
Abstract
The nucleoprotein (NP) binds the viral RNA genome as oligomers assembled with the polymerase in a ribonucleoprotein complex required for transcription and replication of influenza A virus. Novel antiviral candidates targeting the nucleoprotein either induced higher order oligomers or reduced NP oligomerization by targeting the oligomerization loop and blocking its insertion into adjacent nucleoprotein subunit. In this study, we used a different structure-based approach to stabilize monomers of the nucleoprotein by drugs binding in its RNA-binding groove. We recently identified naproxen as a drug competing with RNA binding to NP with antiinflammatory and antiviral effects against influenza A virus. Here, we designed novel derivatives of naproxen by fragment extension for improved binding to NP. Molecular dynamics simulations suggested that among these derivatives, naproxen A and C0 were most promising. Their chemical synthesis is described. Both derivatives markedly stabilized NP monomer against thermal denaturation. Naproxen C0 bound tighter to NP than naproxen at a binding site predicted by MD simulations and shown by competition experiments using wt NP or single-point mutants as determined by surface plasmon resonance. MD simulations suggested that impeded oligomerization and stabilization of monomeric NP is likely to be achieved by drugs binding in the RNA grove and inducing close to their binding site conformational changes of key residues hosting the oligomerization loop as observed for the naproxen derivatives. Naproxen C0 is a potential antiviral candidate blocking influenza nucleoprotein function.
1. Introduction
Protein self-assembly and homo-oligomerization modulate many biological functions associated with gene expression. This is particularly true in a context of host–virus interactions. For example, catalytically active monomers of a human DNA-editing enzyme slowly oligomerize on the viral HIV genome and inhibit HIV reverse transcription (Chaurasiya et al., Citation2014). Nucleoproteins of negative-strand RNA viruses self-assembled on viral RNA to form highly stable nucleocapsids (Ruigrok, Crepin, & Kolakofsky, Citation2011). Moreover, viral genomes are the only RNAs in infected cells that are assembled with NP. It is therefore thought that NPs must also exist in an autoinhibited form to avoid RNA binding or oligomerization in the wrong biological context. This can occur by intra- rather than intermolecular binding of the flexible arms [e.g. La Crosse Orthobunyavirus (Reguera, Malet, Weber, & Cusack, Citation2013)], influenza (Chenavas et al., Citation2013), or chaperoning by another viral or cellular protein [e.g. the rabies phosphoprotein (Mavrakis et al., Citation2006)].
The nucleoprotein (NP) is a highly abundant protein during influenza A virus (IAV) infection. NP covers the segments of genomic RNA and assembles with the three polymerase subunits in a ribonucleoprotein complex (RNP). NP binds to single-stranded RNA; each strand of NP–RNA oligomers are engaged in a double-helical structure (Arranz et al., Citation2012; Moeller, Kirchdoerfer, Potter, Carragher, & Wilson, Citation2012), suggesting an important role of NP in the maintenance of this structure requested for function (Chan et al., Citation2010; Elton, Medcalf, Bishop, & Digard, Citation1999; Mena et al., Citation1999; Ng, Wang, & Shaw, Citation2009; Ng et al., Citation2008; Ye, Krug, & Tao, Citation2006). NP is a highly conserved protein (>90% amino acids sequence identity) among the different viral strains of influenza A virus and has no cellular counterpart. Therefore, the nucleoprotein constitutes a good candidate for inhibition by antiviral compounds (Chenavas, Crepin, Delmas, Ruigrok, & Slama-Schwok, Citation2013). Recent studies have highlighted the interest of targeting proteins within the viral particle as the nucleoprotein NP and the RNP (Ruigrok et al., Citation2011), for which a low probability of resistance has been reported in some cases (Lejal et al., Citation2013; Shih et al., Citation2010). In contrast, resistance to some presently used antivirals emerged in circulating resistant strains of influenza A virus (Butler, Hooper, Petrie, Lee, & Maurer-Stroh, Citation2014).
At present, our structural insight on the nucleoprotein comes from RNA-free NP forming wt trimeric and mutant monomeric structures. The trimer is stabilized by a swapping loop protruding from one monomer to its neighbour (Das, Aramini, Ma, Krug, & Arnold, Citation2010; Krug & Aramini, Citation2009; Ng et al., Citation2008; Ye et al., Citation2006). The swapping loop was considered to be a good target for the design of new NP inhibitors that reduced NP oligomerization (Ng et al., Citation2008; Shen et al., Citation2011; Ye et al., Citation2006). Competing with viral RNA binding to NP constitutes another strategy to develop novel inhibitors of NP.
We recently identified by in silico screening that naproxen is both a known generic drug inhibiting the inducible cyclooxygenase and also an antiviral candidate (Lejal et al., Citation2013). Naproxen competed with RNA for binding to NP (Lejal et al., Citation2013; Slama Schwok, Delmas, Quideau, Bertrand, & Tarus, Citation2012); surface plasmon resonance supported the binding site of naproxen defined by MD simulations. Naproxen protected Madin Darby canine kidney (MDCK) cells against a viral challenge with H1N1 and H3N7 strains of influenza A virus, with IC50 values of ca 10 μM. In addition, we could not detect resistant virus to naproxen treatment throughout eight cell passages, whereas tamiflu-resistant virus were generated after four passages. Naproxen had antiviral effects in a mice model and reduced lung bleeding (Lejal et al., Citation2013).
In the present work, we used a fragment-based approach to extend the lead compound naproxen for improved affinity towards the nucleoprotein. This study was guided by a structure-based design and MD simulations. Molecular dynamics is a powerful method to investigate structural and dynamical properties of macromolecules in atomic details, and used to predict many functional aspects associated with protein dynamics as drug resistance and drug binding (Lopez-Martinez, Ramirez-Salinas, Correa-Basurto, & Barron, Citation2013; Purohit, Citation2014; Purohit, Rajendran, & Sethumadhavan, Citation2011a, Citation2011b; Rajendran, Purohit, & Sethumadhavan, Citation2012; Rajendran & Sethumadhavan, Citation2014; Wang et al., Citation2011). MD simulations are well suited to address protein flexibility, in particular of NP flexibility (Tarus, Chevalier, et al., Citation2012). In this work, two naproxen derivatives were synthesized and tested for their binding and inhibition of NP oligomerization. The novel naproxen derivatives stabilized the monomeric form of the NP protein, likely by providing assistance to folding, in particular of the C-terminal and of flexible loops. This led to inhibition of NP oligomerization in the presence of RNA, a process required for RNP function.
2. Results
2.1. Fragment-based design of novel naproxen derivatives and their binding to NP
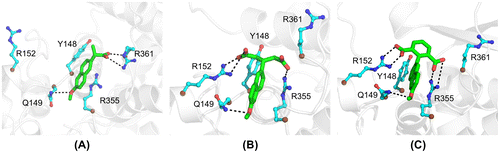
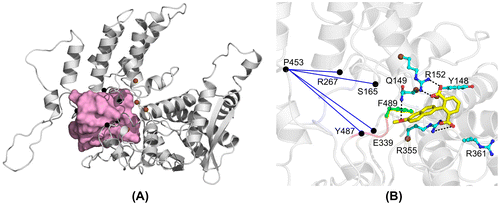
To improve recognition of the nucleoprotein, we generated novel naproxen derivatives by fragment extension from the lead naproxen compound (Lejal et al., Citation2013) using a structure-based approach derived from the structure of one NP subunit (subunit A) within NP trimer (PDB 2IQH (Ye et al., Citation2006)). Naproxen bound at a site located in the putative RNA-binding groove of the nucleoprotein. This site comprised Y148, a residue with proposed interactions with viral RNA nucleobases (Ye et al., Citation2006), that may constitute a binding platform for a NP inhibitor; Y148 was surrounded by basic residues R361, R355 and R152 (Figure (A)). To target additional charged residues from the other face of Y148, potentially R152 and/or nearby R150, the carboxylate moiety of naproxen that recognized R361 was extended (Supplementary Figure 1). We also investigated extension of the methyl moiety of the methoxy group CH3O of naproxen by various short aliphatic groups either polar: naproxen C1: CH3-CH2NH2, naproxen C2: CH3CONH2 or non-polar: naproxen C3: CH2CH3 for increased hydrophobic interactions, but these derivatives were unsuccessful according to MD simulations (see Section 8). The most successful derivatives according to the calculation of their interaction energies with NP (Supplementary Figure 1) were naproxen A and naproxen C0 deriving from naproxen by an additional carboxylate substituent, either aliphatic or aromatic, as compared with a single carboxylate moiety in naproxen (Figure ). Naproxen A and C0 were designed to keep the same stacking interactions with the aromatic residues Y148 and F489 (not shown) as naproxen did. At the end of 10 ns dynamics, one carboxylate of naproxen A and C0 bound to R355. In the naproxen complex, a strong electrostatic interaction between R361 and naproxen carboxylic group, and a cation – π aromatic ring interaction between the guanidium groups of R355 and naproxen were observed (Lejal et al., Citation2013). The additional carboxylate of the naproxen C0 derivative targeted R152 and made transient interactions with R150. In all three compounds (naproxen, naproxen A and C0), the oxygen of the methoxy moiety formed a hydrogen bond with Q149, while the methyl group was in hydrophobic interaction with F489 (not shown).
Figure 1. Structures of naproxen and two of its derivatives bound to nucleoprotein (NP) of influenza A virus. (A) After 10 ns of MD, naproxen converged to a conformation that involves the naphthalene ring in hydrophobic interaction with Y148 and cation–π interaction with R355, while the naproxen carboxylate made a salt bridge with R361 and the methoxy oxygen atom made a hydrogen bond with Q149. (B) The interactions made by the naphthalene ring and methoxy group of naproxen with NP were conserved in the naproxen A derivative. The two carboxylate groups of naproxen A were involved in salt bridges with R152 and R355, respectively. (C) As previously noted, the naphthalene ring and the methoxy of naproxen C0 derivative conserved their interactions with NP. The salt bridges with R152 and R355 were stabilized by the 1,3-dicarboxylated phenyl group of naproxen C0.
We expected that naproxen A and C0 would stabilize monomers of NP and consequently destabilize NP–RNA oligomers required for NP function. We tested these hypotheses by a combination of MD simulations and experimental studies using wt and mutants NP.
An intrinsic property of NP is its flexibility associated with three flexible regions in NP monomer: the basic loop 1 and loop 2 on one face of the protein, and the oligomerization loop 3 on the other face of the protein (swapping domain), highlighted by MD simulations; the role of these flexible loops in promoting RNA binding and oligomerization was suggested from experimental studies (Tarus, Chevalier, et al., Citation2012). To test whether binding of naproxen and its derivatives altered the flexibility of NP, root mean-square fluctuations (rmsf) of the free NP and the ligand-bound protein were calculated. Naproxen A and C0 binding decreased the flexibility of loop 2, shown by its rmsf decrease in Figure . Such a decrease was observed by mutation of the R361 residue and is consistent with naproxen A and C0 binding site being located in the RNA binding groove (Tarus, Chevalier, et al., Citation2012). In addition, the fluctuations of a C-terminal region (residues 428-438 of the linker between helices α19 and α20, called Cf) were markedly reduced by the binding of the naproxen derivatives without significantly affecting the flexibility of the oligomerization loop (swapping domain aa 402-428). This Cf linker may also participate in oligomer formation based on experimental data and our comparison of the fluctuation of NP monomer and trimer by MD simulations (Tarus, Chevalier, et al., Citation2012). The reduced flexibilities of loops 1 and 2 and of Cf (Figure ) suggest that RNA binding and further RNA-induced oligomerization could be impeded by naproxen and its derivatives.
Figure 2. Root mean-square fluctuations (rmsf) of the complexes of NP with naproxen and its derivatives. RMSF show that the positions of flexible domains defined in the free NP are preserved. The flexibility of the loops 1 and 2 are lowered by the interaction with the naproxen and its derivatives. The fluctuations of the oligomerization loop (loop 3) are similar for the free NP and the NP – bound naproxen C0 (red and green curves in B, respectively). Observable decreases of the fluctuations of NP-naproxen and NP-naproxen C0 derivative are seen in the C-terminal domain compared to free NP (blue and green curves of Cf (residues 429-436) in A and B, respectively compared to free NP shown in red in A and B).
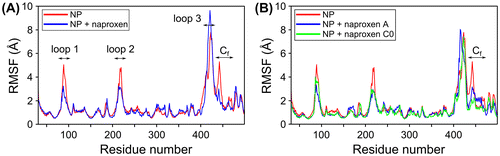
Figure 3. Comparison of the apparent Tm of NP (5 μM) alone and NP complexes with naproxen, naproxen C0 and A. The melting curves were obtained by monitoring the mean diameter as a function of temperature (inset, Figure A). The first derivatives of the melting curves are shown in the presence of: A: 1/1 ligand /NP ratio compared to NP alone (black), green: NP + naproxen, blue: NP + naproxen C0, red NP + naproxen A; B: 3/1 NP ratio, same colour as in A. C: comparison of the stabilization of NP quantified by the difference in Tm value of NP + ligand compared to free NP alone as a function of the ratio ligand over NP (Dose response), green: naproxen, blue: naproxen C0. The dotted lines correspond to fits of the data to a simple dose response curve.
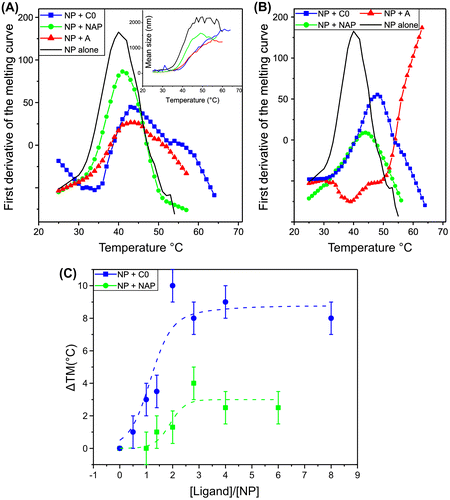
Figure 4. Comparison of the binding of naproxen C0 and A derivatives (100 nM) in competition with RNA binding to NP analysed by SPR.
The RNA fragment was attached by its 5′ biotin end to surface-bound streptavidin. The maximum RU (RUmax) and RU are the signals observed with NP in the absence and presence of naproxen derivatives, respectively. Data are the means of the results of three experiments that could be fitted (blue dashed line) by a simple dose-response isotherm (1/1 complex) for naproxen C0, yielding IC50 = 0.32 ± 0.09 μM. The lines connecting between the points of the naproxen A curve are only guidelines for the eye.
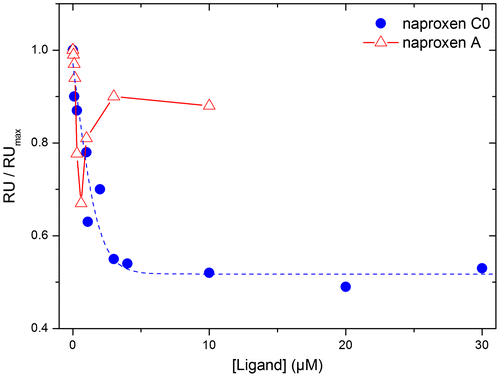
Figure 5. Naproxen and its derivatives perturbed RNA-induced NP oligomerization. (A) Size distribution of RNA-free NP alone in monomeric form and in NP trimer/ tetramers; Blue: monomers; red: trimers/tetramers; ((B)–(E) NP (10 μM) was mixed with various RNA oligonucleotides (3.3 μM) in the presence or absence of the ligands (10 μM). The size distributions are shown after 3 h ((B)–(D)) or 1 h (E) equilibration, corresponding to the end of the oligomerization kinetics. Naproxen and its derivatives interfered with NP oligomerization induced by RNA binding: B: NP + 24mer RNA, C: R361A + 24mer RNA, D: NP + 16mer RNA, E: NP + 48 mer RNA.
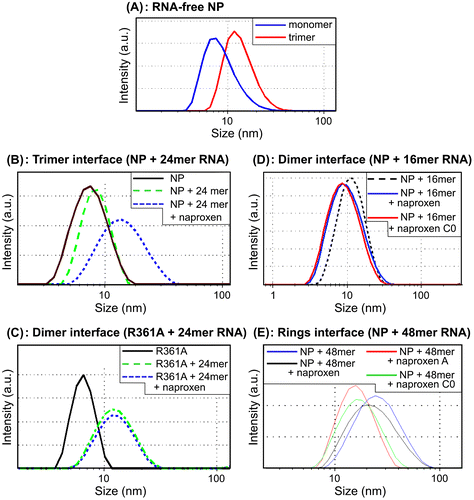
Figure 6. Modifications of the oligomerization pocket (pink volume in A) of NP monomer A in the presence of the naproxen derivatives. The oligomerization pocket is represented as a protomol built around the oligomerization loop of monomer B based on the pdb structure 2IQH. The black points in A and B define distances (blue lines in B) between Cα atoms of residues localized at the edge of the oligomerization domain of monomer A and that are changed by binding of the naproxen C0 derivative. The Cα atoms of targeted residues by the naproxen C0 derivative are represented as brown spheres (A and B). The naproxen C0 derivative binding domain is connected to the oligomerization domain of NP (brown spheres and pink volume in A) through the C-terminal of NP (F489 in B).
To give experimental support to the hypothesis that the naproxen C0 and A derivatives stabilize monomeric NP, we expressed wt NP and Y148A, R152A and R361A mutants. Binding of the naproxen C0 and A derivatives to these proteins and their subsequent thermal stabilization were tested by melting temperature and surface plasmon resonance experiments. The chemical synthesis of these derivatives is described below.
2.2. Synthesis of the naproxen derivatives
2.2.1. Synthesis of naproxen A
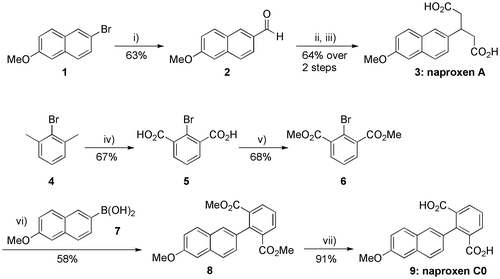
Naproxen A (3) was synthesized in three steps as depicted in Scheme . Commercially available 2-bromo-6-methoxynaphthalene (1) was first submitted to a halogen/metal exchange using n-BuLi in anhydrous tetrahydrofuran (THF) and the resulting lithiated intermediate was then directly formylated by adding dimethylformamide (DMF) into the reaction mixture to furnish 6-methoxy-2-naphthaldehyde (2) in 90% yield (Matsunaga et al., Citation2004). This aldehyde was then reacted with two equivalents of ethyl acetoacetate in ethanol (EtOH) in the presence of a catalytic amount of piperidine (Sharma & Ray, Citation2008), (Sulyok et al., Citation2001). This Knoevenagel condensation/Michael addition reaction sequence gave the expected bis(ethyl acetoacetate) intermediate, which was hydrolysed and decarboxylated using aqueous potassium hydroxide (KOH) in refluxing EtOH (Sharma & Ray, Citation2008), (Sulyok et al., Citation2001) to furnish the desired 3-naphthylglutaric acid derivative 3, i.e. naproxen A, in 64% yield from 2 (Scheme , see the supporting information for details).
Scheme 1. Chemical synthesis of naproxens A (3) and C0 (9). Reagents and conditions: (i) n-BuLi (1.1 equiv.), anhydrous THF, −78 °C, 30 min, then DMF (2.1 equiv.), −78 °C to rt; (ii) Ethyl acetoacetate (2.0 equiv.), EtOH, cat. piperidine, 0 °C to rt, 3 days (70%); (iii) aq. KOH (32 equiv., 24 M), EtOH, reflux, 3.5 h (92%); (iv) KMnO4 (2.0 equiv.), t-BuOH/H2O (1:1), reflux, 4 h, then KMnO4 (2.0 equiv.), reflux, 16 h; (v) MeOH, conc. H2SO4, reflux, 16 h; (vi) 6 (0.9 equiv.), Pd(PPh3)4 (0.03 equiv.), aq. K2CO3 (2.0 equiv.), 1,4-dioxane, reflux, 2 h; (vii) aq. LiOH (10.0 equiv.), THF, reflux, 17 h.
2.2.2. Synthesis of naproxen C0
Naproxen C0 (9) was synthesized in four steps starting from commercially available bromo-m-xylene (4), as shown in Scheme . Oxidation of the two methyl substituents of 4 using potassium permanganate (KMnO4) in a refluxing 1:1 tertio-butanol (t-BuOH)/H2O solvent mixture led to the known isophtalic acid derivative 5 (Courchay, Sworen, Ghiviriga, Abboud, & Wagener, Citation2006), which was esterified with methanol (MeOH) in the presence of concentrated H2SO4. The resulting 2-bromoisophthalate 6 was then coupled with commercially available 6-methoxy-2-naphthaleneboronic acid (7) under Suzuki conditions (Harvey, Dai, Ran, & Penning, Citation2004) to afford the 2-naphthylisophthalate derivative 8 in 58% yield. Naproxen C0 (9) was finally obtained in high yield by saponification of 8 using aqueous lithium hydroxide (LiOH) in refluxing THF (Scheme , see the supporting information for details).
2.3. Experimental Binding of Naproxen C0 and A to NP
2.3.1. Naproxen A and C0 stabilized NP against thermal denaturation
As suggested by MD simulations, we expected that naproxen and its derivatives binding to NP could stabilize NP monomer against thermal denaturation as a result from tight NP–ligand associations. Melting curves were obtained by monitoring the mean size of free NP and NP–ligand complexes as a function of temperature by dynamic light scattering (DLS) (Figure (A), insert). The first derivative of the melting curve was taken as the apparent melting temperature, Tm, being Tm = 40 ± 1 °C for free NP monomer. The Tm was significantly raised by 5–6 °C by adding 5 μM of the naproxen C0 and A derivatives, but only very slightly by 5 μM naproxen, Tm = 46 ± 1 °C, 45 ± 1 °C and 41 ± 1 °C, respectively. Addition of ligands in excess relative to NP further increased the apparent Tm up to a plateau (Figure (B), (C)).
Figure (B) shows that the derivatives were more effective in stabilizing NP than naproxen itself. Naproxen A in excess with respect to NP had an unexpected effect, inducing some aggregation already at low temperatures and presenting a very high apparent melting temperature. The dose response of the apparent Tm as a function of the concentration of naproxen C0 suggests that this derivative formed with NP not only a 1/1 complex as expected and observed in (below) surface plasmon resonance (SPR) experiments but also an additional complex was obtained at a different stoichiometry, likely 2–3 ligands per NP. A similar behaviour was observed for naproxen, in agreement with previous results (Lejal et al., Citation2013).
2.3.2. Affinity of naproxen derivatives for NP determined by SPR experiments
Naproxen and its derivatives were designed to compete with RNA for binding to NP (1/1 complex). Experimentally, this competition can be observed by SPR using immobilized RNA on the sensor surface and adding NP to form the NP–RNA complex in the absence or presence of naproxen derivatives. Naproxen competing with RNA on NP was deduced from the decrease of the NP–RNA complex formed upon co-addition of naproxen with NP on immobilized RNA. SPR signals are proportional to the molecular weight of the added analytes and the respective molecular weights of NP and naproxen are 57 kDa and 252 Da. Therefore, only indirect competition experiment in which NP binds to a surface-bound RNA in the presence or absence of naproxen derivatives can yield a good signal-to-noise ratio. The KD values for NP trimer and monomer binding to RNA were 14 and 40 nM, respectively (Chan et al., Citation2010; Ng et al., Citation2008; Tarus, Bakowiez, et al., Citation2012; Ye et al., Citation2006). Addition of increasing concentrations of naproxen C0 to NP decreased the signal due to the NP–RNA complex. A plateau was obtained at 50% inhibition and the IC50 value of this inhibition by naproxen C0 was 320 ± 90 nM, based on a 1/1 stoichiometry (Figure and Supplementary Figure 2). We could not get evidence for a 1/2 NP–ligand complex by SPR as expected from the Tm data even upon increasing naproxen C0 concentrations up to 50 μM. In this SPR experiment, 300 mM NaCl was used to avoid unspecific binding to the sensorchip, which may also disfavour the complex with 2–3 ligands observed in the DLS experiment at 50 mM NaCl. In these conditions, the IC50 obtained with naproxen was 3.0 ± 0.9 μM (Table ), consistent with an increased affinity of the naproxen C0 derivative as compared to that of naproxen. Repeating these experiments at various NP concentrations in the range 50–300 nM and using the Cheng-Prusoff equation, the inhibitory constant of the C0 derivative Ki = IC50/(1+[NP]/Kd) was found to be Ki = 97 ± 25 nM. The naproxen A derivative exhibited an unexpected biphasic behaviour while it only partly competed with RNA binding to NP. We did not evaluate the IC50 value deriving from the low concentration range due to the small extent of inhibition (Table ).
Table 1. Comparison of the binding of naproxen, Naproxen C0 and Naproxen A derivatives to monomeric wt NP and selected monomeric mutants by competition with NP–RNA complexes.
The SPR experiments were also performed with the R361A and R152A mutants in the presence or absence of naproxen and naproxen C0. These mutants were used based on the expected binding site of the naproxen derivatives (Figure ) and also because they form monomers; in contrast, the derivatives would not bind the R355A or Y148A mutants exclusively forming trimers/tetramers (Tarus, Bakowiez, et al., Citation2012). The R361A mutation was expected to reduce naproxen binding but not binding of its derivatives (Figure ) and indeed naproxen could not bind to this mutant, in contrast with naproxen C0 (Lejal et al., Citation2013; Tarus, Bakowiez, et al., Citation2012). Naproxen binding was unaffected by the R152A mutation and the IC50 was similar to the value determined for naproxen-wt NP interactions. In contrast, competition of naproxen C0 with the R152A–RNA complex was less efficient than with wt NP–RNA, and the IC50 value for interacting with R152A was reduced compared with the value determined for wt NP (Table , Supplementary Figure 2). Altogether, both MD simulations and SPR data were consistent with the tighter binding mode of naproxen C0 to NP compared to that of naproxen. Isothermal dynamic light scattering were then used to test the ability of the naproxen derivatives at destabilizing oligomeric structures of NP as a consequence of their stabilization of the monomeric form. To check if the derivatives may perturb oligomeric interfaces differently, various NP oligomers were generated in the presence of RNA fragments of selected sizes.
2.4. Impeded NP oligomerization by naproxen and its derivatives: experimental evidence and calculation of domain swapping volume
2.4.1. DLS experiments
DLS is a very useful tool to monitor changes in the (self-) assembly of proteins and well suited to follow the oligomerization of monomeric NP (Tarus, Bakowiez, et al., Citation2012). In RNA-free solutions, the apparent diameter of monomeric NP was 6.5 ± 1.0 nm, while its trimeric/tetrameric form had a diameter of 14.6 ± 1.0 nm (Figure (A), blue and red curves, respectively). The RNA-free self-association of NP (10 μM) took place in 17 h at 50 mM NaCl; in these conditions, naproxen stabilized monomeric NP and avoided trimers/tetramers formation (data not shown). Moreover, naproxen could not disrupt already assembled trimers as those obtained with wt NP at high salt concentration (500 mM NaCl) or mutants that only formed trimers–tetramers as Y148A or R355A (Tarus, Bakowiez, et al., Citation2012).
In the presence of short 24-mer RNA oligonucleotides, oligomerization of NP monomers into trimers–tetramers with an apparent size of 15–17 nm occurred within ca 2–3 h (Figure (B), black and blue curves, respectively (Tarus, Bakowiez, et al., Citation2012). Addition of stoichiometric amounts of naproxen to NP stabilized the monomeric form (green curve, Figure (B)). Similar results were obtained with the naproxen C0 and A derivatives (data not shown).
To test whether naproxen could also affect the dimeric interface and compete with dimer formation, two other experiments were performed based on previous findings: (i) the R361A mutant had a limited ability to oligomerize and formed dimers in the presence of 24-mer RNA; however, the R361A mutation markedly reduced binding of naproxen (Lejal et al., Citation2013; Tarus, Chevalier, et al., Citation2012) and (ii) the wt NP only formed dimers with short 16-mer RNA fragments (Tarus, Bakowiez, et al., Citation2012). Naproxen and its derivatives were added in these two conditions to monomeric NP. No stabilization of R361A monomers by naproxen could be observed as expected in the absence of the salt bridge between A361 and naproxen (Figures (A) and (C). In contrast, naproxen and its C0 derivative impeded dimer formation in the presence of rU16 (Figure (D)).
To test the ability of the derivatives to perturb rings formation, we used a longer RNA fragment of 48 nucleotides (nt) that quickly induced NP oligomerization. It yielded species of about 25 nm, shown by electronic microscopy to be rings (Figure (E) and (Chenavas, Estrozi, et al., Citation2013; Tarus, Bakowiez, et al., Citation2012). Naproxen or its derivatives were added to monomeric NP and the formation of rings was partly impeded; while naproxen only slightly decreased the size of the NP–RNA complex to 21–23 nm, naproxen A or C0 further reduced the apparent size of the complexes to 15–17 nm. This suggests that the interface between NP subunits in rings in the presence of RNA is not identical to the interface between NP subunits in trimers–tetramers being formed with 24-mer RNA. Additionally, this difference may arise from larger cooperative effects between NP subunits in rings than in trimers (Tarus, Bakowiez, et al., Citation2012).
2.4.2. Calculation of the swapping pocket of NP with and without naproxen derivatives
The above DLS data showed that the ligands modified NP–RNA oligomerization. To further bring a mechanistic/structural insight on how the naproxen derivatives affected the oligomerization interface, we calculated the swapping pocket, i.e. the volume occupied by the oligomerization loop from monomer B protruding into monomer A by building the protomol obtained from the trimer structure of H1N1 NP (Figure (A), pink volume). We calculated the distances between Cα atoms of all residues localized within that protomol volume and identified those distances that were modified by the binding of naproxen or its derivatives. These distances are listed in Table and compared with the same distances determined in ligand-free NP monomer or trimer. The key residues affected by binding of the naproxen C0 derivative were S165, R267, P453 and Y487. These residues formed a pyramid that became distorted by binding of the naproxen C0 derivative as shown in Figure (B).
Table 2. Selected distances close to C-terminal where the ligand binding site is located showing that structural changes in residues important for trimer formation takes place upon binding of naproxen, naproxen C0 and A derivatives to wt NP; see also Figure .
At the basis of this pyramid, Y487 was located on the same linker than F489, close to E339. The distances P453-Y487and E339-P453 increased in the presence of naproxen and the C0 derivative compared to the same distances found in NP monomer, reaching with naproxen C0 binding values even larger than that of the trimer. In addition, the distances S165-P453 and R267 - P453 were intermediate between the same distances in trimeric and monomeric NP. S165 is a residue that can be phosphorylated and its mimic S165D was found to be exclusively in a monomeric form (Chenavas, Estrozi, et al., Citation2013). In contrast, E339 is involved in the stabilization of NP trimer by a salt bridge with R416 of the next protomer. Taken together, these data suggest that binding of naproxen and its derivatives likely promote structural changes that disfavour domain swapping required for trimer/ oligomer formation.
3. Discussion
3.1. Naproxen and naproxen C0 targeted the RNA-binding groove of NP
In this work, we aimed at inhibiting the binding of the nucleoprotein to RNA and RNA-induced NP oligomerization for interference with NP function. Naproxen, a generic antiinflammatory drug identified by virtual screening, competed with RNA by binding within the RNA-binding groove of NP (Lejal et al., Citation2013). Further extension of naproxen by fragment addition yielded the naproxen A and C0 derivatives designed to form additional salt bridges with basic residues R150 and/or R152 close to Y148 and keep the same stacking interactions with the aromatic residues Y148 and F489 as observed with naproxen (Figures and (B)). The binding site of naproxen C0 suggested by MD simulations was supported by the use of monomeric proteins with point mutations in residues involved of their binding to NP such as R152A and R361A (Table ). Naproxen C0 formed a 1:1 complex with NP that competed with NP–RNA association (Figure ) and showed a better affinity for NP than naproxen did, leading to the inhibition constant Ki = 97 ± 25 nM. R152 participation in the stabilization of the naproxen C0 derivative was supported by an increase of IC50 for R152A compared with IC50 for wt NP; in contrast, R152 did not interfere with naproxen binding, expected from Figure (A) (Figure , Table and supplementary Figure 2). Naproxen C0 and A binding stabilized NP thermal denaturation by 5 °C compared with free NP melting using stoichiometric amount of the ligands compared to NP (1:1 complex), (Figure (A)). Besides the 1/1 complex, it is likely that naproxen C0 as naproxen itself also forms a second type of complex between 2 and 3 ligands per NP (Figure (C)). The binding site of naproxen C0 within this second complex is likely to differ from the first complex being found in the RNA groove (Figure ). This hypothesis accounts for both increased stability (Figure ) and incomplete competition with RNA binding at high naproxen C0 concentration (Figure ), in agreement with alternative naproxen binding site(s) suggested by docking experiments (Lejal et al., Citation2013). The biphasic SPR data and the evidence for aggregation at temperatures lower than the Tm both suggested that naproxen A is likely to adopt multiple binding modes with NP, the conformation shown in Figure not being experimentally the most populated at 20 °C.
3.2 Structural insight
What are the structural features that led to: (i) stabilization of monomeric NP; (ii) destabilization of oligomeric NP–RNA complexes? To answer these questions, we considered several regions of the NP shown in Figure : loops 1 and 2 surrounding the putative RNA-binding groove, the oligomerization loop 3 and the C-terminal (aa 429–498), especially the flexible Cf linker located between α19 and α20.
3.3. Stabilization of monomeric NP at the level of its C-terminal and of the flexible loops
Figure shows the stabilization of the C-terminal of NP by the naproxen derivatives, including F489 and the Cf linker aa 428-438 (Figure ). F489 forming hydrophobic stacking interactions with naproxen C0 (Figure (B)) is part of the C-terminal region shown to be important for monomer stabilization (Chenavas, Estrozi, et al., Citation2013). In agreement with naproxen binding involving interactions with F489, NP was stabilized against proteolytic cleavage of its C-terminal by addition of naproxen (Lejal et al., Citation2013). Interestingly, residues 490–498 were disordered in the original NP trimer crystal structure, and the deletion of residues 491 to 498 had no effect on NP’s ability to form trimers and larger oligomers (Ye et al., Citation2012). The flexibility of the C-terminal of influenza A NP is reminiscent to the flexibility of the C-terminal extension of respiratory syncytial virus nucleoprotein (partly invisible in the atomic structure), while the homologous domains of measles and Sendai virus are intrinsically disordered and bind to P (Longhi et al., Citation2003; Ruigrok et al., Citation2011; Tawar et al., Citation2009). This flexibility would confer NP the ability to adapt to various partners and these interactions could be thus be perturbed by naproxen and naproxen C0. In that respect, taking together that (i) the binding of naproxen C0 involves contribution of both R150 and R152 as suggested by MD simulations and SPR data (Table ) and (ii) R150 being a crucial residue for PB2 polymerase subunit association with NP, it is suggestive that naproxen C0 may be able to interfere with PB2 binding. Work along these lines is currently ongoing.
The binding site of naproxen C0 partially overlapped with the dimeric interface of NPΔ402-428 (Ye et al., Citation2012). This site covers part of the helix-turn-helix motif comprised of residues 148 to 167, including Y148, R152 and the C-terminal tail from residue 482 to the end, in particular F489. Indeed, Figure (C) showed the inhibition of dimer formation by naproxen and the naproxen C0 derivative. In addition to mediating dimer interaction, the C-terminal tail of NP may help regulating RNA binding to NP; it may adopt different conformations in different functional states (e.g. monomeric vs. oligomeric NP and RNA bound vs. RNA-free NP).
Modifications taking place in the RNA-binding groove were linked with a decrease in loop 2 flexibility: binding of the naproxen derivatives to NP reduced the flexibility of loop 2 as previously observed with the R361A mutation located in the RNA groove (Tarus, Chevalier, et al., Citation2012). Moreover, the negative charges introduced by binding of naproxen and its derivatives to NP–RNA groove mimicked the post-translational phosphorylation taking place at S165: substituting S165 by D165 added a negative charge in the RNA binding groove that stabilized the S165D mutant in a monomeric form (Chenavas, Estrozi, et al., Citation2013). Altogether, the reduction of loop 2 movements, the stabilization of the C-terminal and the presence of negative charges close to S165 contributed to monomeric NP stabilization. These features are also involved in the inhibition of oligomers formation by the naproxen derivatives.
3.4. Destabilization of NP-RNA oligomers
A consequence of the competition between naproxen and its derivatives and RNA for binding to NP (Table ) was the inhibition of NP oligomerization or its decrease (Figure ). Only the naproxen derivatives and not naproxen by itself partly impeded the formation of rings. This may be linked with differences in affinity among these derivatives, assuming a higher cooperativity of the NP–RNA complexes in rings being formed faster than trimers/ tetramers (Tarus, Bakowiez, et al., Citation2012).
The protomol shown in Figure (A) describing the pocket occupied by the oligomerization loop of the neighbour monomer in NP varies between NP monomer and trimer, depending on NP homo-oligomerization, as detailed in Table . Three main distances S165-P453, R267-P453 and P453-Y487 were larger in the trimer than in the monomer, suggesting an elongation of the “pyramid” formed by the residues S165, R267, E339, P453 and Y487 required for insertion of the oligomerization tail loop. These distances were also modified in the NP-naproxen derivatives complexes. These data suggest that naproxen C0 and A induced a modification in the tail loop pocket (Figure (B)) that impeded trimer formation and indeed these derivatives kept NP in a monomeric form in the concentration range of 5–50 μM. It is not known yet if the pocket remains the same or is modified in the presence of RNA.
4. Conclusion
The newly designed naproxen derivatives may provide useful tools to probe interactions between NP and viral proteins for mechanistic studies of the RNP. Furthermore, naproxen C0 is an obvious candidate for antiviral therapy, as it presents a larger affinity for NP than naproxen, binds active monomeric NP requested for transcription and replication, and impedes oligomerization. Ongoing cellular experiments are suggestive of its improved activity (Lejal, N, Delmas B and Slama-Schwok A, unpublished data). Interfering with protein self-assembly involved in viral gene expression by structure-based inhibitors of the monomeric nucleoprotein may be extended to other viral nucleoproteins; this work provides new perspectives of design principles for generating such inhibitors.
5. Material and methods
5.1. Molecular modelling
The three-dimensional (3-D) crystal structure of NP monomeric form (PDB ID: 2IQH) was used as a target throughout the virtual screening in a volume centred on Y148. The NP-naproxen structure selected after the virtual screening (Lejal et al., Citation2013) was further used to set the initial coordinates for molecular dynamics simulation. Derivatives of naproxen were designed to additionally target R152, R355, Q149 and F489. The first two residues were targeted by extending the carboxylate moiety of naproxen. The last two residues were targeted by extending the methoxy group of naproxen with short aliphatic or polar groups. The stability of naproxen derivatives in the nucleoprotein binding pocket were investigated through MD simulations of the complex. The flexible groups added to the methoxy moiety increased the entropy of the new derivatives without an enthalpic compensation, resulting in unstable interactions with the nucleoprotein. Consequently, we discarded the compounds with modified methoxy group from further theoretical and experimental analysis. The MD simulations were carried out following a protocol detailed elsewhere (Lejal et al., Citation2013). The programme NAMD (Phillips et al., Citation2005) with the CHARMM27 force field (MacKerell, Banavali, & Foloppe, Citation2000) was employed to perform the MD simulations. The NP-naproxen complex was centred in a cubic cell of pre-equilibrated TIP3P water model (Jorgensen, Chandrasekhar, Madura, Impey, & Klein, Citation1983). The system was electrostatically neutralized and set to an ionic strength of 0.15 M by adding sodium and chloride ions. The electrostatic interactions were calculated using the particle mesh Ewald summation algorithm (Darden, York, & Pedersen, Citation1993). The equations of motion were iterated using the velocity Verlet integrator with a time step of 2 fs. An initial step of system energy minimization was followed by heating to 300 K. Molecular dynamics simulation in NPT ensemble was further employed to equilibrate the system during 1.6 ns and for production run. Five trajectories of 10 ns each were produced for the NP-naproxen and NP-naproxen A derivative complexes and two trajectories of 10 ns for the NP-naproxen C0 derivative (see Supplementary Figure 1C for a convergence test).
5.2. Protein expression and purification of wt NP and the Y148A, R355A and R361A mutants
Proteins expressions were performed as described (Tarus, Bakowiez, et al., Citation2012) with modifications of the purification protocol by replacing the Ni affinity column by Ni magnetic beads (Pure proteome, Millipore), used as recommended by the manufacturer followed by extensive dialysis at 50 mM NaCl, 20 mM Tris buffer at pH 7.5. After purification, the protein concentration was determined by the extinction coefficient ε = 56,200 Μ-1 cm−1 at 280 nm.
5.3. Chemicals and antibodies
Oligonucleotides: biotinylated (5′ end) RNA fragments were purchased from Dharmacon (Thermo-Fischer, France) with HPLC purification; the following sequences were used: rU25 and Flu1: 5′ UUU GUU ACA CAC ACA CAC GCU GUG 3′. Naproxen was purchased from Sigma.
5.4. Chemical synthesis
Reagents and chemicals were purchased from Sigma-Aldrich. Solvents were freshly distilled before use. 1H and 13C NMR spectra were recorded on a Bruker Avance 300 using solvent residuals as internal references. Melting points (MP, uncorrected) were determined on a Büchi Melting Point B-540. TLC analysis was carried out on silica gel (Merck 60F-254) with visualization at 254 and 365 nm. Preparative flash chromatography was carried out with Merck silica gel (Si 60, 40–63 μm). IR spectra were recorded with a FTIR PerkinElmer Spectrum 100 spectrometer. Mass spectrometry low resolution data and high resolution data were obtained from the Centre d’Etudes Structurales et d’Analyses des Molécules Organiques (CESAMO), Université de Bordeaux.
5.5. Dynamic light scattering
The measurements were performed on a Malvern Zetasizer nanoS apparatus thermostated at 20 °C. The size distribution was calibrated with latex particles of 65 and 200 nm radii. The scattering intensity data were processed using the instrumental software to obtain the hydrodynamic diameter (Dh) and the size distribution of scatters in each sample. A total of 10 scans with an overall duration of 5 min were obtained for each sample and time point. All data were analysed in triplicate. Melting curves were obtained by recording the mean size as a function of temperature in the range 20 to 70 to 80 °C, using a temperature gradient of 1 °C/ minute The Tm value was obtained from the maximum of the first derivative curve. The protein concentrations usually were in the range of 5–15 μM. The samples for dynamic light scattering were prepared in 20 mM Tris buffer at pH 7.4 with 50 mM NaCl.
5.6. Surface plasmon resonance experiments
The binding competition kinetics were performed on a Biacore 3000 apparatus using streptavidin-coated sensorchips (SA, Biacore) prepared as indicated by the manufacturer. Immobilization of the biotinylated oligonucleotide on the streptavidin-coated sensorchip was carried out in PBS. The SPR kinetic measurements were performed in 20 mM tris–HCl buffer, pH 7.4 at 20 °C, containing 300 mM NaCl. The oligonucleotides were denaturated at 80 °C and renatured slowly at room temperature for one hour before any experiment. The NP protein or its mutants were injected at concentrations of 100–500 nM in the presence or absence of naproxen derivatives, up to 100 μM in some experiments. Measurements were conducted at 20 °C and samples were injected at 25 μl/min flow rate. The R152A, R361A or Y148A proteins were attached to the surface of the same chip at similar RU and a free flow cell was used for blank subtraction. The data were fitted with a simple dose-response isotherm assuming a 1: 1 NP: naproxen C0 stoichiometry (Supplementary Figure 2).
Supplementary material
The supplementary material for this paper is available online at http://dx.doi.10.1080/07391102.2014.979230.
Disclosure statement
There is no conflict of interest related to this work.
TBSD_A_979230 Supplementary material
Download PDF (1.1 MB)Additional information
Funding
References
- Arranz, R., Coloma, R., Chichon, F. J., Conesa, J. J., Carrascosa, J. L., Valpuesta, J. M., … Martin-Benito, J. (2012). The structure of native influenza virion ribonucleoproteins. Science, 338, 1634–1637.10.1126/science.1228172
- Butler, J., Hooper, K. A., Petrie, S., Lee, R., & Maurer-Stroh, S. (2014). Estimating the fitness advantage conferred by permissive neuraminidase mutations in recent oseltamivir-resistant A(H1N1)pdm09 influenza viruses. PLoS Pathogens, 10, e1004065.10.1371/journal.ppat.1004065
- Chan, W. H., Ng, A. K., Robb, N. C., Lam, M. K., Chan, P. K., Au, S. W., … Shaw, P. C. (2010). Functional analysis of the influenza virus H5N1 nucleoprotein tail loop reveals amino acids that are crucial for oligomerization and ribonucleoprotein activities. Journal of Virology, 84, 7337–7345.10.1128/JVI.02474-09
- Chaurasiya, K. R., McCauley, M. J., Wang, W., Qualley, D. F., Wu, T., Kitamura, S., … Williams, M. C. (2014). Oligomerization transforms human APOBEC3G from an efficient enzyme to a slowly dissociating nucleic acid-binding protein. Nature Chemistry, 6, 28–33.
- Chenavas, S., Crépin, T., Delmas, B., Ruigrok, R. W., & Slama-Schwok, A. (2013). Influenza virus nucleoprotein: Structure, RNA binding, oligomerization and antiviral drug target. Future Microbiology, 8, 1537–1545.10.2217/fmb.13.128
- Chenavas, S., Estrozi, L. F., Slama-Schwok, A., Delmas, B., Di Primo, C., Baudin, F., … Ruigrok, R. W. (2013). Monomeric nucleoprotein of influenza A virus. PLoS Pathogens, 9, e1003275.10.1371/journal.ppat.1003275
- Courchay, F. C., Sworen, J. C., Ghiviriga, I., Abboud, K. A., & Wagener, K. B. (2006). Understanding structural isomerization during ruthenium-catalyzed olefin metathesis: A deuterium labeling study. Organometallics, 25, 6074–6086.10.1021/om0509894
- Darden, T., York, D. M., & Pedersen, L. (1993). Particle mesh Ewald: An N⋅log(N) method for Ewald sums in large systems. The Journal of Chemical Physics, 98, 10089–10092.10.1063/1.464397
- Das, K., Aramini, J. M., Ma, L. C., Krug, R. M., & Arnold, E. (2010). Structures of influenza A proteins and insights into antiviral drug targets. Nature Structural & Molecular Biology, 17, 530–538.
- Elton, D., Medcalf, E., Bishop, K., & Digard, P. (1999). Oligomerization of the influenza virus nucleoprotein: Identification of positive and negative sequence elements. Virology, 260, 190–200.10.1006/viro.1999.9818
- Harvey, R. G., Dai, Q., Ran, C., & Penning, T. M. (2004). Synthesis of the o-Quinones and other oxidized metabolites of polycyclic aromatic hydrocarbons implicated in carcinogenesis. The Journal of Organic Chemistry, 69, 2024–2032.10.1021/jo030348n
- Jorgensen, W. L., Chandrasekhar, J., Madura, J. D., Impey, R. W., & Klein, M. L. (1983). Comparison of simple potential functions for simulating liquid water. The Journal of Chemical Physics, 79, 926–935.10.1063/1.445869
- Krug, R. M., & Aramini, J. M. (2009). Emerging antiviral targets for influenza A virus. Trends in Pharmacological Sciences, 30, 269–277.10.1016/j.tips.2009.03.002
- Lejal, N., Tarus, B., Bouguyon, E., Chenavas, S., Bertho, N., Delmas, B., … Slama-Schwok, A. (2013). Structure-based discovery of the novel antiviral properties of naproxen against the nucleoprotein of influenza A virus. Antimicrobial Agents and Chemotherapy, 57, 2231–2242.10.1128/AAC.02335-12
- Longhi, S., Receveur-Brechot, V., Karlin, D., Johansson, K., Darbon, H., Bhella, D., … Canard, B. (2003). The C-terminal domain of the measles virus nucleoprotein is intrinsically disordered and folds upon binding to the C-terminal moiety of the phosphoprotein. Journal of Biological Chemistry, 278, 18638–18648.10.1074/jbc.M300518200
- Lopez-Martinez, R., Ramirez-Salinas, G. L., Correa-Basurto, J., & Barron, B. L. (2013). Inhibition of influenza A virus infection in vitro by peptides designed in silico. PLoS ONE, 8, e76876.10.1371/journal.pone.0076876
- MacKerell, A. D., Jr, Banavali, N., & Foloppe, N. (2000). Development and current status of the CHARMM force field for nucleic acids. Biopolymers, 56, 257–265.10.1002/(ISSN)1097-0282
- Matsunaga, N., Kaku, T., Ojida, A., Tanaka, T., Hara, T., Yamaoka, M., … Tasaka, A. (2004). C(17,20)-lyase inhibitors. Part 2: Design, synthesis and structure-activity relationships of (2-naphthylmethyl)-1H-imidazoles as novel C(17,20)-lyase inhibitors. Bioorganic & Medicinal Chemistry, 12, 4313–4336.
- Mavrakis, M., Méhouas, S., Réal, E., Iseni, F., Blondel, D., Tordo, N., & Ruigrok, R. W. (2006). Rabies virus chaperone: Identification of the phosphoprotein peptide that keeps nucleoprotein soluble and free from non-specific RNA. Virology, 349, 422–429.10.1016/j.virol.2006.01.030
- Mena, I., Jambrina, E., Albo, C., Perales, B., Ortin, J., Arrese, M., … Portela, A. (1999). Mutational analysis of influenza A virus nucleoprotein: Identification of mutations that affect RNA replication. Journal of Virology, 73, 1186–1194.
- Moeller, A., Kirchdoerfer, R. N., Potter, C. S., Carragher, B., & Wilson, I. A. (2012). Organization of the influenza virus replication machinery. Science, 338, 1631–1634.10.1126/science.1227270
- Ng, A. K., Wang, J. H., & Shaw, P. C. (2009). Structure and sequence analysis of influenza A virus nucleoprotein. Science in China Series C: Life Sciences, 52, 439–449.10.1007/s11427-009-0064-x
- Ng, A. K., Zhang, H., Tan, K., Li, Z., Liu, J. H., Chan, P. K., … Shaw, P. C. (2008). Structure of the influenza virus A H5N1 nucleoprotein: Implications for RNA binding, oligomerization, and vaccine design. The FASEB Journal, 22, 3638–3647.10.1096/fj.08-112110
- Phillips, J. C., Braun, R., Wang, W., Gumbart, J., Tajkhorshid, E., Villa, E., … Schulten, K. (2005). Scalable molecular dynamics with NAMD. Journal of Computational Chemistry, 26, 1781–1802.10.1002/(ISSN)1096-987X
- Purohit, R. (2014). Role of ELA region in auto-activation of mutant KIT receptor: A molecular dynamics simulation insight. Journal of Biomolecular Structure & Dynamics, 32, 1033–1046.
- Purohit, R., Rajendran, V., & Sethumadhavan, R. (2011a). Relationship between mutation of serine residue at 315th position in M. tuberculosis catalase-peroxidase enzyme and isoniazid susceptibility: An in silico analysis. Journal of Molecular Modeling, 17, 869–877.10.1007/s00894-010-0785-6
- Purohit, R., Rajendran, V., & Sethumadhavan, R. (2011b). Studies on adaptability of binding residues and flap region of TMC-114 resistance HIV-1 protease mutants. Journal of Biomolecular Structure & Dynamics, 29, 137–152.
- Rajendran, V., Purohit, R., & Sethumadhavan, R. (2012). In silico investigation of molecular mechanism of laminopathy caused by a point mutation (R482W) in lamin A/C protein. Amino Acids, 43, 603–615.10.1007/s00726-011-1108-7
- Rajendran, V., & Sethumadhavan, R. (2014). Drug resistance mechanism of PncA in Mycobacterium tuberculosis. Journal of Biomolecular Structure & Dynamics, 32, 209–221.
- Reguera, J., Malet, H., Weber, F., & Cusack, S. (2013). Structural basis for encapsidation of genomic RNA by La crosse orthobunyavirus nucleoprotein. Proceedings of the National Academy of Sciences, 110, 7246–7251.10.1073/pnas.1302298110
- Ruigrok, R. W., Crépin, T., & Kolakofsky, D. (2011). Nucleoproteins and nucleocapsids of negative-strand RNA viruses. Current Opinion in Microbiology, 14, 504–510.10.1016/j.mib.2011.07.011
- Sharma, M., & Ray, S. M. (2008). Synthesis and biological evaluation of amide derivatives of (5,6-dimethoxy-2,3-dihydro-1H-inden-1-yl) acetic acid as anti-inflammatory agents with reduced gastrointestinal ulcerogenecity. European Journal of Medicinal Chemistry, 43, 2092–2102.10.1016/j.ejmech.2007.08.008
- Shen, Y. F., Chen, Y. H., Chu, S. Y., Lin, M. I., Hsu, H. T., Wu, P. Y., … Tsai, M. D. (2011). E339 … R416 salt bridge of nucleoprotein as a feasible target for influenza virus inhibitors. Proceedings of the National Academy of Sciences, 108, 16515–16520.10.1073/pnas.1113107108
- Shih, S. R., Horng, J. T., Poon, L. L., Chen, T. C., Yeh, J. Y., Hsieh, H. P., … Hsu, J. T. (2010). BPR2-D2 targeting viral ribonucleoprotein complex-associated function inhibits oseltamivir-resistant influenza viruses. Journal of Antimicrobial Chemotherapy, 65, 63–71.10.1093/jac/dkp393
- Slama Schwok, A., Delmas, B., Quideau, S., Bertrand, H., & Tarus, B. (2012). Antiviral compositions directed against the influenza virus nucleoprotein. International patent extension no PCT/EP2012/001720 deposited April 21, 2012.
- Sulyok, G. A., Gibson, C., Goodman, S. L., Hölzemann, G., Wiesner, M., & Kessler, H. (2001). Solid-phase synthesis of a nonpeptide RGD mimetic library: New selective alphavbeta3 integrin antagonists. Journal of Medicinal Chemistry, 44, 1938–1950.10.1021/jm0004953
- Tarus, B., Bakowiez, O., Chenavas, S., Duchemin, L., Estrozi, L. F., Bourdieu, C., … Slama-Schwok, A. (2012). Oligomerization paths of the nucleoprotein of influenza A virus. Biochimie, 94, 776–785.10.1016/j.biochi.2011.11.009
- Tarus, B., Chevalier, C., Richard, C. A., Delmas, B., Di Primo, C., & Slama-Schwok, A. (2012). Molecular dynamics studies of the nucleoprotein of influenza A virus: Role of the protein flexibility in RNA binding. PLoS ONE, 7, e30038.10.1371/journal.pone.0030038
- Tawar, R. G., Duquerroy, S., Vonrhein, C., Varela, P. F., Damier-Piolle, L., Castagne, N., … Rey, F. A. (2009). Crystal structure of a nucleocapsid-like nucleoprotein-RNA complex of respiratory syncytial virus. Science, 326, 1279–1283.10.1126/science.1177634
- Wang, J., Ma, C., Fiorin, G., Carnevale, V., Wang, T., Hu, F., … DeGrado, W. F. (2011). Molecular dynamics simulation directed rational design of inhibitors targeting drug-resistant mutants of influenza A virus M2. Journal of the American Chemical Society, 133, 12834–12841.10.1021/ja204969m
- Ye, Q., Guu, T. S., Mata, D. A., Kuo, R. L., Smith, B., Krug, R. M., & Tao, Y. J. (2012). Biochemical and structural evidence in support of a coherent model for the formation of the double-helical influenza A virus ribonucleoprotein. MBio, 4, e00467–00412.
- Ye, Q., Krug, R. M., & Tao, Y. J. (2006). The mechanism by which influenza A virus nucleoprotein forms oligomers and binds RNA. Nature, 444, 1078–1082.10.1038/nature05379