

Abstract
AIDS is a global infection involving several complications and its increasing prevalence every year has prioritized our study. Therapy associated with HIV has led to emergence of multidrug resistance and toxicity. Thus, the development of a potent, affordable and safe anti-HIV drug is a global concern. Among the different targets developed, inhibition of non-nucleoside reverse transcriptase (NNRT) is found to be effective and promising. Etravirine, efavirenz, nevirapine, rilpivirine and delavirdine are the marketed NNRTIs available. This study is focused on computational prediction of hit molecules as well as repurposing of various FDA-approved drugs as potential NNRTIs. A synthetic database from ZINCpharmer, publicly available natural databases of coumarins, chromones and chalcones, and two databases of FDA-approved drugs for repurposing were screened to check for the possibility of these compounds to possess anti-HIV activity. Study utilizes a structure-based approach with the generated pharmacophore of target protein (PDB ID: 3MEC), screening of selected datasets is carried out using the Phase tool of Schrodinger. The top filtered compounds with good fitness score were proceeded to molecular docking studies to study their binding affinity to the target. Energy-based calculations using Prime MM-GBSA of Schrodinger was performed to determine free binding energy of the complexes. Prediction of pharmacokinetic parameters of top compounds is further carried out and reported. All the results obtained from different databases are compiled, interpreted and five molecules were subjected to molecular dynamic studies to further confirm the prediction and identified hit molecules for in vitro screening as potential NNRTIs.
Communicated by Ramaswamy H. Sarma
Graphical Abstract
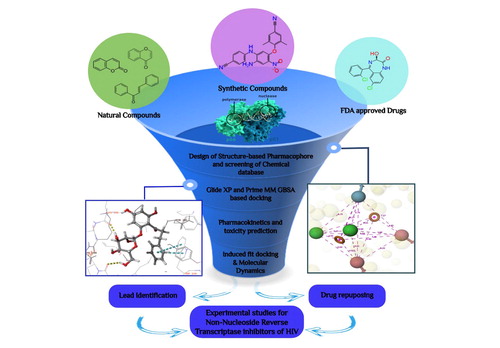
Computationally potential and safe NNRTIs were identified.
Structure-based pharmacophore screening of synthetic, natural and FDA-approved databases and further refinement of databases by Glide XP docking based on docking score and binding interactions at the active site.
Prime MM-GBSA rescoring identified the involvement of two evident contributors: van der Waals free energy (ΔGvdW) and nonpolar solvation terms (ΔGsolLipo) in stabilizing the ligand–receptor complex.
ADMET parameters of top molecules were determined using in silico tools like Schrodinger's Qikprop and a publicly available server pkCSM.
Molecular dynamics confirmed the binding stability and potential hit molecules as NNRTIs.
Highlights
Correction Statement
This article has been republished with minor changes. These changes do not impact the academic content of the article.
Acknowledgments
All the authors are thankful to Prof. Venkataramana C.H.S., Head, Department of Pharmaceutical Chemistry, Faculty of Pharmacy, Dr. Saraswathy G.R., Head, Pharmacological Modelling and Simulation Centre (PMSC), M.S. Ramaiah University of Applied Sciences, Bangalore and Manipal College of Pharmaceutical Sciences (MCOPS), Manipal University for the Molecular Dynamic Simulations support and providing the necessary facilities to carry out this research work.
Disclosure statement
Authors report no conflicts of interest. All the authors alone are responsible for the content and writing of this article.