

Abstract
Apoptosis is a fundamental biological phenomenon, in which anti- or proapoptotic proteins of the Bcl-2 family regulate a committed step. Overexpression of Bcl-2, the prototypical antiapoptotic protein in this family, is associated with therapy resistance in various human cancers. Accordingly, Bcl-2 inhibitors intended for cancer therapy have been developed, typically against the BH3 domain. Recent experimental evidences have shown that the antiapoptotic function of Bcl-2 is not immutable, and that BDA-366, a novel antagonist of the BH4 domain, converts Bcl-2 from a survival molecule to an inducer of cell death. In this study, the underlying mechanisms of this functional conversion were investigated by accelerated molecular dynamics simulation. Results revealed that Pro127 and Trp30 in the BH4 domain rotate to stabilize BDA-366 via π-π interactions, and trigger a series of significant conformational changes of the α3 helix. This rearrangement blocks the hydrophobic binding site (HBS) in the BH3 domain and further prevents binding of BH3-only proteins, which consequently allows the BH3-only proteins to activate the proapoptotic proteins. Analysis of binding free energy confirmed that BDA-366 cross-inhibits BH3-only proteins, implying negative cooperative effects across separate binding sites. The newly identified blocked conformation of the HBS along with the open to closed transition pathway revealed by this study advances the understanding of the Bcl-2 transition from antiapoptotic to proapoptotic function, and yielded new structural insights for novel drug design against the BH4 domain.
Communicated by Ramaswamy H. Sarma
Graphical Abstract
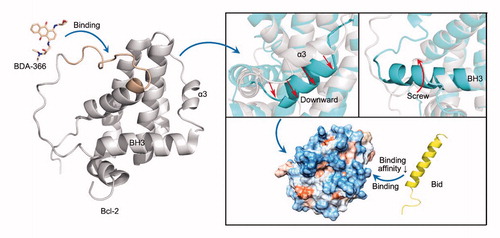
The ability of the small molecule BDA-366 to convert Bcl-2 from an antiapoptotic to a proapoptotic molecule was investigated by accelerated molecular dynamics simulation. Results show that BDA-366 blocks or reduces the affinity of Bcl-2 for BH3-only proteins like Bid via negative cooperative effects, thereby releasing such proteins and unleashing their proapoptotic effects.
Disclosure statement
There are no interest conflicts to declare.
Funding
This work is supported by the National Natural Science Foundation of China [21603013, 31601412]; 100 Talent Program grant and Biological Resources Service Network Initiative [ZSYS-012]; Chinese Academy of Sciences [SKT1604].