
Abstract
The catechin family of molecules that are present in the leaves of green tea has been under investigation since the antioxidant and anti-inflammatory properties of tea were discovered. Among multiple proposed therapeutic targets of these molecules, the direct interaction with nucleic acids has been proposed and experimentally observed but without clear knowledge about the potential binding modes between these ligands and DNA. One of these catechin structures, (–)-epigallocatechin gallate (EGCG), has three aromatic rings that could interact with double-stranded DNA via terminal base-pair stacking, intercalation, or through groove binding. Using enhanced sampling techniques and molecular dynamics simulations, we have found a stable complex between the EGCG ligand and DNA through intercalation of the trihydroxybenzoate aromatic ring and an ApC step. Moreover, we have calculated the absorption spectra of four possible binding modes and compared these to absorption profiles reported in the literature, and explored the possible DNA sequence preference for the EGCG ligand to bind. Our results suggest that an intercalative mode of interaction through the major groove is possible between the EGCG ligands and DNA with apparently very little DNA sequence selectivity.
Introduction
Antioxidant molecules aid in the reduction of chronic disease and are commonly found in fruits and vegetables (Weisburger). Molecules that contain three or more hydroxyl groups in aromatic rings, known as polyphenols, have been shown to possess antioxidative properties. A sub-group of polyphenols consists of a series of catechin molecules that include (–)-epigallocatechin gallate (EGCG), (–)-epigallocatechin (EGC), (–)-epicatechin gallate (ECG), and (–)-epicatechin (EC). These compounds are found in the leaves of Camellia sinensis (tea), and as early as the 1980s their anti-inflammatory and antioxidant properties were evident (Chen et al., Citation2011).Anti-cancer properties of these polyphenols, mainly of EGCG (Du et al., Citation2012), were first evidenced by Want and collaborators using in vivo studies in an animal model (Wang et al., Citation1995). This opened a door to perform further testing and therapeutic assessment of catechin-type molecules found in tea extracts. The effectiveness of EGCG as a tumor suppressor has been routinely observed in hundreds of studies (Bansal, Syan, Mathur, & Choudhary, Citation2011; Beltz, Bayer, Moss, & Simet, Citation2006; Chen et al., Citation2011; Kazi et al., Citation2002; Rahmani, Al Shabrmi, Allemailem, Aly, & Khan, Citation2015; Shankar, Ganapathy, & Srivastava, Citation2007). Progress has been made to understand the mechanism of action for EGCG, which can lead to a limited amount of tumor regression that involves multiple sites and multiple cellular pathways, including the inhibition of kinases, angiogenesis inhibition, antioxidant activity, and modulation of transcription factors (Rahmani et al., Citation2015). The interaction with nucleic acids is then a main focus of research to understand the effectiveness and mechanism of action of EGCG. This is supported by mass spectroscopy studies that have shown direct interaction of EGCG with double-stranded DNA and telomeric quadruplex DNA (Mikutis et al., Citation2013).
Despite the many studies and publications, the reality of a positive pharmacological effect of the EGCG molecule is unsure. In vitro studies using cancer cell lines suggest that natural antioxidant supplements may actually aid in promoting the growth of cancerous cells rather than to prevent it (Lu, Ou, & Lu, Citation2013). Regardless, studies show that EGCG inhibits cell growth and aids in promoting apoptosis, and known potent anti-cancer drugs that interact directly with DNA have been observed to enhance their effect when EGCG is included. The compound cisplatin, which is a transition metal drug routinely included in chemotherapy, showed increased activity when EGCG was included (Chan, Soprano, Weinstein, & Fong, Citation2006; Wang et al., Citation2015), and restored the toxicity to previously desensitized lung cancer cells (Zhang, Wang, Han, Zhou, & Sun, Citation2015). Similar synergistic effects were observed with oxaliplatin, a cisplatin derivative (Hu et al., Citation2015). Cooperative effect was observed when the anti-cancer, DNA binding compound bleomycin was used in conjunction with EGCG, showing a potentiated anti-proliferative effect in pancreatic cancer cells (Bimonte et al., Citation2015). Additionally, interaction of EGCG in single-strand DNA chains showed increased nuclease activity in whole blood and isolated lymphocytes cells (Yuquan, Takeshita, & Morimoto, Citation2001). Using surface plasmon resonance assays and cold spray ionization-mass spectrometry, Fujiki and collaborators found that EGCG binds to DNA and RNA polymers between 1:1 and 3:1 stoichiometry (Kuzuhara, Sei, Yamaguchi, Suganuma, & Fujiki, Citation2006). They additionally detected that multiple binding conformations are present in EGCG–DNA and EGCG–RNA complexes. Electrochemical studies of EGCG when in contact with fish testes DNA revealed a direct interaction with DNA via possible intercalation between the DNA base-pairs (Zheng, Chen, Hoshi, Anzai, & Li, Citation2006). Dasgupta and collaborators performed the analysis on the interaction of EGCG in the presence of copper ions and calf-thymus DNA using several spectroscopic techniques (Ghosh, Sahoo, Jana, & Dasgupta, Citation2008). They observed the fluorescence quenching of ethidium bromide bound to DNA with increasing concentrations of EGCG, which implies that EGCG molecules may interact with double-stranded DNA through the insertion of one of its rings between base-pairs.
Spectroscopic assays that are routinely used to study the interaction of small ligands to DNA include UV-visible absorption and fluorescence quenching (Sirajuddin, Ali, & Badshah, Citation2013). In UV-vis experiments, the changes in the position of the maximum value of the absorption provide information on whether DNA-ligand interactions are occurring. The literature suggests that a hypochromic effect with no absorption wavelength shift is related to an intercalative mode of interaction, whereas a hyperchromic effect combined with shifting in the absorption wavelength (either blue or redshift) points to a groove binding type of interaction (Sirajuddin et al., Citation2013). In lack of a solution structure of the actual EGCG + DNA complex, spectroscopic information currently only provides evidence that an actual interaction between ligand and DNA is occurring, but the actual mode of interaction is speculative at best, since multiple experiments found in the literature point to contradictory experimental observations.
In order to study the modes of interaction and stability of EGCG bound to DNA, we performed molecular dynamics (MD) simulations in which each of the aromatic rings of the ligand structure (A, B and D, Figure ) is manually intercalated between an AT base-step. The intercalation process was studied using equilibrium umbrella sampling (US) with the aid of a biasing force to drive the system between the desired states. The use of this enhanced sampling technique has been used before to study the intercalation process of several DNA binders (Galindo-Murillo, Garcia-Ramos, Ruiz-Azuara, Cheatham, & Cortes-Guzman, Citation2015) and is a convenient methodology to study a particular problem that otherwise would require millisecond sampling rate, which is still not reachable without the use of specialized MD-specific hardware, such as the ANTON supercomputer (Shaw et al., Citation2009).
Figure 1. Structure of (–)-epigallocatechin-3-gallate (EGCG). Rings A and C correspond to the benzopyran moiety, ring B to trihydroxyphenyl, and ring D to trihydroxybenzoate.
Methodology
Ligand parameters
The EGCG molecule was manually built using Gaussview (Millman & Keith, Citation2014). The structure was optimized using density functional theory with the m06–2× level (Zhao & Truhlar, Citation2007) and a 6–311++G (2d, 2p) basis set. We have previously used this combination of functional and basis set to optimize the structure of both organic and transition metal compounds with good approximation to experimental structures (Díaz-Gómez, Galindo-Murillo, & Cortés-Guzmán, Citation2017; Galindo-Murillo & Cheatham, Citation2014; Toledano-Magaña et al., Citation2017). Frequency calculations were performed and no negative values were found, ensuring the convergence of the structure. The resulting optimized structure was compared with experimental structures available from PDB entry codes 3OOB (X-ray, (Urusova et al., Citation2011)) and 2KDH (NMR, (Robertson, Li, & Sykes, Citation2009)). The RMSD using the benzopyran ring between the calculated structure and the X-ray structure was of .53 Å and .82 Å from the NMR data. Charge parameters for the optimized structure were generated using the R.E.D. server (Vanquelef et al., Citation2011). The general amber force field, GAFF (Wang, Wolf, Caldwell, Kollman, & Case, Citation2004), was used to describe the structural parameters.
Umbrella sampling
To study a possible intercalation mechanism of interaction, biased sampling was used by means of umbrella sampling to construct a pathway of intercalation between the DNA and rings A, B, and D of the EGCG ligand. This methodology consists of a series of intermediate steps along a pre-defined reaction coordinate that effectively samples an energy pathway with the aid of a biasing potential. Using a 36-mer with the sequence d(GCCTATAAACGCCTATAA), the EGCG molecule was manually intercalated between base-pairs A9-T28 and C10-G27 in three different configurations, mainly, the intercalations of ring A, B, and D as a starting structure. The separation restraint was centered on the heavy atoms of rings A, B, and D of the EGCG molecule and the center of mass of the two base-pairs forming the intercalation site. Multiple restrains were tested with different anchor points until we ensured that the ligand moved toward the major groove, while leaving the EGCG molecule to reorient freely once it was no longer in close proximity of the DNA chain. Because of the branching structure of the EGCG molecule, it is very unlikely that a similar process could take place through the minor groove, on which multiple steric clashes were observed when we tested a reaction coordinate through the minor groove. The sampling was carried out using .2 Å increment windows from 1 to 20 Å with a 10 kcal/mol Å harmonic restraint constant. Each window was sampled for 100 ns, and five independent copies of each window were performed to ensure statistical validity. The biasing potential was removed using the weighted-histogram analysis method, WHAM (Kumar, Rosenberg, Bouzida, Swendsen, & Kollman, Citation1992; Roux, Citation1995).
Simulation details
The 36-mer double strand DNA sequence used in this work has been previously studied (Lankas, Šponer, Langowski, & Cheatham, Citation2004). The DNA was built using the procedures previously described by the Ascona B-DNA Consortium, ABC (Beveridge, Cheatham, & Mezei, Citation2012; Beveridge et al., Citation2004; Dixit et al., Citation2004; Lavery et al., Citation2009; Pasi et al., Citation2014) and with the TIP3P (Jorgensen, Chandrasekhar, Madura, Impey, & Klein, Citation1983) water model using a truncated octahedral periodic cell and a 12 Å distance between the solute and the edge of the box. A total of 34 sodium ions were added to reach a neutral charge, and an excess of 48 sodium and chlorine ions was added to reach a final concentration of ~150 mM using the Joung-Cheatham set of ion parameters (Joung & Cheatham, Citation2008). After the model was built, the position of the ions was randomized by swapping a water molecule with an ion such that no ion was closer than 4 Å to another and the ions were farther than 6 Å away of the DNA–EGCG complex. This process was repeated four times to create four independent initial geometries, and each copy was run through the same minimization, equilibration, and production phase. The initial minimization process was performed using 1000 steps of steepest descent optimization switching to 1000 steps of conjugate gradients, applying a harmonic restraint of 25 kcal mol−1 Å−1 to the DNA and the EGCG molecules. Then, using a Langevin thermostat (Pastor, Brooks, & Szabo, Citation1988)with a collision frequency value of 1 ps−1and the same restraints, the system was slowly heated from 100 to 300 K in 1000 ps of MD simulation. The restraint was decreased from 25 to .5 kcal mol−1 Å−1 in a series of equilibrium steps, each 2 ns long using constant pressure and temperature. The Berendsen barostat (pressure relaxation time set to 1 ps) was used (Berendsen, Postma, van Gunsteren, DiNola, & Haak, Citationn.d.). A 2 fs time step was used for each equilibration step with a cutoff of 9.0 Å. MD simulations were performed using the CUDA version of pmemd available in AMBER 14(Case et al., Citation2014; Cheatham & Case, Citation2013; Götz et al., Citation2012; Le Grand, Götz, & Walker, Citation2013; Salomon-Ferrer et al., Citation2013). The PME method (Konerding, Cheatham, Kollman, & James, Citation1999) was used to calculate the long-range interaction and trajectory information was saved every 10 ps. Clustering analysis was performed using the hierarchical agglomerative algorithm as implemented in CPPTRAJ with a cutoff value of 3 Å between cluster centroids and using only heavy atoms.
EGCG–DNA binding energy analysis was calculated using the molecular mechanics Poisson-Boltzmann surface area (MM-PBSA) methodology, (Hansson, Marelius, & Aqvist, Citation1998; Kollman et al., Citation2000) through the MMPBSA.py (Miller et al., Citation2012) application available in AmberTools16. A total of 5000 snapshots were used for the MM-PBSA analysis in each case.
Absorption spectra were calculated following a QM TD-DFT methodology with the hybrid functional of Adamo, PBE0(Adamo & Barone, Citation1999). This functional was chosen after running a series of tests calculation with other levels of theory (which included M062x, M11, B3PW91, cam-B3LYP, and PBE0) until we could find a good agreement with the reported experimental data. Additionally, this function has been tested thoroughly elsewhere (del Campo, Gázquez, Trickey, & Vela, Citation2012; Cárdenas-Jirón & Cortez, Citation2013). All QM calculations to obtain the UV-vis spectra were made using 6–311 + G (d, p). Implicit solvation was treated using the SMD dielectric method (Marenich, Cramer, & Truhlar, Citation2009). To reduce computation time, only residues 9–12, 25–28 and the EGCG ligand were used. This selected section of the DNA represents the full binding site of the EGCG molecule. Deleted bonds were capped with hydrogen atoms at both ends of the DNA chain. The remaining bases, sugars, and full backbone atoms were taken into account for the QM calculations. Quantum mechanical calculations were run using the G09-D.01 version of the Gaussian program (Frisch et al., Citation2009). Trajectory analysis was run using CPPTRAJ (Roe & Cheatham, Citation2013) as found in AmberTools 16.
Results and discussion
Intercalation pathway studies using umbrella sampling
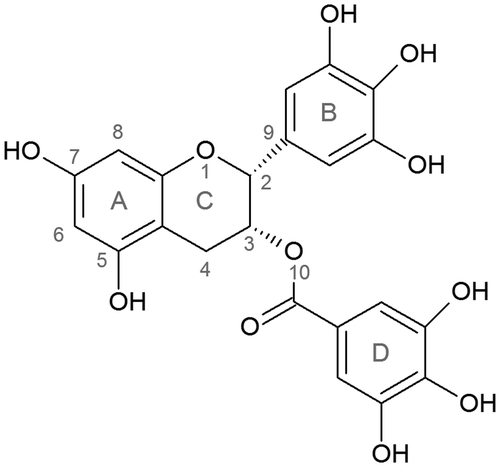
The resulting free energy profile for each system and an average structure obtained from the minimum energy points is presented in Figure . The energy minimum for ring A is reached at 9.8 Å with a free energy value of −2.0 kcal/mol, 10.8 Å for ring B with a free energy value of −1.6 kcal/mol and 11 Å for ring D with a value of −1.2 kcal/mol. The intercalation process of ring A of EGCG decreases steadily as the benzopyran ring is removed from the intercalation site. At 4.8 Å there is a metastable intermediate with an average free energy value of 3.7 kcal/mol, which corresponds to a reorientation of the whole molecule that allows the opposing ring D to interact with the DNA backbone. The energetically stable (energy minima) configuration was found to be a partial stacking interaction for ring A, where the benzopyran moiety is still present between the ApC base-step. For rings B and D, the energy minima are with the corresponding ring outside the stacked bases. Free energy values for the stable interaction in each case show that ring A has preference over the other two conformations (even though with differences of .4 and .6 kcal/mol ,respectively, which is beyond the credibility of any molecular dynamics technique). The de-stacking interaction pathway for ring D also shows a metastable structure when the trihydroxybenzoate ring is at 5.7 Å with a value of −.5 kcal/mol. This structure corresponds to a stacking interaction where ring D is between the aromatic center of A9 and the geometric center between the base-pair of C10 and G27. When in this conformation, both ring A and B are forming hydrogen bonds with the oxygen atoms from the DNA backbone of the 5′-strand. The structural difference between the metastable and stable structures corresponds to ring D of the EGCG ligand shifting outside of the intercalation pocket and the nucleotides resetting to a canonical pairing conformation (Supplementary Figure S1). Visual inspection of the representative structures obtained from a clustering analysis for each case (Figure , bottom), suggested that ring A allowed a reconfiguration of rings B and D, facilitating the formation of non-bonded interactions with the DNA and hence, stabilizing the overall formation of the complex. As an initial test for this hypothesis, we measured the DNA bending angle, using the top, center, and bottom residues as anchor points (residue numbers 2, 35 for top anchor; 8, 9, 10, 28, 29, 30 for center and; 17, 20 for bottom anchor). The angle between the center of mass from these three anchors provides an overall measure of the amount of bending of the DNA. This analysis was applied to the trajectory (~10 000 frames) of the window with the energetically stable value, as mentioned before. The overall bending angle when the ring A is at the 9.8 Å position is 160.6° (±5.8); for ring B in the 10.8 Å position is 162.2° (±6.2), and for ring D in the 11 Å position is 155.3° (±10.6). The angle value for the metastable window for ring D located at 5.7 Å is 166.8 (±6.5).
Figure 2. Top: potential of mean force energy profiles for the intercalation process of rings A, B, and D, respectively. Bottom: most populated structure extracted using clustering analysis from 100 ns of sampling time from window 9.8 Å for ring A, 10.8 Å for ring B, and 11 Å for ring D.
Moreover, using the five independent trajectories corresponding to the lowest energy point in the PMF results, we calculated the number of contacts between the EGCG molecule and the DNA from the 100 ns trajectory information (which is the length of each independent window used for umbrella sampling). The average number of unique contacts using a threshold of 5.0 Å between the EGCG molecule and the DNA chain was 220 for ring A. Rings B and D present a broader population of number of contact between ~75–150 which agrees with our observation of ring A allowing the rest of the EGCG ligand to interact with the DNA.
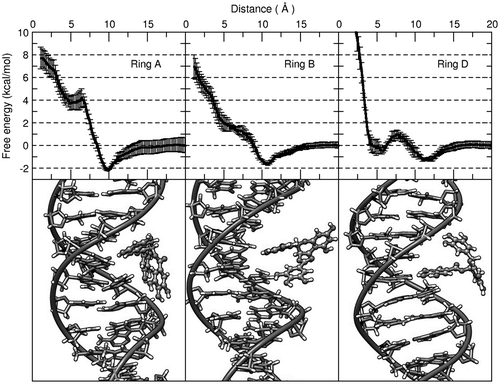
The observation of a metastable partial intercalated state in the case of ring D at ~5 Å raised the question whether this configuration would remain stable in an extended unbiased molecular dynamics simulation. To explore this idea, we started with the same initial intercalated structures we used for the umbrella sampling analysis (each of the EGCG ligands intercalated between an ApCstep) and performed 10 unbiased independent simulations for 1.5 μs using the same equilibration and production protocols as described in the methods section (with randomized ions in each starting structure). To our surprise, the EGCG molecule intercalated with the trihydroxybenzoate moiety (ring D) remained stable in all of the 10 copies, whereas ring A and B detached from the intercalated site less than ~10 ns (Figure ).
Figure 3. Distance values as measured from all the DNA residues to the center of mass of the EGCG ligand over a period of 1.5 µs. The starting structure was with the corresponding ring manually intercalated between base-pairs 9, 28, and 10, 27 (ApC step, the same structures used for the umbrella sampling simulations). Each line corresponds to one of 10 independent, unrestrained simulation.
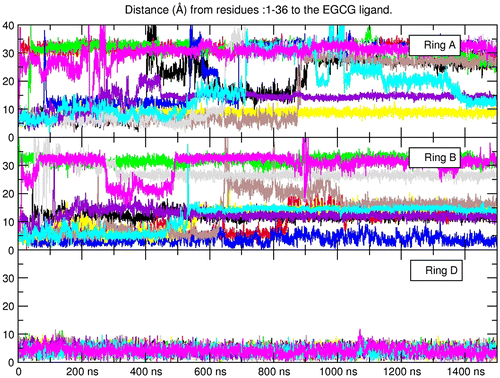
When ring D is in the intercalated position it forms stacking interactions with both A9 and G27, the benzopyran (ring A) positions directly next to the DNA backbone forming hydrogen bonds with the phosphate oxygen atoms from residues 27 and 28, and ring B stabilizes with hydrogen bonds between the hydroxyl oxygen atoms and N6 of adenine 6 (Figure ). The sum of the previously described interactions is possible when ring D is in the intercalated position mainly because the branched structure of the EGCG ligand places both rings A and B in an optimum configuration to interact with the DNA backbone and the base-pairs in the interaction site. When ring A is stacked with the flanking bases, the aromatic ring of the benzopyran forms a stacking interaction with adenine 9 (A9) and guanine 27 (G27) in a similar manner as with the ring D position, although, since the benzopyran is a dual ring structure, rings B and D are facing away from the DNA, which do not allow the formation of stabilizing non-bonded interactions. Because rings B and D are freely rotating aromatic rings, the hindrance of these fluctuations causes ring A to slide away from the intercalation site. In the case of ring B, and in a similar way as ring D, the aromatic ring forms stacking interactions with A9 and G27. The problem in this case is that the aromatic ring B is most stabilized the closer it can get in contacts with the aromatic rings of both adenine and guanine present in the intercalation pocket. This means that the ligand is trying to push itself deeper in the intercalation pocket, but the branching structure of the ligand forms steric clashes that do not allow a proper π–π overlap interaction to occur between adenine – trihydroxyphenyl – guanine. Eventually, the fluctuations of the system overcome the weak formation of the complex and the ligand detaches from the intercalation site. This does not happen with ring D in the intercalated mode since the ketone group present in the benzoate structure allows the aromatic ring to be inserted deeper in the pocket and form stable stacking interactions before steric clashes occur by rings A and B.
Figure 4. View from 5′ → 3′ (left) and 3′ → 5′ (right) of the representative structure from the top populated cluster extracted using the 10 independent simulations of the EGCG ligand with the ring D in the intercalation position.
The binding free energy for the three complexes from the umbrella sampling simulations and the 10 independent unbiased MD simulations of ring D (Figure ) is presented in Table . For each of the umbrella sampling simulations, the binding energy was calculated using a representative structure obtained from the lowest energy window by means of a clustering algorithm as described in the methodology section. Similarly, the binding energy of the unbiased simulations was calculated from a representative structure extracted from the clustering analysis using the 10 independent set of trajectories. The binding energy obtained from the unbiased simulation present the lowest value with a difference of ~20 kcal/mol from the structures obtained from the umbrella sampling studies.
Table 1. Binding energy values between DNA in the four proposed binding configurations and the maximum absorption wavelength as reported in the literature for EGCG and calculated using TD-DFT. The binding energy for the unbiased ‘Complex from MD – ring D’ is the average of the 10 independent simulations. Numbers in parenthesis represent standard deviation.
The energy values obtained from the umbrella sampling simulations are within ~4 kcal/mol within each other, which is expected since their binding modes are similar: In each case, the EGCG molecule is interacting within the major groove of the DNA with both the sugar moiety and the phosphate groups, and little interaction with the actual DNA bases is present. No real binding difference could be proposed based on the umbrella sampling results. Contrary to this, the EGCG–DNA binding mode of the unbiased simulations with the ring D in intercalation presents a binding energy value which is more ~15 kcal/mol stronger than the binding poses observed from the umbrella sampling simulations and is within the range of the computed free binding energy of similar sized molecules interacting within the grooves of DNA (Díaz-Gómez et al., Citation2017; Galindo-Murillo & Cheatham, Citation2014; Galindo-Murillo et al., Citation2015; García-Ramos, Galindo-Murillo, Cortés-Guzmán, & Ruiz-Azuara, Citation2013). It is important to mention that the presented energy values are over-estimates not only due to the PBSA which tends to over-estimate binding free energies, but also the loss of translational and rotational entropy (~12 kcal/mol) were not taken into account in the free energy calculations (Homeyer & Gohlke, Citation2012; Štefl et al., Citation2003).
Spectroscopic calculations
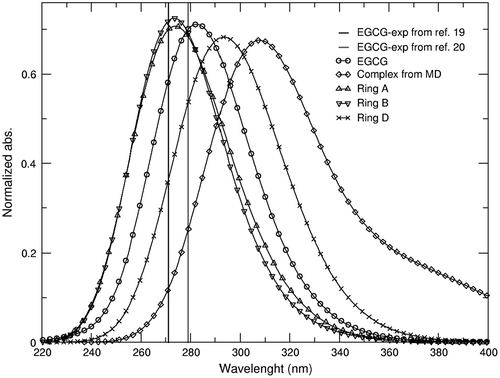
To date, no published X-ray or NMR structure of the interaction of any type of catechin molecule with nucleic acids is available, although there is strong experimental evidence that these molecules do bind with DNA and RNA (Kuzuhara et al., Citation2006). Dasgupta and collaborators reported the experimental absorption profile for EGCG with increasing concentrations of calf-thymus DNA (Supplementary Figure 4 of reference (Ghosh et al., Citation2008)). This experimental titration of the EGCG molecule shows the hypochromic effect as DNA concentration increases, and bathochromic shift of the λmax by ~10 nm. The absorption spectra were computed at the QM TD-DFT level for each of the three different conformation studied using the umbrella sampling methodology. The structures used in the QM calculations are the ones shown in Figure (bottom), which are extracted after a clustering analysis from the lowest energy window. Additionally, we also calculated the spectra for the most populated structure obtained in the unbiased MD simulation (with ring D in intercalation mode). Comparison between the experimental and calculated UV-vis absorption information is presented in Figure . The calculated absorption’s maxima obtained for the EGCG molecule is at 279 nm, which is off by 5 nm from the reported experimental values at ~272–275 nm (in water). Ring A and ring B structures both show a λmax = 274 (Table ) with a reduction in the absorbance of ~.005. In contrast, ring D (from US) and the intercalated complex showed hypochromic effect of ~.04 and redshift of 19 and 34 nm, respectively. The shifts of λmax and absorption from the complex structure obtained from the MD trajectories are observed in the experimental EGCG–DNA titration experiments, suggesting that the experimental binding mode is with the trihydroxybenzoate intercalating between the DNA base-pairs (Figure ).
Figure 5. Normalized absorbance spectra for the EGCG molecule (circle). DNA–EGCG complex structure (diamonds) using the representative structure from a clustering analysis from the simulation where ring D is in the intercalated position within the DNA (Figure ) and the structure from the PMF, ring D (cross ticks). Spectra for the PMF’s from ring A (up triangle tick) and ring B (down triangle tick) are extracted from a clustering analysis using the lowest energy window. Absorption spectra were calculated using the PBE0 functional with the SMD implicit solvation model in water. Experimental values of the EGCG molecule are shown as lines from references: (19, 20).
Sequence dependence analysis
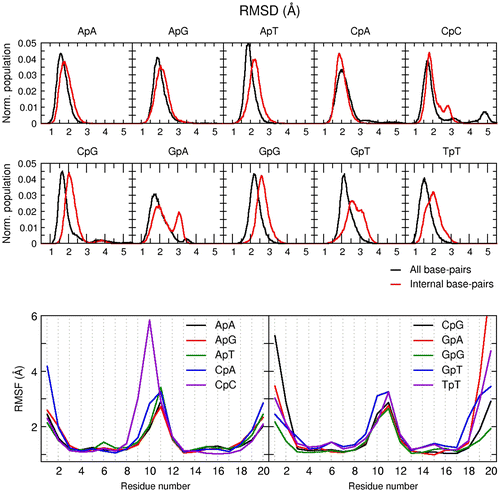
The stable intercalation configuration from the trihydroxybenzoate (ring D) moiety of the EGCG molecule at the ApC base-step opened a new set of ideas regarding possible DNA sequence selectivity. To test this, we used the most populated structure of our previous simulation presented in Figure as starting structure and manually switched the top and bottom base-pairs of the intercalation site with every possible combination of base-steps. The performed simulations are shown in Table .The resulting structures were minimized following the same procedure as described in the methods section. Three independent simulations for each base-step were sampled for 5 μs. The DNA–EGCG major groove intercalation complex was stable for the entire sampling time-tested, regardless of the sequence and overall, structural deformation of the DNA was only detected to a significant amount as fraying events on both ends of the DNA which are routinely observed for double-stranded DNA simulations (Galindo-Murillo, Roe, & Cheatham, Citation2014a; Galindo-Murillo, Roe, & Cheatham, Citation2014b; Zgarbová, Otyepka, Šponer, Lankaš, & Jurečka, Citation2014). A normalized histogram of the RMSD for the three aggregated trajectories of each system is shown in Figure , top. The simulations with the base-steps ApA, ApG, ApT, CpA, and GpG show a stable intercalation complex populated between ~2–3 Å. The additional structures that are being populated for CpC, GpA, GpT between ~2.5–4 Å and to a lesser extent with TpT and CpG are due to long-lived frayed event deformations present at both ends of the terminal base-pairs. This can be confirmed by means of a root mean square fluctuations (RMSF) analysis on each residue (Figure bottom). In the figure, we notice that residues 5, 6, 15, and 16 show fluctuations between ~.1–.5 Å for all the tested systems, whereas residues present at both ends of the DNA chain show fluctuations in the ~4–8 Å range. This observation also indicates a stable complex is present regardless the sequence, and the different populations present in the RMSD analysis are not involved in the DNA–EGCG interaction.
Table 2. Simulations using different base-steps in the intercalation site. The original sequence corresponds to d(GCCTATAA-X-I-Y-GCCTATAA), where the X and Y positions are the swapped base-pairs and I marks the intercalation site. The sequence column describes the switched base-step.
Figure 6. Top: normalized histogram of the RMSD values using the first frame as reference over the three aggregated independent copies for each system. Black line includes all residues; red line is RMSD without the terminal base-pairs at each end of the DNA. Bottom: RMSF by residue of each system using the full aggregated trajectory.
Energy analysis between the EGCG and the DNA using the MM-PBSA protocol and different sequences does not provide a clear trend (Table ). The results all indicate an energetically favorable complex as observed in the RMSD and RMSF data. Among the data-set, the binding energy values of stable structure as suggested by the binding values are observed with the GpG, CpG, and ApG present the lowest energy value and TpT, GpA, and CpA the highest values, although, the average difference between these six simulations is ~2.8 kcal/mol. Hydrogen bonding analysis using the 10 simulations showed that interactions with the backbone provide the main stabilization point for the complex. The interaction between the hydroxyl group of ring A (position 5, Figure ) with the phosphate group of residue 15 is present in all simulations (Supplementary Table S2). Additionally, the hydroxyl groups of ring B also form hydrogen bonding with the phosphate group on the opposite side located in residue 5. The most populated structure for each individual system using the full aggregated trajectory shows the EGCG ligand in the intercalation pocket in each case (Supplementary information S2).
Visual inspection of the EGCG molecule in the intercalation site indicates that the ligand does not show significant conformational changes throughout the simulations regardless of the sequence. This observation was further tested by measuring the flexible dihedral angles between the trihydroxyphenyl, trihydroxybenzoate, and the benzopyran rings (the bonds between ring B to C and D to C, Supplementary Figure S3). The dihedral angle between the trihydroxyphenyl moiety and the benzopyran structure is populated in a balanced range between 101° and 270° for the entire aggregated simulations that corresponds to the expected free rotation of the aromatic ring. For the trihydroxybenzoate ring, two dihedrals were monitored located in the methyl ester bond connecting the aromatic trihydroxyphenyl ring with the benzopyran (Supplementary Figure S3). Both dihedral bonds populate values close to ~167° and ~187°, respectively, which correspond to the conformation previously observed (Figure ) and no major shifts are observed confirming a lack of conformational shifts of the EGCG ligand.
Conclusions
The branched structure of the EGCG molecule limits the way it can interact with double-stranded DNA. Numerous steric clashes make interaction of the ligand through the minor groove of the DNA energetically unfavorable hence we hypothesized that the interaction would take place through the major groove. We have then proposed three possible binding configurations based on the number of aromatic rings present in the ligand. Using enhanced sampling molecular dynamics, we explored the possibility of an intercalation interaction between each of the aromatic sites of the EGCG ligand between an ApC DNA step. The results showed energetically stable configurations outside the intercalation pockets in all the cases, however, the EGCG–DNA interaction using the trihydroxybenzoate (ring D) showed an additional stable configuration at a distance of ~5 Å between the center of mass of both EGCG ligand and DNA axis. Upon visual examination of this structure, we confirmed that the ligand was in full intercalation mode between the DNA base-pairs. Unbiased, unrestrained simulations of the DNA-ligand complex starting from the manual intercalation of each of the EGCG aromatic rings yielded a stable complex structure of the trihydroxybenzoate moiety with DNA after 10 independent trajectories (each > 1.5 μs in length), whereas intercalation structures using the trihydroxyphenyl or the benzopyran moiety were unstable and the intercalation interaction was lost in ~50–100 ns of simulation time in all of the 10 independent runs.
It is important to mention that the stable intercalation binding mode observed between the trihydroxybenzoate and the DNA could potentially be biased by the known over-stacking effect present in the parm99-bsc0 AMBER force field as reported by Garcia and collaborators (Chen & Garcia, Citation2013). A follow-up and more recent work performed by Elcock and collaborators studying the stacking energies of dinucleotide monophosphates mention the possibility of a more favorable for RNA than DNA, although they conclude that this observation is dependent of the presence of T/U bases and the experimental databases on which comparison is made. We hypothesize that even though the over estimation of stacking interactions is in fact, present, and potentially facilitating the intercalation of the EGCG with DNA, this biasing was not observed on two of the three binding poses we explored. In fact, when manually intercalating rings A and B of the EGCG molecule, the intercalation pose was lost in a ~200 ns time scale, even though, 10 independent simulations were run for each of the systems. Further research in this particular stacking subject will be addressed in future work, noting that in more recent work on dinucleotides by our lab with the various AMBER force fields (work under review), the over-stacking observed by Elcock is less profound than previously reported.
The QM calculation of absorption spectra of the stable intercalation complex showed a hypochromic effect with a redshift of ~10 nm, which is consistent with what is observed in experiments of the EGCG molecule in the presence of calf-thymus absorption experiments. Possible DNA sequence preference by the ligand was studied, although no clear trend was observed. Our observations have generated atomistic details about the major groove intercalation process of a known antioxidant molecule, moving forward the studies of how DNA interacts with molecules.
Disclosure statement
No potential conflict of interest was reported by the authors.
Funding
This work was supported by the Blue Waters sustained-petascale computing project of National Science Foundation [grant number NSF OCI 07-25070], [grant number PRAC OCI-1036208]; the NSF Extreme Science and Engineering Discovery Environment [grant number XSEDE, OCI-1053575], allocation [grant number MCA01S027P]; the Center for High Performance Computing at the University of Utah.
Supplementary material
The supplementary material for this paper is available online at https://doi.org/10.1080/07391102.2017.1389306.
Inter atomic distances. For atom descriptions, refer to Figure 1 in the main manuscript
Download MS Word (1.8 MB)Acknowledgments
We would like to thank James Robertson for his comments about the manuscript.
References
- Adamo, C., & Barone, V. (1999). Toward reliable density functional methods without adjustable parameters: The PBE0 model. The Journal of Chemical Physics, 110(13), 6158. doi:10.1063/1.478522
- Bansal, S., Syan, N., Mathur, P., & Choudhary, S. (2011). Pharmacological profile of green tea and its polyphenols: A review. Medicinal Chemistry Research, 21(11), 3347–3360. doi:10.1007/s00044-011-9800-4
- Beltz, L. A., Bayer, D. K., Moss, A. L., & Simet, I. M. (2006). Mechanisms of cancer prevention by green and black tea polyphenols. Anti-Cancer Agents in Medicinal Chemistry, 6(5), 389–406. Retrieved from http://www.ncbi.nlm.nih.gov/pubmed/1701785010.2174/187152006778226468
- Berendsen, H. J. C., Postma, J. P. M., van Gunsteren, W. F., DiNola, A., & Haak, J. R. (n.d.). Molecular dynamics with coupling to an external bath. Journal of Chemical Physics, 81(8), 3684. doi:10.1063/1.448118
- Beveridge, D. L., Barreiro, G., Byun, K. S., Case, D. A., Cheatham, T. E., 3rd, Dixit, S., … Young, M. A. (2004). Molecular dynamics simulations of the 136 unique tetranucleotide sequences of DNA oligonucleotides. I. Research design and results on d(CpG) steps. Biophysical Journal, 87(6), 3799–3813. 10.1529/biophysj.104.045252
- Beveridge, D. L., Cheatham, T. E., 3rd, & Mezei, M. (2012). The ABCs of molecular dynamics simulations on B-DNA, circa 2012. Journal of Biosciences, 37(3), 379–397. doi:10.1007/s12038-012-9222-6
- Bimonte, S., Leongito, M., Barbieri, A., Del Vecchio, V., Barbieri, M., Albino, V., … Izzo, F. (2015). Inhibitory effect of (–)-epigallocatechin-3-gallate and bleomycin on human pancreatic cancer MiaPaca-2 cell growth. Infectious Agents and Cancer, 10, 22. doi:10.1186/s13027-015-0016-y
- Cárdenas-Jirón, G. I., & Cortez, L. (2013). Assessment of the photosensitization properties of cationic porphyrins in interaction with DNA nucleotide pairs. Journal of Molecular Modeling, 19(7), 2913–2924. doi:10.1007/s00894-013-1822-z
- Case, D. A., Darden, T. A., Cheatham, T. E., 3rd, Simmerling, C. L., Wang, J., Duke, R. E., … Kollman, P. A. (2014). AMBER14. San Francisco: University of California.
- Chan, M. M., Soprano, K. J., Weinstein, K., & Fong, D. (2006). Epigallocatechin-3-gallate delivers hydrogen peroxide to induce death of ovarian cancer cells and enhances their cisplatin susceptibility. Journal of Cellular Physiology, 207(2), 389–396. doi:10.1002/jcp.20569
- Cheatham, T. E., 3rd, & Case, D. A. (2013). Twenty-five years of nucleic acid simulations. Biopolymers, 12, 969–977. doi:10.1002/bip.22331
- Chen, A., & Garcia, A. (2013). High-resolution reversible folding of hyperstable RNA tetraloops using molecular dynamics simulations. Proceedings of the National Academy of Sciences of the United States of America, 110(42), 16820–16825. doi:10.1073/pnas.1309392110
- Chen, D., Wan, S. B., Yang, H., Yuan, J., Chan, T. H., & Dou, Q. P. (2011). EGCG, green tea polyphenols and their synthetic analogs and prodrugs for human cancer prevention and treatment. Advances in Clinical Chemistry, 53, 155–177. Retrieved from http://www.pubmedcentral.nih.gov/articlerender.fcgi?artid=3304302&tool=pmcentrez&rendertype=abstract10.1016/B978-0-12-385855-9.00007-2
- del Campo, J. M., Gázquez, J. L., Trickey, S. B., & Vela, A. (2012). Non-empirical improvement of PBE and its hybrid PBE0 for general description of molecular properties. The Journal of Chemical Physics, 136(10), 104108. doi:10.1063/1.3691197
- Díaz-Gómez, D. G., Galindo-Murillo, R., & Cortés-Guzmán, F. (2017). The role of the DNA backbone in minor-groove ligand binding. ChemPhysChem, 18(14), 1909–1915. doi:10.1002/cphc.201700260
- Dixit, S. B., Beveridge, D. L., Case, D. A., Cheatham, T. E., 3rd, Giudice, E., Lankas, F., … Varnai, P. (2004). Molecular dynamics simulations of the 136 unique tetranucleotide sequences of DNA oligonucleotides. II: Sequence context effects on the dynamical structures of the 10 unique dinucleotide steps. Biophysical Journal, 89(6), 3721–3740. 10.1529/biophysj.105.067397
- Du, G.-J., Zhang, Z., Wen, X.-D., Yu, C., Calway, T., Yuan, C.-S., & Wang, C.-Z. (2012). Epigallocatechin gallate (EGCG) is the most effective cancer chemopreventive polyphenol in green tea. Nutrients, 4(11), 1679–1691. doi:10.3390/nu4111679
- Frisch, M. J., Trucks, G. W., Schlegel, H. B., Scuseria, G. E., Robb, M. A., Cheeseman, J. R., … Pople, J. A. (2009). Gaussian 09, Revision D.01. Wallingford, CT: Gaussian Inc.
- Galindo-Murillo, R., & Cheatham, T. E., 3rd (2014). DNA binding dynamics and energetics of cobalt, nickel, and copper metallopeptides. ChemMedChem, 9(6), 1252–1259. doi:10.1002/cmdc.201402020
- Galindo-Murillo, R., Garcia-Ramos, J. C., Ruiz-Azuara, L., Cheatham, T. E., 3rd, & Cortes-Guzman, F. (2015). Intercalation processes of copper complexes in DNA. Nucleic Acids Research, 43(11), 5364–5376. doi:10.1093/nar/gkv467
- Galindo-Murillo, R., Roe, D. R., & Cheatham, T. E., 3rd (2014). Convergence and reproducibility in molecular dynamics simulations of the DNA duplex d(GCACGAACGAACGAACGC). Biochimica et Biophysica Acta, 1850(5), 1041–1058. doi:10.1016/j.bbagen.2014.09.007
- Galindo-Murillo, R., Roe, D. R., & Cheatham, T. E., 3rd (2014). On the absence of intra-helical DNA dynamics on the μs to ms timescale. Nature Communications, 5, 5152. doi:10.1038/ncomms6152
- García-Ramos, J. C., Galindo-Murillo, R., Cortés-Guzmán, F., & Ruiz-Azuara, L. (2013). Metal-based drug–DNA interactions. Journal of the Mexican Chemical Society, 57(3), 245–259. Retrieved from http://www.jmcs.org.mx/PDFS/V57/3/12.-GarciaRamos.pdf
- Ghosh, K. S., Sahoo, B. K., Jana, D., & Dasgupta, S. (2008). Studies on the interaction of copper complexes of (–)-epicatechin gallate and (–)-epigallocatechin gallate with calf thymus DNA. Journal of Inorganic Biochemistry, 102(9), 1711–1718. doi:10.1016/j.jinorgbio.2008.04.008
- Götz, A. W., Williamson, M. J., Xu, D., Poole, D., Le Grand, S., & Walker, R. C. (2012). Routine microsecond molecular dynamics simulations with AMBER on GPUs. 1. Generalized born. Journal of Chemical Theory and Computation, 8(5), 1542–1555. doi:10.1021/ct200909j
- Hansson, T., Marelius, J., & Aqvist, J. (1998). Ligand binding affinity prediction by linear interaction energy methods. Journal of Computer-Aided Molecular Design, 12(1), 27–35. Retrieved from http://www.ncbi.nlm.nih.gov/pubmed/957008710.1023/A:1007930623000
- Homeyer, N., & Gohlke, H. (2012). Free energy calculations by the molecular mechanics Poisson−Boltzmann surface area method. Molecular Informatics, 31(2), 114–122. doi:10.1002/minf.201100135
- Hu, F., Wei, F., Wang, Y., Wu, B., Fang, Y., & Xiong, B. (2015). EGCG synergizes the therapeutic effect of cisplatin and oxaliplatin through autophagic pathway in human colorectal cancer cells. Journal of Pharmacological Sciences, 128(1), 27–34. doi:10.1016/j.jphs.2015.04.003
- Jorgensen, W. L., Chandrasekhar, J., Madura, J. D., Impey, R. W., & Klein, M. L. (1983). Comparison of simple potential functions for simulating liquid water. Journal of Chemical Physics, 79(2), 926. doi:10.1063/1.445869
- Joung, I. S., & Cheatham, T. E., 3rd (2008). Molecular dynamics simulations of the dynamic and energetic properties of alkali and halide ions using water-model-specific ion parameters. The Journal of Physical Chemistry B, 113(40), 13279–13290. doi:10.1021/jp902584c
- Kazi, A., Smith, D. M., Daniel, K., Zhong, S., Gupta, P., Bosley, M. E., & Dou, Q. P. (2002). Potential molecular targets of tea polyphenols in human tumor cells: Significance in cancer prevention. Vivo, 16(6), 397–403. Retrieved from http://www.ncbi.nlm.nih.gov/pubmed/12494882
- Kollman, P. A., Massova, I., Reyes, C. M., Kuhn, B., Huo, S., Chong, L., … Cheatham 3rd, T. E. (2000). Calculating structures and free energies of complex molecules: Combining molecular mechanics and continuum models. Accounts of Chemical Research, 33(12), 889–897. doi:10.1021/ar000033j
- Konerding, D. E., Cheatham, T. E., 3rd, Kollman, P. A., & James, T. L. (1999). Restrained molecular dynamics of solvated duplex DNA using the particle mesh Ewald method. Journal of Biomolecular NMR, 13(2), 119–131. Retrieved from http://www.ncbi.nlm.nih.gov/pubmed/1007075310.1023/A:1008353423074
- Kumar, S., Rosenberg, J. M., Bouzida, D., Swendsen, R. H., & Kollman, P. A. (1992). THE weighted histogram analysis method for free-energy calculations on biomolecules. I. The method. Journal of Computational Chemistry, 13(8), 1011–1021. doi:10.1002/jcc.540130812
- Kuzuhara, T., Sei, Y., Yamaguchi, K., Suganuma, M., & Fujiki, H. (2006). DNA and RNA as new binding targets of green tea catechins. The Journal of Biological Chemistry, 281(25), 17446–17456. doi:10.1074/jbc.M601196200
- Lankas, F., Šponer, J., Langowski, J., & Cheatham, T. E., 3rd (2004). DNA deformability at the base pair level. Journal of the American Chemical Society, 126(13), 4124–4125. doi:10.1021/ja0390449
- Lavery, R., Zakrzewska, K., Beveridge, D. L., Bishop, T. C., Case, D. A., Cheatham, T. E., 3rd, … Sponer, J. (2009). A systematic molecular dynamics study of nearest-neighbor effects on base pair and base pair step conformations and fluctuations in B-DNA. Nucleic Acids Research, 38(1), 299–313. doi:10.1093/nar/gkp834
- Le Grand, S., Götz, A. W., & Walker, R. C. (2013). SPFP: Speed without compromise – A mixed precision model for GPU accelerated molecular dynamics simulations. Computer Physics Communications, 184(2), 374–380. doi:10.1016/j.cpc.2012.09.022
- Lu, L. Y., Ou, N., & Lu, Q.-B. (2013). Antioxidant induces dna damage, cell death and mutagenicity in human lung and skin normal cells. Scientific Reports, 3, 3169. doi:10.1038/srep03169
- Marenich, A. V., Cramer, C. J., & Truhlar, D. G. (2009). Universal solvation model based on solute electron density and on a continuum model of the solvent defined by the bulk dielectric constant and atomic surface tensions. The Journal of Physical Chemistry B, 113(18), 6378–6396. doi:10.1021/jp810292n
- Mikutis, G., Karaköse, H., Jaiswal, R., LeGresley, A., Islam, T., Fernandez-Lahore, M., & Kuhnert, N. (2013). Phenolic promiscuity in the cell nucleus–epigallocatechingallate (EGCG) and theaflavin-3,3′-digallate from green and black tea bind to model cell nuclear structures including histone proteins, double stranded DNA and telomeric quadruplex DNA. Food & Function, 4(2), 328–337. doi:10.1039/c2fo30159 h
- Miller, B. R., McGee, T. D., Swails, J. M., Homeyer, N., Gohlke, H., & Roitberg, A. E. (2012). MMPBSA.py : An efficient program for end-state free energy calculations. Journal of Chemical Theory and Computation, 8(9), 3314–3321. doi:10.1021/ct300418 h
- Millman, J., & Keith, T. A. (2014). GAUSSVIEW. Shawnee mission. Shawnee, KS: Semichem.
- Pasi, M., Maddocks, J. H., Beveridge, D. L., Bishop, T. C., Case, D. A., Cheatham, T. E., 3rd, … Lavery, R. (2014). μABC: A systematic microsecond molecular dynamics study of tetranucleotide sequence effects in B-DNA. Nucleic Acids Research, 42(19), 12272–12283. doi:10.1093/nar/gku855
- Pastor, R. W., Brooks, B. R., & Szabo, A. (1988). An analysis of the accuracy of Langevin and molecular dynamics algorithms. Molecular Physics, 65(6), 1409–1419. doi:10.1080/00268978800101881
- Rahmani, A. H., Al Shabrmi, F. M., Allemailem, K. S., Aly, S. M., & Khan, M. A. (2015). Implications of green tea and its constituents in the prevention of cancer via the modulation of cell signalling pathway. BioMed Research International, 2015, 925640. doi:10.1155/2015/925640
- Robertson, I. M., Li, M. X., & Sykes, B. D. (2009). solution structure of human cardiac troponin c in complex with the green tea polyphenol, (−)-epigallocatechin 3-gallate. The Journal of Biological Chemistry, 284(34), 23012–23023. doi:10.1074/jbc.M109.021352
- Roe, D. R., & Cheatham, T. E., 3rd (2013). PTRAJ and CPPTRAJ: Software for processing and analysis of molecular dynamics trajectory data. Journal of Chemical Theory and Computation, 9(7), 3084–3095. doi:10.1021/ct400341p
- Roux, B. (1995). The calculation of the potential of mean force using computer simulations. Computer Physics Communications, 91(1–3), 275–282. doi:10.1016/0010-4655(95)00053-I
- Salomon-Ferrer, R., Götz, A. W., Poole, D., Grand, S. Le, Walker, R. C., Le Grand, S., & Walker, R. C. (2013). Routine microsecond molecular dynamics simulations with AMBER on GPUs. 2. Explicit solvent particle mesh Ewald. Journal of Chemical Theory and Computation, 9(9), 3878–3888. doi:10.1021/ct400314y
- Shankar, S., Ganapathy, S., & Srivastava, R. K. (2007). Green tea polyphenols: Biology and therapeutic implications in cancer. Frontiers in Bioscience: A Journal and Virtual Library, 12, 4881–4899. Retrieved from http://www.ncbi.nlm.nih.gov/pubmed/1756961710.2741/2435
- Shaw, D. E., Bowers, K. J., Chow, E., Eastwood, M. P., Ierardi, D. J., Klepeis, J. L., … Batson, B. (2009). Millisecond-scale molecular dynamics simulations on Anton. In Proceedings of the Conference on High Performance Computing Networking, Storage and Analysis – SC 09 (p. 1). New York, NY: ACM Press. 10.1145/1654059.1654099
- Sirajuddin, M., Ali, S., & Badshah, A. (2013). Drug–DNA interactions and their study by UV–Visible, fluorescence spectroscopies and cyclic voltametry. Journal of Photochemistry and Photobiology B: Biology, 124, 1–19. doi:10.1016/j.jphotobiol.2013.03.013
- Štefl, R., Cheatham, T. E., 3rd, Špačková, N., Fadrná, E., Berger, I., Koča, J., & Šponer, J. (2003). Formation pathways of a guanine-quadruplex DNA revealed by molecular dynamics and thermodynamic analysis of the substates. Biophysical Journal, 85(3), 1787–1804. doi:10.1016/S0006-3495(03)74608-6
- Toledano-Magaña, Y., García-Ramos, J. C., Torres-Gutiérrez, C., Vázquez-Gasser, C., Esquivel-Sánchez, J. M., Flores-Alamo, M., … Ruiz-Azuara, L. (2017). Water-soluble ruthenium (II) chiral heteroleptic complexes with amoebicidal in vitro and in vivo activity. Journal of Medicinal Chemistry, 60(3), 899–912. doi:10.1021/acs.jmedchem.6b00795
- Urusova, D. V, Shim, J.-H., Kim, D. J., Jung, S. K., Zykova, T. A., Carper, A., … Dong, Z. (2011). Epigallocatechin-gallate suppresses tumorigenesis by directly targeting Pin1. Cancer Prevention Research (Philadelphia, Pa.), 4(9), 1366–77. 10.1158/1940-6207.CAPR-11-0301
- Vanquelef, E., Simon, S., Marquant, G., Garcia, E., Klimerak, G., Delepine, J. C., … Dupradeau, F. (2011). R.E.D. server: A web service for deriving RESP and ESP charges and building force field libraries for new molecules and molecular fragments. Nucleic Acids Research, 39(Web Server issue), W511–W517. 10.1093/nar/gkr288
- Wang, J., Wolf, R. M., Caldwell, J. W., Kollman, P. A., & Case, D. A. (2004). Development and testing of a general amber force field. Journal of Computational Chemistry, 25(9), 1157–1174. doi:10.1002/jcc.20035
- Wang, X., Jiang, P., Wang, P., Yang, C. S., Wang, X., & Feng, Q. (2015). EGCG enhances cisplatin sensitivity by regulating expression of the copper and cisplatin influx transporter CTR1 in ovary cancer. PLOS ONE, 10(4), e0125402. doi:10.1371/journal.pone.0125402
- Wang, Z. Y., Wang, L. D., Lee, M. J., Ho, C. T., Huang, M. T., Conney, A. H., & Yang, C. S. (1995). Inhibition of N-nitrosomethylbenzylamine-induced esophageal tumorigenesis in rats by green and black tea. Carcinogenesis, 16(9), 2143–2148. Retrieved from http://www.ncbi.nlm.nih.gov/pubmed/755406710.1093/carcin/16.9.2143
- Weisburger, J. H. (1999, September-October). Mechanisms of action of antioxidants as exemplified in vegetables, tomatoes and tea. Food and Chemical Toxicology, 37(9–10), 943–948. doi:10.1016/S0278-6915(99)00086-1
- Yuquan, L., Takeshita, T., & Morimoto, K. (2001). Effects of (–)-epigallocatechin gallate (EGCG) on DNA strand breaks as evaluated by single-cell gel electrophoresis (SCG) in human lymphocytes. Environmental Health and Preventive Medicine, 5(4), 150–154. doi:10.1007/BF02918291
- Zgarbová, M., Otyepka, M., Šponer, J., Lankaš, F., & Jurečka, P. (2014). Base pair fraying in molecular dynamics simulations of DNA and RNA. Journal of Chemical Theory and Computation, 10(8), 3177–3189. doi:10.1021/ct500120v
- Zhang, Y., Wang, X., Han, L., Zhou, Y., & Sun, S. (2015). Green tea polyphenol EGCG reverse cisplatin resistance of A549/DDP cell line through candidate genes demethylation. Biomedicine & Pharmacotherapy = Biomédecine & Pharmacothérapie, 69, 285–290. 10.1016/j.biopha.2014.12.016
- Zhao, Y., & Truhlar, D. G. (2007). The M06 suite of density functionals for main group thermochemistry, thermochemical kinetics, noncovalent interactions, excited states, and transition elements: Two new functionals and systematic testing of four M06-class functionals and 12 other function. Theoretical Chemistry Accounts, 120(1–3), 215–241. doi:10.1007/s00214-007-0310-x
- Zheng, X., Chen, A., Hoshi, T., Anzai, J., & Li, G. (2006). Electrochemical studies of (–)-epigallocatechin gallate and its interaction with DNA. Analytical and Bioanalytical Chemistry, 386(6), 1913–1919. doi:10.1007/s00216-006-0752-3