject and $350,000 for subsequent injection annually until the patient survives. No healthcare system can afford to pay for these treatments. In this background, the need for biosimilars to shoulder the burden of RA and of most cancers is a necessity. It is imperative that academics as well as companies understand the costs and stages associated with biologics processing. This article brings these costs to the forefront with a focus on the biosimilars being developed for RA. Introduction of low cost biosimilars may change the clinical pattern for targeting RA and other such inflammatory diseases. There has been an incentive during the last 3 years on the FDA to set a lower bar for demonstrating biosimilarity and a limit on the variability of branded biologics. In Europe, patients have started to take advantage of lower prices as compared to the originator. Pharmaceutical companies with originator molecules had to resort to pervasive discounting to maintain the market share and in some instances had to lower the price up to 70%. Low cost, no doubt, is the main aim of introducing biosimilars in the drug market, but this must not be at the cost of safety and efficacy of the drug. Extensive studies aiming at the bio-comparability particularly for safety and adverse effects are required. A critical evaluation after the launch of the biosimilar should also be conducted to compare its long term pharmacokinetic and pharmacodynamics to avoid any complications resulting from batch-to-batch variations. The selection of the most suitable TNF inhibitor, originator or biosimilar is of great consideration. The pharmacodynamics properties of the inhibitor should be considered along with its efficacy, safety profile, tolerance, and cost. Patients should be clinically investigated through screening tests for associated problems such as infections, malignancies, heart diseases, tuberculosis and osteoporosis before and during the period of treatment. Any adverse events or any deviation from biocomparability should be reported to achieve the best outcomes. Strict surveillance by the regulating bodies and firm exercise of the legislative guidelines and regulations will help in the production of reliable low cost biosimilars.
Acknowledgements
MSA is thankful to Research England Connecting Capability Fund (CCF), THYME Project (Teesside, Hull and York–Mobilising Bioeconomy Knowledge Exchange) ccf14-7167 for providing opportunity to interacting with several industries, which has been instrumental to confirm the manufacturing costs in this article. I am also thankful to THYME funding manager Claire Adams and also to Dr. Jonathan Haigh for interesting discussions.
Disclosure statement
No potential conflict of interest was reported by the author(s).
Notes
1 Personal correspondence and discussion with various Contract Development Manufacturing Organisations (CDMOs) including Fujifilm Diosynth Biotechnologies, CPI and IntherVax.
References
- Carillo S, Mittermayr S, Farrell A, et al. Glycosylation analysis of therapeutic glycoproteins produced in CHO cells. Methods Mol Biol. 2017;1603:227–241.
- Research AM. World biosimilars/follow on biologics market – opportunities and forecasts. 2014.
- Levine HL, Cooney BR. The development of therapeutic monoclonal antibody products. Waltham (MA): BioProcess Technology Consultants, Inc; 2017.
- Emily Capra JS, Guang Y. Gene therapy coming of age: opportunities and challenges to getting ahead. Pharmaceuticals and Medical Products Practice. 2019.
- Hanna, C., Uccello, C. Prescription drug spending in the U.S. Healthcare System. Am Academy of Actuaries 2018. 1-12. Available from: www.actuary.org.
- Isaacs J, Gonçalves J, Strohal R, et al. The biosimilar approval process: how different is it? Considerations Med. 2017;1(1):3–6.
- Kogan LA. US biologics price competition and innovation Act of 2009 triggers public debates, regulatory/policy risks, and international trade concerns. Global Trade & Cust J. 2011;(6):513.
- Kirchhoff CF, Wang XM, Conlon HD, et al. Biosimilars: key regulatory considerations and similarity assessment tools. Biotechnol Bioeng. 2017;114(12):2696–2705.
- Reusch D, Haberger M, Falck D, et al. Comparison of methods for the analysis of therapeutic immunoglobulin G Fc-glycosylation profiles-Part 2: mass spectrometric methods. MAbs. 2015;7(4):732–742.
- Christl L. FDA’s overview of the regulatory guidance for the development and approval of biosimilar products in the US. USA: US Food and Drug Administration. 2016.
- Olech E. Biosimilars: rationale and current regulatory landscape. Semin Arthritis Rheum. 45(5 Suppl):S1–S10.
- Alten R, Cronstein BN. Clinical trial development for biosimilars. Semin Arthritis Rheum. 44(6 Suppl):S2–S8.
- Laimer J, Hofer H, Fritz M, et al. MAESTRO-multi agent stability prediction upon point mutations. BMC Bioinformatics. 2015;16(1):116.
- Weise M, Bielsky M-C, De Smet K, et al. Biosimilars—why terminology matters. Nat Biotechnol. 2011;29(8):690–693.
- Hamm M, Wang Y, Rustandi RR. Characterization of N-linked glycosylation in a monoclonal antibody produced in NS0 cells using capillary electrophoresis with laser-induced fluorescence detection. Pharmaceuticals. 2013;6(3):393–406.
- Schiestl M, Stangler T, Torella C, et al. Acceptable changes in quality attributes of glycosylated biopharmaceuticals. Nat Biotechnol. 2011;29(4):310–312.
- Mann M, Jensen ON. Proteomic analysis of post-translational modifications. Nat Biotechnol. 2003;21(3):255–261.
- Beck A, Sanglier-Cianférani S, Van Dorsselaer A. Biosimilar, biobetter, and next generation antibody characterization by mass spectrometry. Anal Chem. 2012;84(11):4637–4646.
- Food U, Administration D. Guidance for industry: scientific considerations in demonstrating biosimilarity to a reference product. Rockville (MD), US Food and Drug Administration; 2012.
- Sha S, Agarabi C, Brorson K, et al. N-Glycosylation design and control of therapeutic monoclonal antibodies. Trends Biotechnol. 2016;34(10):835–846.
- Shinkawa T, Nakamura K, Yamane N, et al. The absence of fucose but not the presence of galactose or bisecting N-acetylglucosamine of human IgG1 complex-type oligosaccharides shows the critical role of enhancing antibody-dependent cellular cytotoxicity. J Biol Chem. 2003;278(5):3466–3473.
- St Amand MM, Tran K, Radhakrishnan D, et al. Controllability analysis of protein glycosylation in CHO cells. PLoS One. 2014;9(2):e87973.
- Wooding KM, Peng W, Mechref Y. Characterization of pharmaceutical IgG and biosimilars using miniaturized platforms and LC-MS/MS. Curr Pharm Biotechnol. 2016;17(9):788–801.
- Hong J, Lee Y, Lee C. et al. Physicochemical and biological characterization of SB2, a biosimilar of Remicade®(infliximab). MAbs. 2017;9(2):364–382.
- Cho IH, Lee N, Song D. et al. Evaluation of the structural, physicochemical, and biological characteristics of SB4, a biosimilar of etanercept. MAbs. 2016;8(6):1136–1155.
- Kozlowski S, Swann P. Current and future issues in the manufacturing and development of monoclonal antibodies. Adv Drug Deliv Rev. 2006;58(5–6):707–722.
- Dumont J, Euwart D, Mei B, et al. Human cell lines for biopharmaceutical manufacturing: history, status, and future perspectives. Crit Rev Biotechnol. 2016;36(6):1110–1122.
- Lam SK, Ng TB. Lectins: production and practical applications. Appl Microbiol Biotechnol. 2011;89(1):45–55.
- Dobson CL, Devine PW, Phillips JJ, et al. Engineering the surface properties of a human monoclonal antibody prevents self-association and rapid clearance in vivo. Sci Rep. 2016;6:38644.
- Wouters OJ, McKee M, Luyten J. Estimated research and development investment needed to bring a new medicine to market, 2009–2018. JAMA. 2020;323(9):844–853.
- Farid SS, Baron M, Stamatis C, et al. Benchmarking biopharmaceutical process development and manufacturing cost contributions to R&D. Mabs. 2020;12(1):1754999.
- Burchiel SW, Aspbury R, Munday J. The search for biosimilars and biobetters. Drug Discov Today. 2019;24(5):1087–1091.
- Dan N, Setua S, Kashyap VK, et al. Antibody-drug conjugates for cancer therapy: chemistry to clinical implications. Pharmaceuticals. 2018;11(2):32.
- European Medicines Agency. (2005). ICH Topic Q5E, comparability of Biotechnological/Biological products. Step 5, CPMP/ICH/5721/03, 1–13.
- Neubauer S, Cifaldi M, Mittendorf T, et al. Biologic TNF inhibiting agents for treatment of rheumatoid arthritis: persistence and dosing patterns in Germany. Health Econ Rev. 2014;4(1):32.
- Smolen JS, Aletaha D, Koeller M, et al. New therapies for treatment of rheumatoid arthritis. Lancet. 2007;370(9602):1861–1874.
- Malemud CJ, Blumenthal DE. Protein kinase small molecule inhibitors for rheumatoid arthritis: Medicinal chemistry/clinical perspectives. World J Orthop. 2014;5(4):496–503.
- Pesu M, Laurence A, Kishore N, et al. Therapeutic targeting of Janus kinases. Immunol Rev. 2008;223:132–142.
- Ozminkowski RJ, Burton WN, Goetzel RZ, et al. The impact of rheumatoid arthritis on medical expenditures, absenteeism, and short-term disability benefits. J Occup Environ Med. 2006;48(2):135–148.
- Albers JM, Kuper HH, van Riel PL, et al. Socio-economic consequences of rheumatoid arthritis in the first years of the disease. Rheumatology. 1999;38(5):423–430.
- Birnbaum H, Pike C, Kaufman R, et al. Societal cost of rheumatoid arthritis patients in the US. Curr Med Res Opin. 2010;26(1):77–90.
- Zhang W, Anis AH. The economic burden of rheumatoid arthritis: beyond health care costs. Clin Rheumatol. 2011;30(S1):25–32.
- Boonen A, Severens JL. The burden of illness of rheumatoid arthritis. Clin Rheumatol. 2011;30(S1):3–8.
- Cooper NJ. Economic burden of rheumatoid arthritis: a systematic review. Rheumatology. 2000;39(1):28–33.
- Yoon SJ, Bae SC, Lee SI, et al. Measuring the burden of disease in Korea. J Korean Med Sci. 2007;22(3):518–523.
- Koenders MI, van den Berg WB. Novel therapeutic targets in rheumatoid arthritis. Trends Pharmacol Sci. 2015;36(4):189–195.
- Foundation A. Arthritis By The Numbers Atlanta, USA. 2018; [cited 2019 Dec 22]. Available from: https://www.arthritis.org/Documents/Sections/About-Arthritis/arthritis-facts-stats-figures.pdf.
- Aj LB. The first description of rheumatoid arthritis. Unabridged text of the doctoral dissertation presented in 1800. Joint Bone Spine. 2001;68(2):130–143.
- Fauci A. Harrison’s principle of internal medicine. 18 ed. United States: McGraw Hill; 2011.
- Silman AJ, Hochberg MC. Epidemiology of the rheumatic diseases. 2nd ed. New York: Oxford University Press; 2001.
- McInnes IB, Schett G. The pathogenesis of rheumatoid arthritis. N Engl J Med. 2011;365(23):2205–2219.
- Smolen JS, Steiner G. Therapeutic strategies for rheumatoid arthritis. Nat Rev Drug Discov. 2003;2(6):473–488.
- Duke O, Panayi GS. The pathogenesis of rheumatoid arthritis. In Vivo. 1988;2(1):95–103.
- Jimenez-Boj E, Redlich K, Türk B, et al. Interaction between synovial inflammatory tissue and bone marrow in rheumatoid arthritis. J Immunol. 2005;175(4):2579–2588.
- McInnes IB, Schett G. Cytokines in the pathogenesis of rheumatoid arthritis. Nat Rev Immunol. 2007;7(6):429–442.
- Maini RN, Taylor PC. Anti-cytokine therapy for rheumatoid arthritis. Annu Rev Med. 2000;51:207–229.
- McInnes IB, Liew FY. Cytokine networks–towards new therapies for rheumatoid arthritis. Nat Clin Pract Rheumatol. 2005;1(1):31–39.
- Gaujoux-Viala C, Nam J, Ramiro S, et al. EULAR recommendations for the management of rheumatoid arthritis with synthetic and biological disease-modifying antirheumatic drugs: 2019 update. Ann Rheum Dis. 2014;73(3):510–515.
- Smolen JS, Landewe R, Bijlsma J, et al. EULAR recommendations for the management of rheumatoid arthritis with synthetic and biological disease-modifying antirheumatic drugs: 2016 update. Ann Rheum Dis. 2017;76(6):960–977.
- Emery P, Breedveld FC, Hall S, et al. Comparison of methotrexate monotherapy with a combination of methotrexate and etanercept in active, early, moderate to severe rheumatoid arthritis (COMET): a randomised, double-blind, parallel treatment trial. Lancet. 2008;372(9636):375–382.
- Smolen JS, Kay J, Doyle MK, et al. Golimumab in patients with active rheumatoid arthritis after treatment with tumour necrosis factor alpha inhibitors (GO-AFTER study): a multicentre, randomised, double-blind, placebo-controlled, phase III trial. Lancet. 2009;374(9685):210–221.
- Emery P, Gottenberg JE, Rubbert-Roth A, et al. Rituximab versus an alternative TNF inhibitor in patients with rheumatoid arthritis who failed to respond to a single previous TNF inhibitor: SWITCH-RA, a global, observational, comparative effectiveness study. Ann Rheum Dis. 2015;74(6):979–984.
- Gottenberg JE, Morel J, Perrodeau E, et al. Comparative effectiveness of rituximab, abatacept, and tocilizumab in adults with rheumatoid arthritis and inadequate response to TNF inhibitors: prospective cohort study. BMJ. 2019;364:l67.
- Gottenberg JE, Brocq O, Perdriger A, et al. Non-TNF-targeted biologic vs a second anti-TNF drug to treat rheumatoid arthritis in patients with insufficient response to a first anti-TNF drug: a randomized clinical trial. JAMA. 2016;316(11):1172–1180.
- Teitsma XM, Marijnissen AK, Bijlsma JW, et al. Tocilizumab as monotherapy or combination therapy for treating active rheumatoid arthritis: a meta-analysis of efficacy and safety reported in randomized controlled trials. Arthritis Res Ther. 2016;18(1):211.
- Emery P, Burmester GR, Bykerk VP, et al. Evaluating drug-free remission with abatacept in early rheumatoid arthritis: results from the phase 3b, multicentre, randomised, active-controlled AVERT study of 24 months, with a 12-month, double-blind treatment period. Ann Rheum Dis. 2015;74(1):19–26.
- Klareskog L, van der Heijde D, de Jager JP, et al. Therapeutic effect of the combination of etanercept and methotrexate compared with each treatment alone in patients with rheumatoid arthritis: double-blind randomised controlled trial. Lancet. 2004;363(9410):675–681.
- Breedveld FC, Weisman MH, Kavanaugh AF, et al. The PREMIER study: a multicenter, randomized, double-blind clinical trial of combination therapy with adalimumab plus methotrexate versus methotrexate alone or adalimumab alone in patients with early, aggressive rheumatoid arthritis who had not had previous methotrexate treatment. Arthritis Rheum. 2006;54(1):26–37.
- Jansen JP, Buckley F, Dejonckheere F, et al. Comparative efficacy of biologics as monotherapy and in combination with methotrexate on patient reported outcomes (PROs) in rheumatoid arthritis patients with an inadequate response to conventional DMARDs–a systematic review and network meta-analysis. Health Qual Life Outcomes. 2014;12(1):102.
- Kalliolias GD, Liossis SN. The future of the IL-1 receptor antagonist anakinra: from rheumatoid arthritis to adult-onset Still’s disease and systemic-onset juvenile idiopathic arthritis. Expert Opin Invest Drugs. 2008;17(3):349–359.
- Schiff M, Keiserman M, Codding C, et al. Efficacy and safety of abatacept or infliximab vs placebo in ATTEST: a phase III, multi-centre, randomised, double-blind, placebo-controlled study in patients with rheumatoid arthritis and an inadequate response to methotrexate. Ann Rheum Dis. 2008;67(8):1096–1103.
- Cabral VP, Andrade CA, Passos SR, et al. Severe infection in patients with rheumatoid arthritis taking anakinra, rituximab, or abatacept: a systematic review of observational studies. Rev Bras Reumatol Engl Ed. 2016;56(6):543–550.
- Salliot C, Dougados M, Gossec L. Risk of serious infections during rituximab, abatacept and anakinra treatments for rheumatoid arthritis: meta-analyses of randomised placebo-controlled trials. Ann Rheum Dis. 2009;68(1):25–32.
- Fleischmann RM, Genovese MC, Enejosa JV, et al. Safety and effectiveness of upadacitinib or adalimumab plus methotrexate in patients with rheumatoid arthritis over 48 weeks with switch to alternate therapy in patients with insufficient response. Ann Rheum Dis. 2019;78(11):1454–1462.
- Vinson D, Molet-Benhamou L, Degboe Y, et al. Impact of tapering targeted therapies (bDMARDs or JAKis) on the risk of serious infections and adverse events of special interest in patients with rheumatoid arthritis or spondyloarthritis: a systematic analysis of the literature and meta-analysis. Arthritis Res Ther. 2020;22(1):97.
- Ramiro S, Sepriano A, Chatzidionysiou K, et al. Safety of synthetic and biological DMARDs: a systematic literature review informing the 2016 update of the EULAR recommendations for management of rheumatoid arthritis. Ann Rheum Dis. 2017;76(6):1101–1136.
- Dreyer L, Cordtz RL, Hansen IMJ, et al. Risk of second malignant neoplasm and mortality in patients with rheumatoid arthritis treated with biological DMARDs: a Danish population-based cohort study. Ann Rheum Dis. 2018;77(4):510–514.
- Burmester GR, Pope JE. Novel treatment strategies in rheumatoid arthritis. Lancet. 2017;389(10086):2338–2348.
- Verden A, Dimbil M, Kyle R, et al. Analysis of spontaneous postmarket case reports submitted to the FDA regarding thromboembolic adverse events and JAK inhibitors. Drug Saf. 2018;41(4):357–361.
- Kaufman MB. Pharmaceutical approval update. P T. 2018;43(9):528–530.
- EMA. Xeljanz 2019. [updated 5-7-2019; cited 2020 Oct 15]. Available from: https://www.ema.europa.eu/en/medicines/human/EPAR/xeljanz.
- FDA. FDA approves Boxed Warning about increased risk of blood clots and death with higher dose of arthritis and ulcerative colitis medicine tofacitinib (Xeljanz, Xeljanz XR). 2019; [updated 28-8-2019; cited 2019 Oct 15]. Available from: https://www.fda.gov/drugs/drug-safety-and-availability/fda-approves-boxed-warning-about-increased-risk-blood-clots-and-death-higher-dose-arthritis-and
- Becede M, Alasti F, Gessl I, et al. Risk profiling for a refractory course of rheumatoid arthritis. Semin Arthritis Rheum. 2019;49(2):211–217.
- Tracey D, Klareskog L, Sasso EH, et al. Tumor necrosis factor antagonist mechanisms of action: a comprehensive review. Pharmacol Ther. 2008;117(2):244–279.
- Azevedo VF, Galli N, Kleinfelder A, et al. Etanercept biosimilars. Rheumatol Int. 2015;35(2):197–209.
- Hess A, Axmann R, Rech J, et al. Blockade of TNF-alpha rapidly inhibits pain responses in the central nervous system. Proc Natl Acad Sci USA. 2011;108(9):3731–3736.
- Feldmann M, Brennan FM, Maini RN. Rheumatoid arthritis. Cell. 1996;85(3):307–310.
- Vervoordeldonk MJ, Tak PP. Cytokines in rheumatoid arthritis. Curr Rheumatol Rep. 2002;4(3):208–217.
- Feldmann M, Brennan FM, Maini RN. Role of cytokines in rheumatoid arthritis. Annu Rev Immunol. 1996;14:397–440.
- Zhang JM, An J. Cytokines, inflammation, and pain. Int Anesthesiol Clin. 2007;45(2):27–37.
- Raza K, Falciani F, Curnow SJ, et al. Early rheumatoid arthritis is characterized by a distinct and transient synovial fluid cytokine profile of T cell and stromal cell origin. Arthritis Res Ther. 2005;7(4):R784–R795.
- Feldmann M, Brennan F, Bondeson J, et al. Analysis of cytokine expression in rheumatoid synovium has provided new insights into the pathogenesis of rheumatoid arthritis and new therapeutic opportunities. Transplant Proc. 2001;33(3):2085–2086.
- Rossi JF, Lu ZY, Jourdan M, et al. Interleukin-6 as a therapeutic target. Clin Cancer Res. 2015;21(6):1248–1257.
- Feldmann M, Maini RN, Bondeson J, et al. Cytokine blockade in rheumatoid arthritis. Adv Exp Med Biol. 2001;490:119–127.
- Feldmann M, Maini RN. Anti-TNF alpha therapy of rheumatoid arthritis: what have we learned? Annu Rev Immunol. 2001;19(1):163–196.
- Taylor PC, Peters AM, Paleolog E, et al. Reduction of chemokine levels and leukocyte traffic to joints by tumor necrosis factor alpha blockade in patients with rheumatoid arthritis. Arthritis Rheum. 2000;43(1):38–47.
- Toussirot E, Wendling D. The use of TNF-alpha blocking agents in rheumatoid arthritis: an overview. Expert Opin Pharmacother. 2004;5(3):581–594.
- Raychaudhuri SP, Nguyen CT, Raychaudhuri SK, et al. Incidence and nature of infectious disease in patients treated with anti-TNF agents. Autoimmun Rev. 2009;9(2):67–81.
- Saklatvala J. Tumour necrosis factor alpha stimulates resorption and inhibits synthesis of proteoglycan in cartilage. Nature. 1986;322(6079):547–549.
- Bathon JM, Martin RW, Fleischmann RM, et al. A comparison of etanercept and methotrexate in patients with early rheumatoid arthritis. N Engl J Med. 2000;343(22):1586–1593.
- Maini R, St Clair EW, Breedveld F, et al. Infliximab (chimeric anti-tumour necrosis factor alpha monoclonal antibody) versus placebo in rheumatoid arthritis patients receiving concomitant methotrexate: a randomised phase III trial. ATTRACT Study Group. Lancet. 1999;354(9194):1932–1939.
- Maini RN, Elliott M, Brennan FM, et al. TNF blockade in rheumatoid arthritis: implications for therapy and pathogenesis. APMIS. 1997;105(4):257–263.
- Bhatnagar R, Penfornis H, Mauviel A, et al. Interleukin-1 inhibits the synthesis of collagen by fibroblasts. Biochem Int. 1986;13(4):709–720.
- Smolen JS, Weinblatt ME. When patients with rheumatoid arthritis fail tumour necrosis factor inhibitors: what is the next step? Ann Rheum Dis. 2008;67(11):1497–1498.
- Goekoop-Ruiterman YP, de Vries-Bouwstra JK, Allaart CF, et al. Clinical and radiographic outcomes of four different treatment strategies in patients with early rheumatoid arthritis (the BeSt study): a randomized, controlled trial. Arthritis Rheum. 2005;52(11):3381–3390.
- Gerlag DM, Tak PP. Novel approaches for the treatment of rheumatoid arthritis: lessons from the evaluation of synovial biomarkers in clinical trials. Best Pract Res Clin Rheumatol. 2008;22(2):311–323.
- Brennan FM, Chantry D, Jackson A, et al. Inhibitory effect of TNF alpha antibodies on synovial cell interleukin-1 production in rheumatoid arthritis. Lancet. 1989;334(8657):244–247.
- Bystrom J, Clanchy FI, Taher TE, et al. Response to treatment with TNFalpha inhibitors in rheumatoid arthritis is associated with high levels of GM-CSF and GM-CSF(+) T lymphocytes. Clin Rev Allergy Immunol. 2017;53(2):265–276.
- Polzer K, Baeten D, Soleiman A, et al. Tumour necrosis factor blockade increases lymphangiogenesis in murine and human arthritic joints. Ann Rheum Dis. 2008;67(11):1610–1616.
- Szekanecz Z, Pakozdi A, Szentpetery A, et al. Chemokines and angiogenesis in rheumatoid arthritis. Front Biosci. 2009;1:44–51.
- Bradfield PF, Amft N, Vernon-Wilson E, et al. Rheumatoid fibroblast-like synoviocytes overexpress the chemokine stromal cell-derived factor 1 (CXCL12), which supports distinct patterns and rates of CD4+ and CD8+ T cell migration within synovial tissue. Arthritis Rheum. 2003;48(9):2472–2482.
- Maini RN, Feldmann M. How does infliximab work in rheumatoid arthritis? Arthritis Res. 2002;4 (Suppl 2):S22–S28.
- Lim H, Lee SH, Lee HT, et al. Structural biology of the TNFalpha antagonists used in the treatment of rheumatoid arthritis. Int J Mol Sci. 2018;19(3):768.
- Chen X, DuBois DC, Almon RR, et al. Interrelationships between Infliximab and recombinant tumor necrosis factor-alpha in plasma using minimal physiologically based pharmacokinetic models. Drug Metab Dispos. 2017;45(7):790–797.
- Frenzel A, Schirrmann T, Hust M. Phage display-derived human antibodies in clinical development and therapy. MAbs. 2016;8(7):1177–1194.
- Jespers LS, Roberts A, Mahler SM, et al. Guiding the selection of human antibodies from phage display repertoires to a single epitope of an antigen. Nat Biotechnol. 1994;12(9):899–903.
- Grazio S. [Adalimumab (Humira)-efficacy in rheumatoid arthritis treatment with particular reference to working ability]. Reumatizam. 2008;55(2):62–67.
- Guo Q, Wang Y, Xu D, et al. Rheumatoid arthritis: pathological mechanisms and modern pharmacologic therapies. Bone Res. 2018;6:15.
- Keystone E, Haraoui B. Adalimumab therapy in rheumatoid arthritis. Rheum Dis Clin North Am. 2004;30(2):349–364.
- Goel N, Stephens S. Certolizumab pegol. MAbs. 2010;2(2):137–147.
- Veronese FM, Pasut G. PEGylation, successful approach to drug delivery. Drug Discov Today. 2005;10(21):1451–1458.
- Chapman AP. PEGylated antibodies and antibody fragments for improved therapy: a review. Adv Drug Deliv Rev. 2002;54(4):531–545.
- Chapman AP, Antoniw P, Spitali M, et al. Therapeutic antibody fragments with prolonged in vivo half-lives. Nat Biotechnol. 1999;17(8):780–783.
- Nesbitt A, Fossati G, Bergin M, et al. Mechanism of action of certolizumab pegol (CDP870): in vitro comparison with other anti-tumor necrosis factor alpha agents. Inflamm Bowel Dis. 2007;13(11):1323–1332.
- Keystone E, Heijde D, Mason D, Jr, et al. Certolizumab pegol plus methotrexate is significantly more effective than placebo plus methotrexate in active rheumatoid arthritis: findings of a fifty-two-week, phase III, multicenter, randomized, double-blind, placebo-controlled, parallel-group study. Arthritis Rheum. 2008;58(11):3319–3329.
- Smolen J, Landewe RB, Mease P, et al. Efficacy and safety of certolizumab pegol plus methotrexate in active rheumatoid arthritis: the RAPID 2 study. A randomised controlled trial. Ann Rheum Dis. 2009;68(6):797–804.
- Palframan R, Airey M, Moore A, et al. Use of biofluorescence imaging to compare the distribution of certolizumab pegol, adalimumab, and infliximab in the inflamed paws of mice with collagen-induced arthritis. J Immunol Methods. 2009;348(1–2):36–41.
- Mease PJ, Armstrong AW. Managing patients with psoriatic disease: the diagnosis and pharmacologic treatment of psoriatic arthritis in patients with psoriasis. Drugs. 2014;74(4):423–441.
- Bykerk VP, Cush J, Winthrop K, et al. Update on the safety profile of certolizumab pegol in rheumatoid arthritis: an integrated analysis from clinical trials. Ann Rheum Dis. 2015;74(1):96–103.
- Inman RD, Davis JC, Jr., Heijde D, et al. Efficacy and safety of golimumab in patients with ankylosing spondylitis: results of a randomized, double-blind, placebo-controlled, phase III trial. Arthritis Rheum. 2008;58(11):3402–3412.
- Kavanaugh A, McInnes I, Mease P, et al. Golimumab, a new human tumor necrosis factor alpha antibody, administered every four weeks as a subcutaneous injection in psoriatic arthritis: Twenty-four-week efficacy and safety results of a randomized, placebo-controlled study. Arthritis Rheum. 2009;60(4):976–986.
- Hutas G. Golimumab, a fully human monoclonal antibody against TNFalpha. Curr Opin Mol Ther. 2008;10(4):393–406.
- Emery P, Fleischmann RM, Strusberg I, et al. Efficacy and safety of subcutaneous golimumab in methotrexate-naive patients with rheumatoid arthritis: five-year results of a randomized clinical trial. Arthritis Care Res. 2016;68(6):744–752.
- Xu Z, Vu T, Lee H, et al. Population pharmacokinetics of golimumab, an anti-tumor necrosis factor-alpha human monoclonal antibody, in patients with psoriatic arthritis. J Clin Pharmacol. 2009;49(9):1056–1070.
- Caporali R, Caprioli M, Bobbio-Pallavicini F, et al. DMARDS and infections in rheumatoid arthritis. Autoimmun Rev. 2008;8(2):139–143.
- Zidi I, Mestiri S, Bartegi A, et al. TNF-alpha and its inhibitors in cancer. Med Oncol. 2010;27(2):185–198.
- Keystone EC, Genovese MC, Klareskog L, et al. Golimumab, a human antibody to tumour necrosis factor {alpha} given by monthly subcutaneous injections, in active rheumatoid arthritis despite methotrexate therapy: the GO-FORWARD Study. Ann Rheum Dis. 2009;68(6):789–796.
- Emery P, Fleischmann RM, Moreland LW, et al. Golimumab, a human anti-tumor necrosis factor alpha monoclonal antibody, injected subcutaneously every four weeks in methotrexate-naive patients with active rheumatoid arthritis: twenty-four-week results of a phase III, multicenter, randomized, double-blind, placebo-controlled study of golimumab before methotrexate as first-line therapy for early-onset rheumatoid arthritis. Arthritis Rheum. 2009;60(8):2272–2283.
- Acevedo-Vasquez E, Ponce de Leon D, Gamboa-Cardenas R. Latent infection and tuberculosis disease in rheumatoid arthritis patients. Rheum Dis Clin North Am. 2009;35(1):163–181.
- Dumitru RB, Horton S, Hodgson R, et al. A prospective, single-centre, randomised study evaluating the clinical, imaging and immunological depth of remission achieved by very early versus delayed Etanercept in patients with Rheumatoid Arthritis (VEDERA). BMC Musculoskelet Disord. 2016;17:61.
- Moreland LW, Schiff MH, Baumgartner SW, et al. Etanercept therapy in rheumatoid arthritis. A randomized, controlled trial. Ann Intern Med. 1999;130(6):478–486.
- Madhusudan S, Muthuramalingam SR, Braybrooke JP, et al. Study of etanercept, a tumor necrosis factor-alpha inhibitor, in recurrent ovarian cancer. J Clin Oncol. 2005;23(25):5950–5959.
- Mears JB, Huynh-Duc L, Fiechtner JJ. A patient with rheumatoid arthritis on methotrexate and etanercept who developed Pasteurella multocida Bacteremia. J Clin Rheumatol. 2015;21(8):457. Dec
- Neovius M, Arkema EV, Olsson H, et al. Drug survival on TNF inhibitors in patients with rheumatoid arthritis comparison of adalimumab, etanercept and infliximab. Ann Rheum Dis. 2015;74(2):354–360.
- McCamish M, Woollett G. The state of the art in the development of biosimilars. Clin Pharmacol Ther. 2012;91(3):405–417.
- Kay J, Smolen JS. Biosimilars to treat inflammatory arthritis: the challenge of proving identity. Ann Rheum Dis. 2013;72(10):1589–1593.
- Beck A, Reichert JM. Approval of the first biosimilar antibodies in Europe: a major landmark for the biopharmaceutical industry. MAbs. 2013;5(5):621–623.
- Rinaudo-Gaujous M, Paul S, Tedesco ED, et al. Review article: biosimilars are the next generation of drugs for liver and gastrointestinal diseases. Aliment Pharmacol Ther. 2013;38(8):914–924.
- Hlavaty T, Letkovsky J. Biosimilars in the therapy of inflammatory bowel diseases. Eur J Gastroenterol Hepatol. 2014;26(6):581–587.
- Genazzani A, Altomare G, Balato N, et al. Biosimilar infliximab: an expert view. G Ital Dermatol Venereol. 2015;150(4):449–459.
- FDA. FDA approves first biosimilar product Zarxio: FDA; 2015; [cited 2016 Feb 16]. Available from: http://www.fda.gov/NewsEvents/Newsroom/PressAnnouncements/ucm436648.htm.
- Ledford H. Biosimilar’ drugs poised to penetrate market. Nature. 2010;468(7320):18–19.
- Jung SK, Lee KH, Jeon JW, et al. Physicochemical characterization of Remsima. MAbs. 2014;6(5):1163–1177.
- Tang H, Hsueh P, Kletter D, et al. The detection and discovery of glycan motifs in biological samples using lectins and antibodies: new methods and opportunities. Adv Cancer Res. 2015;126:167–202.
- Zhang P, Woen S, Wang T, et al. Challenges of glycosylation analysis and control: an integrated approach to producing optimal and consistent therapeutic drugs. Drug Discov Today. 2016;21(5):740–765.
- Ahmadi S, Davami F, Davoudi N, et al. Monoclonal antibodies expression improvement in CHO cells by PiggyBac transposition regarding vectors ratios and design. PLoS One. 2017;12(6):e0179902.
- Krambeck FJ, Bennun SV, Andersen MR, et al. Model-based analysis of N-glycosylation in Chinese hamster ovary cells. PLoS One. 2017;12(5):e0175376.
- Magnelli PE, Bielik AM, Guthrie EP. Identification and characterization of protein glycosylation using specific endo- and exoglycosidases. J Vis Exp. 2011;26(58):e3749.
- Jefferis R. Recombinant antibody therapeutics: the impact of glycosylation on mechanisms of action. Trends Pharmacol Sci. 2009;30(7):356–362.
- Cao M, De Mel N, Shannon A, et al. Charge variants characterization and release assay development for co-formulated antibodies as a combination therapy. MAbs. 2019;11(3):489–499.
- Duivelshof BL, Fekete S, Guillarme D, et al. A generic workflow for the characterization of therapeutic monoclonal antibodies-application to daratumumab. Anal Bioanal Chem. 2019;411(19):4615–4627.
- Gomes RA, Almeida C, Correia C, et al. Exploring the analytical power of the QTOF MS platform to assess monoclonal antibodies quality attributes. PLoS One. 2019;14(7):e0219156.
- Lee JH, Paek K, Moon JH, et al. Biological characterization of SB3, a trastuzumab biosimilar, and the influence of changes in reference product characteristics on the similarity assessment. BioDrugs. 2019;33(4):411–422.
- Lippold S, Nicolardi S, Dominguez-Vega E, et al. Glycoform-resolved FcRIIIa affinity chromatography-mass spectrometry. MAbs. 2019;11(7):1191–1196.
- FDA. FDA approves Inflectra, a biosimilar to Remicade: FDA; 2016; [cited 2016 Jun 28]. Available from: http://www.fda.gov/NewsEvents/Newsroom/PressAnnouncements/ucm494227.htm.
- Blair HA, Deeks ED. Infliximab biosimilar (CT-P13; Infliximab-dyyb): a review in autoimmune inflammatory diseases. BioDrugs. 2016;30(5):469–480.
- Jung YS, Park DI, Kim YH, et al. Efficacy and safety of CT-P13, a biosimilar of infliximab, in patients with inflammatory bowel disease: a retrospective multicenter study. J Gastroenterol Hepatol. 2015;30(12):1705–1712.
- Park W, Hrycaj P, Jeka S, et al. A randomised, double-blind, multicentre, parallel-group, prospective study comparing the pharmacokinetics, safety, and efficacy of CT-P13 and innovator infliximab in patients with ankylosing spondylitis: the PLANETAS study. Ann Rheum Dis. 2013;72(10):1605–1612.
- Ahrweiler S. ACR Response Criteria: PhuseWiki; 2012; [cited 2020 Oct 03]. Available from: https://www.phusewiki.org/wiki/index.php?title=ACR_Response_Criteria.
- Yoo DH, Hrycaj P, Miranda P, et al. A randomised, double-blind, parallel-group study to demonstrate equivalence in efficacy and safety of CT-P13 compared with innovator infliximab when coadministered with methotrexate in patients with active rheumatoid arthritis: the PLANETRA study. Ann Rheum Dis. 2013;72(10):1613–1620.
- Emery P, Vencovsky J, Sylwestrzak A, et al. A phase III randomised, double-blind, parallel-group study comparing SB4 with etanercept reference product in patients with active rheumatoid arthritis despite methotrexate therapy. Ann Rheum Dis. 2017;76(1):51–57.
- Bae SC, Kim J, Choe JY, et al. A phase III, multicentre, randomised, double-blind, active-controlled, parallel-group trial comparing safety and efficacy of HD203, with innovator etanercept, in combination with methotrexate, in patients with rheumatoid arthritis: the HERA study. Ann Rheum Dis. 2017;76(1):65–71.
- Castaneda-Hernandez G, Szekanecz Z, Mysler E, et al. Biopharmaceuticals for rheumatic diseases in Latin America, Europe, Russia, and India: innovators, biosimilars, and intended copies. Joint Bone Spine. 2014;81(6):471–477.
- EMA. FLIXABI: EUROPEAN MEDICINE AGENCY; 2016; [cited 2016 Jun 26]. Available from: http://www.ema.europa.eu/ema/index.jsp?curl=pages/medicines/human/medicines/004020/human_med_001980.jsp&mid=WC0b01ac058001d124.
- Takeuchi T, Yamanaka H, Tanaka Y, et al. Evaluation of the pharmacokinetic equivalence and 54-week efficacy and safety of CT-P13 and innovator infliximab in Japanese patients with rheumatoid arthritis. Mod Rheumatol. 2015;25(6):817–824.
- EMA. Benepali: EUROPEAN MEDICINES AGENCY; 2016; [cited 2016 June 18]. Available from: http://www.ema.europa.eu/ema/index.jsp?curl=pages/medicines/human/medicines/004007/human_med_001944.jsp&mid=WC0b01ac058001d124.
- Chadwick L, Zhao S, Mysler E, et al. Review of biosimilar trials and data on etanercept in rheumatoid arthritis. Curr Rheumatol Rep. 2018;20(12):84.
- Schneider CK. Biosimilars in rheumatology: the wind of change. Ann Rheum Dis. 2013;72(3):315–318.
- Castaneda-Hernandez G, Gonzalez-Ramirez R, Kay J, et al. Biosimilars in rheumatology: what the clinician should know. RMD Open. 2015;1(1):e000010–e000010.
- Zhao S, Chadwick L, Mysler E, et al. Review of biosimilar trials and data on adalimumab in rheumatoid arthritis. Curr Rheumatol Rep. 2018;20(10):57.
- Papp K, Bachelez H, Costanzo A, et al. Clinical similarity of the biosimilar ABP 501 compared with adalimumab after single transition: long‐term results from a randomized controlled, double‐blind, 52‐week, phase III trial in patients with moderate‐to‐severe plaque psoriasis. Br J Dermatol. 2017;177(6):1562–1574.
- Markus R, McBride HJ, Ramchandani M, et al. A review of the totality of evidence supporting the development of the first adalimumab biosimilar ABP 501. Adv Ther. 2019;36(8):1818–1833.
- Dorner T, Kay J. Biosimilars in rheumatology: current perspectives and lessons learnt. Nat Rev Rheumatol. 2015;11(12):713–724.
- Papamichael K, Van Stappen T, Jairath V, et al. Review article: pharmacological aspects of anti-TNF biosimilars in inflammatory bowel diseases. Aliment Pharmacol Ther. 2015;42(10):1158–1169.
- Geiler J, Buch M, McDermott MF. Anti-TNF treatment in rheumatoid arthritis. Curr Pharm Des. 2011;17(29):3141–3154.
- Lai Z, La Noce A. Key design considerations on comparative clinical efficacy studies for biosimilars: adalimumab as an example. RMD Open. 2016;2(1):e000154.
- Jayaraman K. India’s Cipla sets sights on avastin, herceptin and enbrel. Nat Biotechnol. 2010;28(9):883–884.
- Roy PS, John S, Karankal S, et al. Comparison of the efficacy and safety of Rituximab (Mabthera™) and its biosimilar (Reditux™) in diffuse large B-cell lymphoma patients treated with chemo-immunotherapy: a retrospective analysis. Indian J Med Paediatr Oncol. 2013;34(4):292.
- Midha V, Mahajan R, Mehta V, et al. Efficacy and safety of the adalimumab biosimilar Exemptia as induction therapy in moderate-to-severe ulcerative colitis. Intest Res. 2018;16(1):83–89.
- Statista. Top 10 pharmaceutical products by sales worldwide in 2015 (in billion U.S. dollars): The Statistics Portal; 2016. [cited 2016 Jun 26]. Available from: http://www.statista.com/statistics/258022/top-10-pharmaceutical-products-by-global-sales-2011/.
- Meier FM, McInnes IB. Small-molecule therapeutics in rheumatoid arthritis: scientific rationale, efficacy and safety. Best Pract Res Clin Rheumatol. 2014;28(4):605–624.
- Simmons DL. Targeting kinases: a new approach to treating inflammatory rheumatic diseases. Curr Opin Pharmacol. 2013;13(3):426–434.
- Cohen MD, Keystone EC. JAK inhibitors for rheumatoid arthritis. Curr Treat Options in Rheum. 2015;1(4):305–319.
- Kavanaugh A, Smolen JS, Emery P, et al. Effect of certolizumab pegol with methotrexate on home and work place productivity and social activities in patients with active rheumatoid arthritis. Arthritis Rheum. 2009;61(11):1592–1600.
- Scheinberg MA, Azevedo VF. Biosimilars in rheumatology: perspective and concerns. Rheumatology. 2014;53(3):389–390.
- Expensive treatments for genetic disorders are arriving. But who should foot the bill? Nature Editorial. 2019:576:7–8.