
 ?Mathematical formulae have been encoded as MathML and are displayed in this HTML version using MathJax in order to improve their display. Uncheck the box to turn MathJax off. This feature requires Javascript. Click on a formula to zoom.
?Mathematical formulae have been encoded as MathML and are displayed in this HTML version using MathJax in order to improve their display. Uncheck the box to turn MathJax off. This feature requires Javascript. Click on a formula to zoom.Abstract
The collagen protein provides tensile strength to the extracellular matrix in addition to localising cells, proteins and protein cofactors. Collagen is susceptible to a build up of glycation modifications as a result of an exceptionally long half-life. Glucosepane is a collagen cross-linking advanced glycation end product; the structural and mechanical effects of glucosepane are still the subjects of much debate. With the prospect of an ageing population, the management and treatment of age-related diseases is becoming a pressing concern. One area of interest is the isolation of hydrated glucosepane, which has yet to be reported at an atomistic level. This study presents a series of glucosepane–water complexes within an implicit aqueous environment. Electronic structure calculations were performed using density functional theory and a high level basis set. Hydrogen bonds between glucosepane and explicit water were identified by monitoring changes to covalent bonds, calculating levels of electron donation from Natural Bonding Orbital analysis and the detection of bond critical points. Hydrogen bond strength was calculated using second-order perturbation calculations. The combined results suggest that glucosepane is very hydrophilic, with the imidazole feature being energetically more attractive to water than either hydroxyl group, although all hydrogen bonds, regardless of bond strength, were electrostatic in nature. Our results are in growing support of an earlier hypothesis that cross-links may result in an increase in interstitial water retention, which would permit the collagen fibril to swell, thereby potentially affecting the tensile and compression properties and biological function of connective tissues.
Introduction
The protein collagen makes up 70–80% of the dry weight of tissues such as tendons, ligaments and cartilage (Ottani, Raspanti, & Ruggeri, Citation2001; Bhattacharjee & Bansal, Citation2005) and provides the tissues with their mechanical strength. With an exceptionally long lifetime (>100 years in articular cartilage Maroudas, Palla, & Gilav, Citation1992) and a half-life of about 200 years in equine tendon (Thorpe et al., Citation2010), collagen is vulnerable to irreversible modifications such as racemisation, isomerisation, deamination, oxidation and glycation. The addition of sugars (glycation) can result in a series of spontaneous non-enzymatic reactions, also known as the Maillard reaction (Maillard, Citation1913), leading to the formation of covalent cross-link advanced glycation end (AGE) products (Bulterijs & Sjöberg, Citation2009). The reaction between free glucose and collagen-bound lysine and arginine side chains results in an AGE product known as glucosepane (Bulterijs & Sjöberg, Citation2009). Although there are a number of cross-linking AGE products found in the human ECM, including pentosidine (Nomoto, Yagi, Hamada, Naito, & Yonei, Citation2013), deoxyglucosone lysine dimer (DOGDIC), methylglyoxal lysine dimer (MOLD) and glyoxal lysine dimer (GOLD) (Avery & Bailey, Citation2005, Citation2006), glucosepane has been found to be the most abundant in human skin and glomerular basement membrane (Sell et al., Citation2005).
Due to the dense collagen fibrillar environment, a slow reaction rate and a reaction mechanism that is still unconfirmed, very little is known about glucosepane at the atomistic level. To understand the impact of glucosepane on the collagen mechanical properties (tensile and compression strength) and biological function, a full understanding of molecular characteristics is required. At the macroscopic level, the presence of AGE products affects the stiffness and the solubility of the collagen by increasing its resistance to digestion by proteases (Kent, Light, & Bailey, Citation1985). In vitro studies have shown that an increase in covalent cross-links mediated by riboflavin results in increased retention of interstitial water in addition to an increase in fibril diameter (Rich, Odlyha, Cheema, Mudera, & Bozec, Citation2014), and as suggested, cross-links may permit a swelling of the fibril. These changes may be caused by a change in the hydrophilicity of collagen due to glucosepane but this remains a relatively unexplored area, and to date, the link between hydrophilicity and AGE product cross-linked collagen remains unclear. Our understanding of the relationship between collagen cross-linking and hydration in context of the extracellular matrix is also relevant to collagen applications within industry, since many surgical applications of collagen-based materials, for example, involve dried collagen (Klopper, Citation1986).
This study presents the first in-depth quantum mechanical analysis, using a number of complementary electronic structure techniques, of hydrogen bond formation between glucosepane and explicit water molecules within an implicit aqueous environment. We aim to identify potential sites of water association, characterise the nature of the intermolecular bond and measure its strength.
Theoretical methods
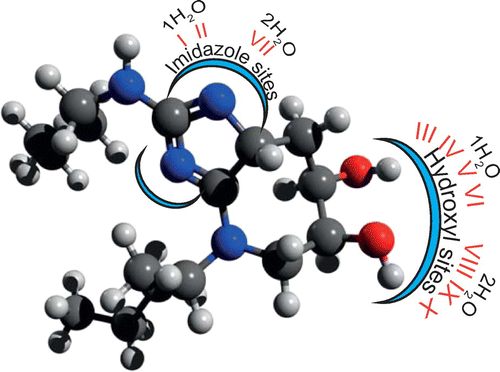
To understand the interaction between glucosepane and water, the imidazole group and hydroxyl groups were identified as two potential sites for the formation of hydrogen bonds (see Figure ). The imidazole section of glucosepane derives from the guanidine functional group of the arginine side chain, whilst both hydroxyl groups on the seven-membered ring originate from glucose. Ten models of water coordination were proposed, of which I and II represent single water coordination with each nitrogen of the imidazole site in turn, III to VI denote single water coordination at the hydroxyl groups, VII denotes water coordination to both nitrogen atoms of the imidazole site simultaneously and VIII to X denote simultaneous coordination of water molecules at the hydroxyl groups. An initial glucosepane starting geometry was taken from the last time frame of a 60 ns explicitly solvated all-atom MD simulation from a previous study (Collier, Nash, Birch, & de Leeuw, Citation2015). Single and double water molecules were added in turn within approximate hydrogen bond distance from the two proposed sites. The two aliphatic side chains were held fixed and a steepest descent energy minimisation was performed using the Universal Force Field (as implemented in Avogadro (Hanwell et al., Citation2012)). This was repeated until 10 unique water coordination models were generated, encompassing initial orientations of water to the oxygen and hydrogen of each hydroxyl group, respectively, and water with each nitrogen atom of the imidazole site. Structures were saved into Cartesian coordinate geometry (pdb file format) in preparation for electronic structure calculations.
Figure 1. An illustration of glucosepane depicting the imidazole region and hydroxyl groups. Each glucosepane–water bound complex label along with the respective number of explicit water molecules is also included.
Geometry optimisation was performed using Density Functional Theory (DFT) calculations, followed by complementary single-point energy (SPE) calculations. The infrared (IR) spectra from vibrational frequency analysis calculations were inspected for signs of a shift in the peak associated with the O–H bond, indicative of weak interactions, whilst any changes to the O–H bond length were reported. Electron occupancy and second-order permutation of donor–acceptor Lewis structures were recorded to elucidate individual lone pair contributions. Finally, Quantum Theory Atoms in Molecules (QTAIM) (Bader, Citation1991) was used to provide further evidence of hydrogen bonding by locating bond critical points (BCP) with electron density and electron Laplacian values characteristic to hydrogen bonds.
Electronic structure calculations were performed using GAUSSIAN-09 (Frisch et al., Citation2010). Using the integral equation formalism polarisable continuum model (IEF-PCM), all structure calculations were performed in an implicit water solvent model. The modern DFT hybrid meta-generalised gradient approximation wb97xd functional (Chai & Head-Gordon, Citation2008), which contains empirical dispersion terms and long-range corrections, in conjunction with the 6-311++g(2df,2p) basis set was used for all electronic structure optimisation calculations. The large basis set should compensate for the basis set superposition error (BSSE). A convergence criterion of maximum force, RMS force, maximum displacement and RMS displacement was adopted throughout, set to, 10 × 10−6, 6 × 10−5, 4 × 10−5, respectively, along with an ultra-fine integration grid. Vibrational frequency analysis was performed for each optimised structure to verify that an energy minimum had in fact been found. The DFT energy calculations were complemented by SPE calculations using the post-Hartree Fock ab initio Møller–Plesset perturbation theory to second order (MP2) (Møller & Plesset, Citation1934), in conjunction with the double-zeta Dunning’s correlation consistent basis set augmented with diffusion functions (aug-cc-pVDZ). Efforts were made to use a triple-zeta basis set for improved accuracy, but given the size of our molecular fragments this was not possible. Zero-point energies (ZPE) from the DFT frequency calculations were used to adjust the MP2 calculations to aid comparison between the different chemical methods. Natural Orbital Theory (NBO) population analysis was performed at the wb97xd/6-311++g(2df,2p) level. Donor–acceptor interactions in the NBO basis were evaluated using the second-order permutation method. Finally, the electronic landscape was analysed for bond critical points and bond critical paths using QTAIM via the Multiwfn software suite version 3.3.7 (Lu & Chen, Citation2012).
Results and discussion
Initial structure and electronic structure optimisation
An initial glucosepane starting structure was extracted from the previous work on a cross-linked collagen peptide MD study (Collier et al., Citation2015). All solvent molecules were removed and substituted by nH2O (n = 1, 2) explicit molecules. Water was manually positioned 1.8 Å from the participating glucosepane atom as an initial approximation for a weak intermolecular interaction. To prevent an interaction between water molecules and the protein backbone, lysine and arginine side chains were truncated at the α-carbon atom with a methyl group. This also helped to reduce the computational load.
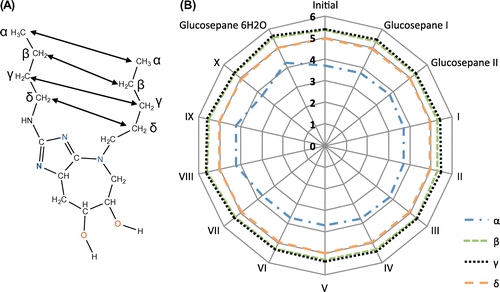
We first used frozen α-carbon coordinates to avoid further optimisation of the initial structure of the truncated protein backbone, but these calculations consistently failed to converge to an energy minimum. By removing the constraints, each structure converged to the specified criteria with very little deviation to the interatomic distance between respective carbon atoms along the aliphatic side chain (Figure ). Slight flexibility can be seen in between α-carbon atoms in complexes IV, V and glucosepane coordinated with six water molecules. The comparative interatomic distances between β-, γ- and δ-carbons are within close proximity of those in the initial structure and therefore of little concern to the forthcoming relative interaction energy calculations.
Figure 2. A distance comparison between interatomic carbon atoms along the aliphatic chain of lysine and arginine. (A) The structure of glucosepane (with methyl capping in place of the α-carbon atoms). Carbon atoms along the length of each aliphatic chain are denoted α, β, γ and δ. The ε carbon on lysine lacks comparison given the shorter length of arginine. (B) A measure of interatomic carbon distances per complex, per glucosepane structure and for the initial MD simulation structure.
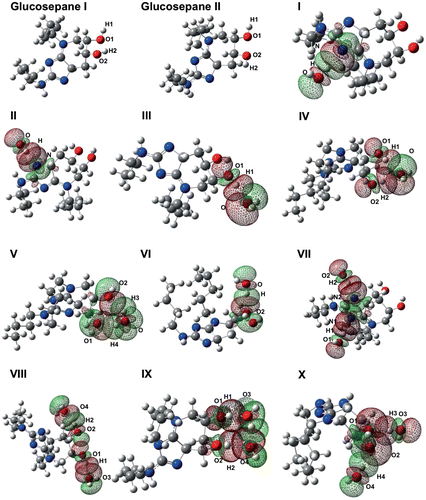
Optimisation of the first unbound glucosepane structure, denoted Glucosepane I, revealed a weak intramolecular interaction between the hydroxyl atoms H2 and O1. Disruption to this intramolecular weak bond by the presence of water would add a bias to the calculation of relative interaction energies for structures where this particular bond remains intact. Therefore, a second unbound glucosepane structure, denoted Glucosepane II, was prepared by orienting the hydrogen atoms H1 and H2 away from the oxygen atoms of the neighbouring hydroxyl group. After successfully performing geometry optimisation, it was obvious that the hydrogen atoms were not coordinating with neighbouring oxygen.
Electronic spectra and bond distances
IR spectroscopy is an established technique used to probe the molecular structure of compounds. It is well known that weak bonds can be detected from the visible shift in peaks at certain wavelengths. In particular, the O–H bond stretching vibration is known to decrease in the presence of a hydrogen bond (Pimentel & McClellan, Citation1960). Computationally calculated IR absorption of the molecular structure may serve as a means of reliably detecting the presence of hydrogen bonds. The IR spectra of glucosepane structures, hydrated glucosepane structures and finally free water were reported from vibrational frequency analysis. Frequency and IR intensities along with a red-shift (a drop in frequency) with respect to a free glucosepane structure are presented in Table . The IR spectra from structures made up of hydrogen-bonded and non-hydrogen-bonded O–H groups would yield two sub-bands from the O–H bond stretch. The sub-band corresponding to non-hydrogen-bonded O–H groups occurs around 3640–3610 cm−1, which is considerably higher than O–H groups involved in hydrogen bonding. Interatomic bond lengths have been reported to an accuracy of .001 Å, given that changes to bond lengths were only calculated between structures optimised using the same computational framework, and are not being compared with either different levels of computational chemistry or experimental results.
Table 1. IR frequency calculations of optimised electronic structure calculations.
From the presented set of calculations, the two O–H bonds in free water showed absorption at 3881.25 cm−1. This is typically quite high for fundamental O–H bond stretching frequency. It is worth noting at this stage, that in most cases chemical model spectra are scaled. However, the vibrational frequencies of weakly bound and/or non-covalent species are generally not available and therefore the peak frequencies and red-shifts of contributing hydrogen bond O–H remain unadjusted (Alecu, Zheng, Zhao, & Truhlar, Citation2010).
Glucosepane II demonstrates a band within the wavelength associated with O–H bond vibration, suggesting the absence of a weak bond associated with the hydroxyl groups. Interestingly, the O2–H2 bond in Glucosepane I not only shows a visible red-shift by 76.70 cm−1 but an increase in d(O–H) by .005 Å. The donor–proton–acceptor angle in a hydrogen bond is typically at least 90° and as such this shift in band intensity is not surprising given the intermolecular angle O1H1O2 of 114.6°, as seen in Figure . The remaining hydroxyl group O1–H1 in Glucosepane I and the hydroxyl groups of Glucosepane II remain unperturbed.
Figure 3. Optimised electronic structure calculations using wb97xd/6-311++g(2df,2p) of both glucosepane controls, and all water-bound glucosepane structures. The molecular orbitals involved in hydrogen water binding are labelled.
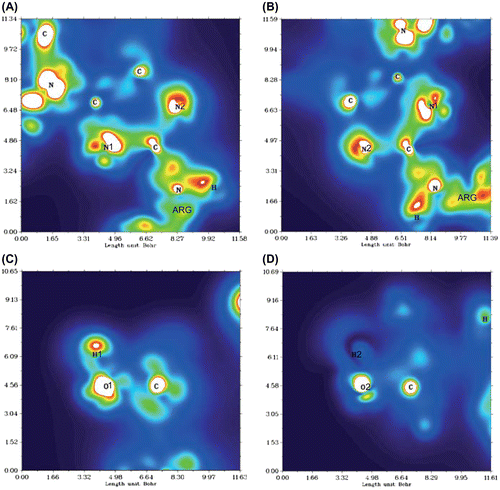
When comparing the IR spectrum of a solitary glucosepane molecule to one coordinated with water, we would expect some degree of red-shift in the O–H peak in the spectrum of the coordinated structure. Between the two sets of IR spectra and bond distances associated with the imidazole ring, complex II demonstrated the greatest degree in red-shift and IR intensity when water is coordinated with N2. Interestingly, coordination with N1, complex I, did not yield as great a shift in the peak wavelength, with a difference of 210.99 cm−1. Both water molecules experienced the expected increase in the O–H bond length and an N⋯H distance well within an expected hydrogen bond distance (see Table ). Simultaneous water binding to the imidazole ring, complex VII and VIII, clearly show how the hydrogen H2 is drawn closer to N2 by .107 Å in comparison with N1 to H1. The water coordinating with N2 also yields the greater red-shift, 550.42 cm−1, compared with the red-shift, 355.53 cm−1, of the water coordinating with N1. In addition, the O–H bond of water coordinating with N2 is stretched further by .009 Å. Investigation of charge distribution of complexes I, II, VI and VII (electron density of all four complexes can be found in Figure ) confirmed that N2 has a Mulliken charge of approximately −.687 and N1 has a Mulliken charge of approximately −.550. The difference in charge is due to the proximity of N1 to the lysine nitrogen, which helps explain the difference in water affiliation.
Table 2. Calculated bond distances (Å) of glucosepane O–H groups and coordinated water intermolecular H⋯O bonds.
Figure 4. Electron density plot of glucosepane I. Centred over the plane defined according to (A) C–N1–C, (B) C–N2–C, (C) C–O1–H1 and (D) C–O2–H2.
The next sites considered as recipients of water association are the hydroxyl groups. Not only must one consider the proximity between the hydroxyl groups and how they may have an effect on water coordination, but also whether the hydroxyl groups will act as hydrogen bond donors or acceptors. Due to this additional consideration, the binding of a single water molecule was investigated first, followed by two molecules.
The first of the optimised hydroxyl water-bound models, complex III, demonstrates the water molecule acting as an electron donor to the hydrogen of O1–H1. Although the water molecule is thought to be acting in isolation from other regions of the glucosepane molecule, it is highly likely that an additional weak interaction, one between the hydroxyl groups themselves, is contributing in part to the stability of this structure. Several attempts were made to isolate an association between water and the hydroxyl group O2–H2, but all attempts resulted in a double coordination of the oxygen from the water molecule with both hydroxyl hydrogen atoms, as seen in complex IV, Figure . Vibrational analysis revealed a single peak at 3769.40 cm−1 for the simultaneous stretching of both water bonds. In a similar vein, attempts at isolated O1 as the sole electron donor yielded a twin association between each hydroxyl group oxygen with hydrogen from the water molecule, denoted in Figure as complex V. Both covalent OH bonds within the single water molecule experience the same IR intensity and red-shift, yet it is interesting to note that the intramolecular bond in H⋯O2 is shorter by .144 Å. Finally, the coordination of a water molecule to O2 of the glucosepane hydroxyl group can be seen in Figure , denoted complex VI. The O–H peak experiences a red-shift similar to the other complexes in addition to an elongation of the covalent bond, both indicative of an electron donor–acceptor intermolecular bond.
In order to elucidate variations in water–glucosepane and water–water coordination, an additional water molecule beside the hydroxyl groups of glucosepane was introduced. We were able to isolate water associating only with their respective hydroxyl group, as seen in complex VIII, and subsequently removing the inherent interaction between hydroxyl groups similar to those seen in glucosepane II. The O–H peak associated with O1–H1 shifts 42.90 cm−1 further than the O2–H2 peak. In addition, consideration of the bond distances confirms that the O1–H1 is elongated by a further .001 Å with respect to the bond distance of O2–H2, and that the water molecule is drawn closer via the intermolecular attraction between H1⋯O3 compared with the attraction between O2⋯H4.
Complex IX shows the coordination of water molecules acting as hydrogen bond donors with the hydroxyl group hydrogen, whilst yielding a water–water arrangement that may result in an additional weak intermolecular interaction. Of the two intermolecular associations, the O1–H1 peaks yield a greater red-shift than O2–H2 by 42.90 cm−1, whilst also demonstrating a longer distortion to the O–H bond by .004 Å. A shorter intermolecular distance of 1.839 Å accompanies the stretching of the hydroxyl group between H1 and O3, compared with intermolecular distance 1.935 Å between H2 and O4. Although O1–H1 is shown to associate more closely with water, the bond distance of O2–H2 is still .009 Å longer compared with O2–H2 of glucosepane II, indicating a distortion to the bond length due to the presence of a neighbouring water molecule. Efforts were made to utilise the oxygen from the glucosepane hydroxyl groups as a hydrogen bond donor with a water molecule as hydrogen acceptor, as seen in complex X. However, the free hydrogen from the hydroxyl group O1–H1 was able to orient itself towards the oxygen, O2, resulting in three weak associations. Both O–H bonds within the water molecule, associated with the glucosepane oxygen donors, demonstrate a visible red-shift to the O–H peak. The bond O4–H4 associating with the glucosepane oxygen O2 is shown to shift further than O3–H3 by 118.45 cm−1, in addition to having an increase in IR intensity by 36.39. Both coordinating water O–H bonds are longer than in an isolated water molecule, suggesting an intermolecular influence.
Interaction energies and molecular orbital analysis
It is well established that hydrogen bond strength is dependent on the types of bond donor and acceptors, ranging from as little as .2 kcal mol−1 in almost complete electrostatic interactions to approximately 40 kcal mol−1 in systems of relatively high charge transfer (Steiner, Citation2002). A relative interaction energy value was used as a measure of the strength of the hydrogen bonds (Table ).
Table 3. DFT and MØller–Plesset ΔE rel. Measurements were taken from wb97xd/6-311++g(2df,2p) optimised electronic structures.
Energy values in the text are denoted as DFT/MP2. The relative interaction energy, , was calculated according to:
where E
comp is the total energy of the glucosepane explicit water complex, is the total energy of the explicit water molecules (n = 1, 2) in isolation and E
AGE is the total energy of glucosepane in isolation. All energy calculations were adjusted for an implicit water environment. DFT interaction energy calculations were complemented with a set of post-Hartree Fock MP2 SPE calculations. The ZPE from the DFT frequency calculations was used to adjust each respective MP2 SPE.
NBO population analysis was conducted using the DFT chemistry model to yield Lewis-like electron donor–acceptor interactions. The donor–acceptor (bond–anti-bond) interactions in the NBO basis were monitored from occupancy exchange between lone pair orbital occupation to the σ*O–H anti-bond (Table ). The strength of the interaction due to delocalisation in the complex was calculated using the second-order perturbation () theory analysis of the Fock matrix in the NBO basis. The calculation of relative interaction energies for complexes I, II, III, VI and VII took into account the intramolecular hydrogen bond between hydroxyl groups. The intramolecular bond was disrupted through water coordination in complexes IV, V, VIII, IX and X. Relative interaction energies for these complexes were adjusted according to the total energy of glucosepane I or II, respectively.
Table 4. Natural Bond Orbital population analysis showing the occupancy of molecular orbitals of hydrogen bond donors, LP and unfilled orbital acceptors, BD*, between glucosepane and coordinating water molecules.
DFT studies of intramolecular association between hydroxyl groups in d-glucose indicate that hydrogen bonds are responsible for the counter-clockwise arrangement of hydroxyl groups, although such is not the case in its α-D-glucopyranose derivative (Silla, Cormanich, Rittner, & Freitas, Citation2014; Çarçabal et al., Citation2005), which is thought to be stabilised by hyperconjugative effects (Silla et al., Citation2014). NBO orbital and second-order perturbation theory of glucosepane I did not yield delocalisation between the oxygen of either hydroxyl group with the opposite σ*C–O anti-bond. Rather, the lone pair on O1 had a delocalisation strength of 2.67 kcal mol−1 with the σ*O2–H2 anti-bond, indicating a weak hydrogen bond.
It was clear, given the discrepancy between interatomic hydrogen bond distances, orbital occupations and second-order perturbation energies that each site contributed uniquely to water association. The O–H⋯N2 interaction between a single water molecule and the nitrogen in the imidazole group furthest from the lysine side chain (complex II) yields the lowest , −6.73/−6.93 kcal mol−1, when compared with the other single water bound complexes. Interestingly, when the water molecule binds to the imidazole nitrogen N1 (complex I), separated from the lysine nitrogen by one carbon atom, the
weakens by 1.40/.34 kcal mol−1. The difference in
can be supported by a difference of 10.47 kcal mol−1 in favour of the bond–anti-bond delocalisation of O–H⋯N2. Although both nitrogen imidazole atoms yield the strongest association with water, their proximity to the complete collagen backbone may interfere with water binding.
In complex III, an O1–H1⋯O hydrogen bond was found to contribute with a of −3.94/−4.67 kcal mol−1. Although there is no obvious observable interaction between the two hydroxyl groups of glucosepane, similar to glucosepane I, NBO analysis suggests a marginal delocalisation of 3.87 kcal mol−1. This interaction would contribute ever so slightly to the degree of
when the total energy of glucosepane II was taken into account. The double coordination of water with the hydrogen from each hydroxyl group, as seen in complex IV, leads to a significantly stable complex, with a
of −5.87/−6.55 kcal mol−1. NBO analysis revealed a fraction of a difference between the paired interactions in anti-bond electron occupation, whilst second-order perturbation theory analysis revealed a stronger interaction between the combined lone pairs of oxygen with O2–H2 by .9 kcal mol−1. Conversely, the coordination of both hydrogen atoms of a single water molecule with the oxygen of each hydroxyl group, as seen in complex V, results in a weaker but stable
of −4.16/−4.42 kcal mol−1. There was low electron occupancy of .0804 in the O–H4 anti-bond with a combined strength of 3.03 kcal mol−1. Yet, the O–H3 anti-bond occupancy of .01779 with the combined delocalisation strength of 8.07 kcal mol−1 significantly contributes to the stability of the water interaction. With the weakest
of −3.33/−4.25 kcal mol−1, NBO analysis of complex VI revealed a combined delocalisation strength of 10.97 kcal mol−1 along with an anti-bond O–H occupancy of .02266.
The coordination of two water molecules begins with complex VII, where a water molecule coordinates to each nitrogen (N1 and N2) of the imidazole ring yielding a of −11.60/−12.95 kcal mol−1. The intramolecular hydrogen bond formed between hydroxyl groups was accounted for using the isolated glucosepane I structure in the relative interaction energy calculations. NBO analysis indicates that there is a greater degree of electron delocalisation between N2 and the O–H of water, which is backed up by
24.55 kcal mol−1 compared with N1 with a
value of 14.73 kcal mol−1. The coordination of oxygen from a water molecule with the hydrogen from each hydroxyl group, complex VIII, resulted in a ΔE
rel of −7.19/−8.94 kcal mol−1. An observable intramolecular interaction between the hydroxyl groups of the glucosepane structure was not detected. NBO analysis clearly indicates that O1–H1...O3 is the strongest interaction, with a greater degree of electron delocalisation and a combined
of 15.46 kcal mol−1 compared to a combined
of 12.92 kcal mol−1 for the O2–H2⋯O4 interaction. Complex IX utilises the hydroxyl hydrogen as electron acceptors, but due to an additional association between water molecules, this complex yields a ΔE
rel of −8.45/−9.47 kcal mol−1, resulting in a far more stable glucosepane–water association. Both glucosepane–water intermolecular interactions are shown to equally contribute to the stability of the complex, with
summed over both lone pair donor–acceptor interactions equating to 11.06 kcal mol−1 and 11.49 kcal mol−1, for the O1–H1⋯O3 and O2–H2⋯O4 interaction, respectively. There is a greater degree of delocalisation in the anti-bond for O2–H2, than with O1–H1. It is interesting to note that although the NBO analysis would suggest that O2–H2⋯O4 has the strongest delocalisation in the complex, the intermolecular distance of O1–H1⋯O3 is not only shorter, but it also experiences a greater degree of red-shift; the increase in stability of complex IX compared to complex VIII can be attributed to the added delocalisation between neighbouring water molecules as indicated by a
of 7.89 kcal mol−1. The final doubly coordinated water complex, complex X, yields the least stable structure with a ΔE
rel of −5.24/−8.49 kcal mol−1. Although the red-shift attributed to the IR peak of O3–H3 is smaller than that of O4–H4, the intermolecular distance is shorter. Delocalisation of the O3–H3⋯O1 interaction by as much as .00841 and a difference in
by as much as 5.91 kcal mol−1 would suggest that the strongest intermolecular interaction lies between O3–H3⋯O1.
QTAIM electron density topology analysis
From very strong [F⋯H⋯F]- hydrogen bonds, predominately derived from charge transfer, to those based almost entirely on electrostatic and Van der Waals interactions, such as very weak C–H⋯X hydrogen bonds, it is possible to differentiate between them by the topology of the electron density (Bader, Citation1991). According to the Quantum Theory of Atoms in Molecules by Bader, elucidation of a bonded structure can be derived from the charge distribution landscape of a molecular geometry. The line of maximum charge distribution linking the nuclei is called a bond path (BP) and the critical point along a BP is referred to as a bond critical point (BCP) (Bader & Essén, Citation1984). The Laplacian of the charge density determines where the function is locally concentrated, where ∇2 ρ(r) < 0, and locally depleted, where ∇2 ρ(r) > 0. Shared interactions (covalent bonds) are associated with relatively large electron density values and a negative Laplacian, whilst closed-shell interactions (ionic and Van der Waals interactions) are associated with small values of electron density and a positive Laplacian (a positive curvature of the density along the bond path). The isolation of hydrogen bond critical points for every complex was not possible without compromising on convergence criteria. As suggested by Lane et al., the absence of a BCP should not be viewed as evidence against hydrogen bonding but rather more simply as the absence of one piece of evidence for hydrogen bonding, and that the BCP criterion of AIM theory is in some cases too stringent (Lane, Contreras-García, Piquemal, Miller, & Kjaergaard, Citation2013).
Depending on the type of donor–acceptor pair, hydrogen bond interactions can be almost entirely electrostatic, almost covalent or some degree of both. The combination of a positive ρ(r c ) and a positive ∇2 ρ(r) for each BCP would suggest that the intermolecular interactions between glucosepane and water were predominately electrostatic in nature (see Table ). It is interesting to note the difference in bond electron topology between complexes III, V and VIII, yielding a very low ∇2 ρ(r), yet still fundamentally electrostatic.
Table 5. The electron density,  , and Laplacian, , of hydrogen bond (−3, −1) critical points between coordinating water molecules and glucosepane calculated using BCP topology analysis.
, and Laplacian, , of hydrogen bond (−3, −1) critical points between coordinating water molecules and glucosepane calculated using BCP topology analysis.
Conclusion
Stable intermolecular configurations between water and glucosepane were successfully identified using DFT electronic structure calculations and verified using post-Hartree Fock ab initio SPE calculations. Both vibrational frequency analysis and Natural Bond Orbital analysis demonstrated characteristics indicative of hydrogen bonds, whilst Quantum Theory of Atoms in Molecules was able to identify hydrogen bond critical points in most of the models presented here. The coordination between water and the nitrogen furthest from the lysine–nitrogen resulted in a greater affinity for an intermolecular bond compared with the opposite nitrogen. A single water molecule was shown to associate with a hydroxyl group, whilst coordination with both hydroxyl groups through delocalisation of the water oxygen lone pairs resulted in further stability.
Our models show clear signs of hydration, with water binding favourably to both nitrogen atoms in the imidazole site and both hydroxyl groups off the seven-membered ring. The presence of glucosepane could potentially increase the retention of water compared with a non-glycated collagen fibril. Such a hypothesis is currently beyond the scope of electronic structure calculations, but the authors are using the results presented in this study to parameterise models of hydrated glucosepane for molecular dynamics simulations.
Collagen-rich tissues where glucosepane forms with increasing age may result in an increase in water content. The localisation of water at a molecular and nanoscale has a significant impact on fibril swelling and hence the function of the tissues. Determining interactions between AGE product cross-links such as glucosepane and water is an important step towards understanding the mechanisms of ageing and age-related diseases.
Disclosure statement
No potential conflict of interest was reported by the authors.
Funding
This work was supported by the Biotechnology and Biological Sciences Research Council [grant number BB/K007785].
Acknowledgements
The authors would like to thank the BBSRC for funding and the Research Computing Platform, UCL, for the use of Legion.
Related Research Data
References
- Alecu, I. M. , Zheng, J. , Zhao, Y. , & Truhlar, D. G. (2010). Computational thermochemistry: Scale factor database and scale factors for vibrational frequencies obtained from electronic model chemistries. Journal of Chemical Theory and Computation , 6 , 2872–2887.10.1021/ct100326h
- Avery, N. C., & Bailey, A. J. (2005). Enzymic and non-enzymic cross-linking mechanisms in relation to turnover of collagen: Relevance to aging and exercise. Scandinavian Journal of Medicine and Science in Sports , 15 , 231–240.10.1111/sms.2005.15.issue-4
- Avery, N. C., & Bailey, A. J. (2006). The effects of the Maillard reaction on the physical properties and cell interactions of collagen. Pathologie Biologie , 54 , 387–395.10.1016/j.patbio.2006.07.005
- Bader, R. F. W. (1991). A quantum theory of molecular structure and its applications. Chemical Reviews , 91 , 893–928.10.1021/cr00005a013
- Bader, R. F. E., & Essén, H. J. (1984). The characterization of atomic interactions. Chem Phys , 80 , 1943–1960.
- Bhattacharjee, A., & Bansal, M. (2005). Collagen Structure: The Madras Triple Helix and the Current Scenario. IUBMB Life (International Union of Biochemistry and Molecular Biology: Life) , 57 , 161–172.10.1080/15216540500090710
- Bulterijs, S., & Sjöberg, J. S. (2009). Characteristics, formation and pathophysiology of glucosepane, a major advanced glycation end product. Rejuvenation Res , 12 , 137–148.
- Çarçabal, P. , Jockusch, R. A. , Hünih, I. , Snoek, L. C. , Kroemer, R. T. , Davis, B. G. , … Simons, J. P. (2005). Hydrogen bonding and cooperativity in isolated and hydrated sugars: Mannose, galactose, glucose and lactose. Journal of the American Chemical Society , 127 , 11414–11425.
- Chai, J. D., & Head-Gordon, M. (2008). Long-range corrected hybrid density functionals with damped atom-atom dispersion corrections. Physical Chemistry Chemical Physics , 10 , 6615–6620.10.1039/b810189b
- Collier, T. , Nash, A. , Birch, H. L. , & de Leeuw, N. H. (2015). Preferential sites for intramolecular glucosepane cross-link formation in type I collagen: A thermodynamic study. Matrix Biology , 48 , 78–88.10.1016/j.matbio.2015.06.001
- Frisch, M. J. , Trucks, G. W. , Schlegel, H. B. , Scuseria, G. E. , Robb, M. A. , Cheeseman, J. R. , … Fox, D. J. (2010) Gaussian 09, Revision B.01, Gaussian Inc, Wallingford CT.
- Hanwell, M. D. , Curtis, D. E. , Lonie, D. C. , Vandermeersch, T. , Zurek, E. , & Hutchison, G. R. (2012). Avogadro: An advanced semantic chemical editors, visualization, and analysis platform. Journal of Cheminformatics , 4 , 17.10.1186/1758-2946-4-17
- Kent, M. J. , Light, N. D. , & Bailey, A. J. (1985). Evidence for glucose-mediated covalent cross-linking of collagen after glycosylation in vitro . Biochemical Journal , 225 , 745–752.10.1042/bj2250745
- Klopper, P. J. (1986). Collagen in surgical research. European Surgical Research , 18 , 218–223.10.1159/000128529
- Lane, J. R. , Contreras-García, J. , Piquemal, J.-P. , Miller, B. J. , & Kjaergaard, H. G. (2013). Are bond critical points really critical for hydrogen bonding? Journal of Chemical Theory and Computation , 9 , 3263–3266.10.1021/ct400420r
- Lu, T., & Chen, F. (2012). Multiwfn: A multifunctional wavefunction analyser. Journal of Computational Chemistry , 33 , 580–592.10.1002/jcc.v33.5
- Maillard, L. C. (1913). Genèse des Matieres Protéiques et des Matieres Humiques: Action de la Glycérine et des Sugres sur les Acides α- amines[Genesis of protean and humic materials: Action of glycerine and sugars on α-amino acids], C. R de la Société de Biologie , Masson & Cie, Paris.
- Maroudas, A. , Palla, G. , & Gilav, E. (1992). Racemization of aspartic acid in human articular cartilage. Connective Tissue Research , 28 , 161–169.10.3109/03008209209015033
- Møller, C., & Plesset, M. S. (1934). Note on an approximation treatment for many-electron systems. Physical Review , 46 , 618–622.10.1103/PhysRev.46.618
- Nomoto, K. , Yagi, M. , Hamada, U. , Naito, J. , & Yonei, Y. (2013). Identification of advanced glycation endproducts derived fluorescence spectrum in vitro and human skin. Anti-Aging Medicine , 10 , 92–100.
- Ottani, V. , Raspanti, M. , & Ruggeri, A. (2001). Collagen structure and functional implications. Micron , 32 , 251–260.10.1016/S0968-4328(00)00042-1
- Pimentel, G. C., & McClellan, A. L. (1960). The hydrogen bond . San Francisco: Freeman.
- Rich, H. , Odlyha, M. , Cheema, U. , Mudera, V. , & Bozec, L. (2014). Effects of photochemical riboflavin-mediated crosslinks on the physical properties of collagen constructs and fibrils. Journal of Materials Science: Materials in Medicine , 25 , 11–21.10.1007/s10856-013-5038-7
- Sell, D. R. , Biemel, K. M. , Reihl, O. , Lederer, M. O. , Strauch, C. M. , & Monnier, V. M. (2005). Glucosepane is a major protein cross-link of the senescent human extracellular matrix. Relationship with diabetes. Journal of Biological Chemistry , 280 , 12310–12315.
- Silla, J. M. , Cormanich, R. A. , Rittner, R. , & Freitas, M. P. (2014). Does intramolecular hydrogen bond play a key role in the stereochemistry of α- and β-d-glucose? Carbohydrate Research , 396 , 9–13.10.1016/j.carres.2014.06.013
- Steiner, T. (2002). The Hydrogen Bond in the Solid State. Angewandte Chemie International Edition , 41 , 48–76.10.1002/1521-3773(20020104)41:1<>1.0.CO;2-5
- Thorpe, C. T. , Streeter, I. , Pinchbeck, G. L. , Goodship, A. E. , Clegg, P. D. , & Birch, H. L. (2010). Aspartic acid racemization and collagen degradation markers reveal an accumulation of damage in tendon collagen that is enhanced with aging. Journal of Biological Chemistry , 285 , 15674–15681.10.1074/jbc.M109.077503