Currently, there is an intense discussion about protein folding (Ramalho & Da Cunha, Citation2011). Studies have shown the relevance of hydrophobic interactions in protein cavities performing an equilibrium force with solvent effects and electrostatic interactions (Ramalho & Da Cunha, Citation2011).
Recently, however, Ben-Naim reported that hydrophobic interactions can be neglected and the force induced for the protein folding is determined just by the gradient of the solvation Gibbs free energy of the protein at a given conformation (Ben-Naim, Citation2012). In the last three decades, in order to address this question, molecular simulation studies have been used for the development of the modern perspective on protein folding. However, it is also well known that hydrophobic interaction computations by conventional computational procedures based on force fields are not a simple task (Ben-Naim, Citation2012). Hydrophobic effects among aromatic amino acids can take place by π–π and T-stacking interactions, which involve high electronic correction. In this case, to achieve accuracy, it is crucial to employ highly correlated electronic structure methods for ground states of molecules, such as Density Functional Theory (DFT) and Møller–Plesset perturbation theory (MP) approaches (Wheeler, Citation2011). In fact, those theoretical approaches can, in principle, provide the most fundamental understanding of chemical phenomenon involving hydrophobic interactions and help experimentalists in the interpretation of their measurements. To date, considering the size of a protein, electronic structure calculations of a whole protein still remain unfeasible to carry out it. In this context, the docking calculation using conventional force fields are invoked in general. It should be kept in mind, however, that detailed aspects of protein folding due to hydrophobic interactions are not easily accessible by theoretical calculation using conventional force fields. Thus, the use of inappropriate interaction potentials can lead to some errors when quantum effects take place.
It is well known that quantum effects can have crucial importance for the protein-ligand recognition (Ying-Qi & Ke-Li, Citation2010). For instance, when the electronic polarization is gradually included, the binding affinity between protein and ligands can change significantly. Besides polarization effects, the electron and hydrogen tunneling which have been uncovered in enzymatic reactions involving quantum tunneling effects are being explored (Glowacki, Harvey, & Mulholland, Citation2012; Nagel, Meadows, Dong, Bahnson, & Klinman, Citation2012). Those effects cannot be modeled by molecular mechanics (MM) calculations, hence, quantum mechanics (QM) or QM/MM approaches are indicated for introducing those electronic effects in the active site of proteins (de la Lande et al., Citation2012).
In order to evaluate the electronic effect for hydrophobic interaction, we have constructed potential energy curves (PEC) for model systems, which could simulate hydrophobic interactions in proteins. In fact, the concept of the PEC plays a crucial role in the understanding of chemical reactions, as well as description, simulation, and modeling of molecular systems.
In line with that the goal of this study is to evaluate the hydrophobic/electrostatic interactions on the model system inosine + phenylalanine dimmer.
The calculations were carried out at the DFT level. Thus, the B3LYP/6-31G∗∗ level has been used to obtain the molecular geometry. The minimum global energy was obtained by conformational analysis in Spartan Pro. The potential energy surface (PES) was evaluated by single point calculations using MM (force field – universal force field [UFF]), DFT, and MP2 methods. The most stable dimmer conformations are displayed in Figure .
Figure 1 (a) Parallel eclipsed, (b) Parallel staggered, (c) Parallel displaced, (d) T-shaped, (e) Angular.
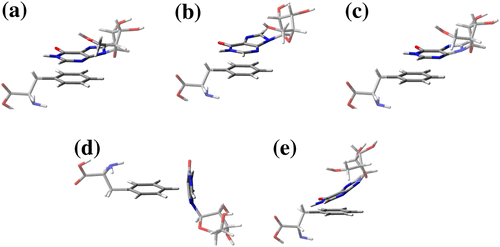
π–π stacking interactions
Firstly, four conformations were evaluated for the dimmer. Those first three conformations represent π–π stacking interactions: A and B, which can, in principle, highlight higher hyperconjucative effects; C with a little bend, which decreases the hyperconjucative effects. On the other hand, conformations D and E simulate T-stacking interactions. Figure shows distinct behavior among the studied conformations. From our results (Figure ), the most and least stable studied dimmer conformations were C and D, respectively. Our findings point out quite a difference between MM (UFF) and pos-Hartree-Fock (MP2) methods for the π–π∗ systems (A, B, and C). The electronic structure method revealed some attractive effects, which are not computed by the empirical methods. As a consequence, the lowest binding energy has been moved for the dimmer conformations A and D, which can significantly affect the interaction between a ligand and a hydrophobic active site of a protein. This information is represented in Figure .
Figure 2 PEC comparing MP2 and UFF results for dimmer conformations. (a) Parallel eclipsed, (b) Parallel staggered, (c) Parallel displaced, (d) Angular.
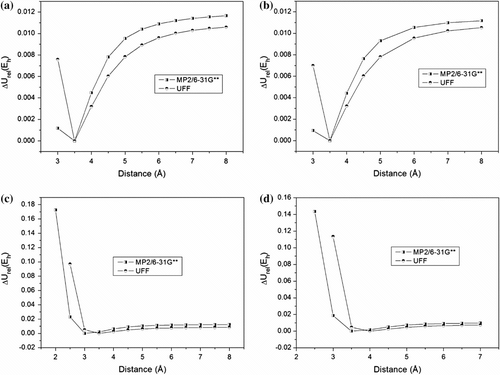
Figure 3 Potential energy columns comparing results of MP2 and UFF methods each conformation. (a) Parallel eclipsed, (b) Parallel staggered, (c) Parallel displaced, (d) Angular.
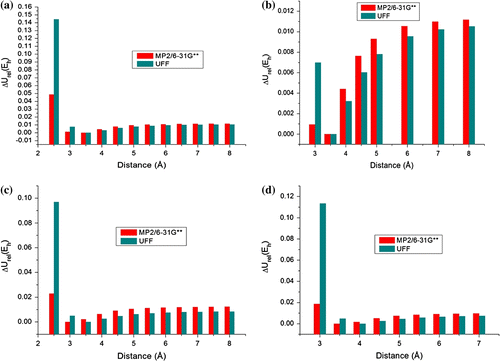
By analyzing Figure , we can observe high discrepancies in relative interaction energy (ΔU rel) when the dimmer gets closer beyond the most stable configuration. At these distances, quantum effects and attractive forces are crucial to stabilize the dimmer, which are not computed completely in the UFF force field. In fact, the discrepancy is higher in A and D conformations. Both methods seem to agree at very long distances, at which the relative energy values are close. Nevertheless, at local minimums (Figure ), MM and QM data diverge significantly. Thus, simple force field methods can not accurately predict π–π interactions due to hyperconjugative and electronic correlation effects at shorter distances.
T-stacking interactions
Another possible dimmer conformation is the T-shaped. Our findings clearly show divergences between MM and QM methods for this conformation as well (Figure ).
Figure 4 PEC and columns comparing MP2 and UFF methods for T-shaped conformation.
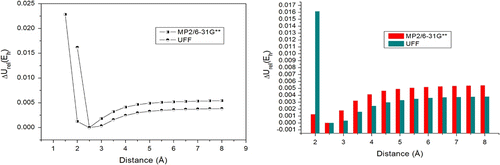
In this case, the local minimum is localized at shorter distances as compared to π–π∗ systems and angular conformation (2.5 Å for T-shaped and 3.0–3.5 for conformations A, B, and C – parallel interactions). It was expected, whereas the atomic repulsion between the aromatic rings is less. This conformation assumes high relative energy values in both methods, leading us to believe that hyperconjugative effects play an important role in this kind of conformation. According to our results, the UFF force field could not compute equivalent relative energy values in relation to the MP2 method. Electronic structure methods reveal lower ΔU rel values than MM methods.
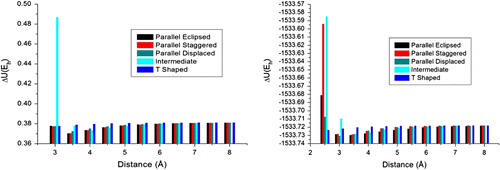
To evaluate the magnitude of hyperconjugative effects on dimmer conformations, we have compared the MM data to DFT results with deletion of hyperconjugative effects. Firstly, we have validated the DFT method using MP2 data, in order to make sure that DFT results are as accurate as the MP2 results in terms of relative interaction energy values
From Figure , it is clear that B97D is in good agreement with MP2 method, in contrast to MM results, which show divergent results with ΔU rel values as compared to DFT data with delection of hyperconjugative effects. Thus, intermolecular calculations need to be carefully performed using higher accurate electronic structure methods to reveal reliable results mainly at shorter distances among ligands. Therefore, the weakness of MM methods for specific interactions at shorter distances does not seem to come from hyperconjugative effects, but it is related to other quantum effects, such as electronic correlation (Figure ).
Figure 5 Potential curves for parallel displaced conformation: (a) Validation of DFT B97D/6-31G∗∗ method. (b) The balance of deletion energy obtained and UFF method.
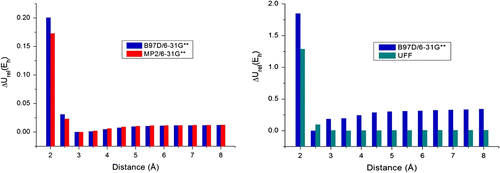
Figure 6 Potential energy graphic for each conformation: (a) UFF, (b) MP2/6-31G∗∗.
MM methods prefer the eclipsed conformation, while a more accurate calculation method highlights the shifting of the most stable configuration along the PES (Figure ). It is clear from our theoretical data that it is necessary to include correction terms for conventional force fields to compute the electronic effects for hydrophobic interactions with higher accuracy. In this perspective, the creation of hydrophobic cavities in the solvent environment could then maximize the hydrophobic/electrostatic interactions among the amino acids, resulting in the protein folding for the model system B.
In the balance of forces between hydrophobic and hydrophilic effects, it is known from literature that polar forces are stronger (Ben-Naim, Citation1991). It should be kept in mind, however, that hydrophobic forces can also play an important role in the protein folding process, because if a hypothetical protein only contains polar amino acids residues in its primary sequence, that protein shows stronger interactions with water molecules and the solvation Gibbs free energy is quite negative, decreasing the probability the protein folding occurring. Nevertheless, if the protein has interplay between non-polar and polar amino acid sequences, initially, hydrophobic cavities can be created, thus favoring a kind of specific hydrogen bond network between polar amino acid residues and water solvent molecules toward starting the folding process. Connecting now the Darwin theory, it is possible that specific codes, selected along evolution, containing some hydrophobic/hydrophilic character can lead to a specific folding, generating stable 3D structures for those proteins. In this context, the paper of Mittal and coworkers points out that “preferential interactions” between amino acids do not drive protein folding. On the other hand, a simple occurrence of stoichiometric ratios of individual amino acids, based on Chargaff’s-like rule, can significantly contribute for the protein folding.
Chargaff’s rules for protein folding
From the Mittal-Jayaram paper, crystal structures of several thousand proteins were analyzed. The results show that protein folding can be closely related to frequencies of occurrence of amino acids in the primary sequence, that is, its stoichiometry. Thus, the Mittal-Jayaram thesis (Mittal, Jayaram, Shenoy, & Bawa, Citation2010) focused on Chargaff’s rules for protein folding. It is known that the Chargaff’s rule can currently be derived from a thermodynamic framework, because GC and AT base pairs are the most stable complexes energetically formed (Furmanchuk, Isayev, Shishkin, Gorb, & Leszczynski, Citation2010). The Gibbs energy solvation gradient of the protein at a given conformation can drive the protein folding. But, at the first moment, the creation of “hydrophobic cavities” might take in the solvent environment, which could then maximize the hydrophobic/electrostatic interactions among the amino acids resulting in the protein folding. Perhaps, Chargaff’s rules might be applied in conjunction with the solvent effect in order to contribute to elucidation of the protein folding problem. In fact, the specific occurrences of amino acids in primary sequences pointed out by Mittal et al. put in evidence that both polar and nonpolar amino acids residues are essential for the protein folding, and their specific stoichiometry leads to hydrophobic/electrostatic intra and intermolecular interactions with solvent molecules.
Concluding remarks
Certainly, the use of the solvent-induced Gibbs free energy concept proposed by Ben-Naim is very useful and interesting to evaluate the folding process from a thermodynamic point of view. It is worth noting, however, that the statistical analysis performed by Mittal and coworkers, along several thousand of crystal structure of proteins, also reveal a defined relation among the amino acid residues for folded proteins. This result encourages us to think about the significant role of hydrophobic/electrostatic interactions involved in this process as well as in an interplay between Ben-Naim and Mittal’s proposals. Our current contribution, in this context, tries to connect both proposals. In the light of our data, it is a hard task to take into account the π–π and T-stacking interactions by theoretical calculations using conventional force fields due to quantum effects of hyperconjugation and electronic correlation, which are essential to compute the hydrophobic interactions in proteins. In this sense, theoretical calculation using standard force fields could lead to questionable conclusions. Therefore, the development of new empirical potentials from interpolated and analytical PESs (Bytautas, Bowman, Huang, & Varandas, Citation2012; Morón et al., Citation2012) can, in principle, be used to take into account the electronic effects, which can play a critical role in hydrophobic interactions in the active site of proteins. Thus, the balance between hydrophobic and electrostatic interactions and its role in the protein folding process can be better evaluated. The protein folding process is a complex phenomena influenced by a great number of different timescales including the translation rate. The Mittal-Jayaram and Ben-Naim papers point out a reliable and modern perspective on protein folding. We strongly feel, then, that those studies might be helpful for exploring protein engineering as well.
Related Research Data
References
- Ben-Naim , A. 1991 . Strong forces between hydrophilic macromolecules; implications in biological systems . Journal of Chemical Physics , 93 : 8196 – 8210 .
- Ben-Naim , A. 2012 . Levinthal’s question revisited, and answered . Journal of Biomolecular Structure & Dynamics , 30 : 113 – 124 .
- Bytautas , L. , Bowman , J. M. , Huang , X. and Varandas , A. J. C. 2012 . Accurate potential energy surfaces and beyond: Chemical reactivity, binding, long-range interactions, and spectroscopy . Advances in Physical Chemistry , 2012 : 1 – 4 .
- de la Lande , A. , Babcock , N. S. , Rezac , J. , Levy , B. , Sanders , B. C. and Salahub , D. R. 2012 . Quantum effects in biological electron transfer . Physical Chemistry Chemical Physics , 14 : 5902 – 5918 .
- Furmanchuk , A. , Isayev , O. , Shishkin , O. V. , Gorb , L. and Leszczynski , J. 2010 . Hydration of nucleic acid bases: A Car-Parrinello molecular dynamics approach . Physical Chemistry Chemical Physics , 12 : 3363 – 3375 .
- Glowacki , D. R. , Harvey , J. N. and Mulholland , A. J. 2012 . Protein dynamics and enzyme catalysis: The ghost in the machine? . Biochemical Society Transactions , 40 : 515 – 521 .
- Mittal , A. , Jayaram , B. , Shenoy , S. and Bawa , T. S. 2010 . A stoichiometry driven universal spatial organization of backbones of folded proteins: Are there Chargaff’s rules for protein folding? . Journal of Biomolecular Structure and Dynamics , 28 : 133 – 142 .
- Morón , V. , Martin-Gondre , L. , Crespos , C. , Larregaray , P. , Gamallo , P. and Sayós , R. 2012 . Classical dynamics study of atomic oxygen over graphite (0 0 0 1) with new interpolated and analytical potential energy surfaces . Computational and Theoretical Chemistry , 990 : 132 – 143 .
- Nagel , Z. D. , Meadows , C. W. , Dong , M. , Bahnson , B. J. and Klinman , J. P. 2012 . Active site hydrophobic residues impact hydrogen tunneling differently in a thermophilic alcohol dehydrogenase at optimal versus nonoptimal temperatures . Biochemistry , 51 : 4147 – 4156 .
- Ramalho , T. C. and Da Cunha , E. F. F. 2011 . Thermodynamic framework of the interaction between protein and solvents drives protein folding . Journal of Biomolecular Structure & Dynamics , 28 : 645 – 646 .
- Wheeler , S. E. 2011 . Local nature of substituent effects in stacking interactions . Journal of the American Chemical Society , 133 : 10262 – 10274 .
- Ying-Qi , J. and Ke-Li , H. 2010 . Quantum mechanical effect in protein–ligand interaction . Expert Opinion on Drug Discovery , 5 : 33 – 49 .