

Abstract
The therapeutic potential of PPARs antagonists extends beyond diabetes. PPARs antagonists represent a new drug class that holds promise as a broadly applicable therapeutic approach for cancer treatment. Thus, there is a strong need to develop a rational design strategy for creating PPARs antagonists. In this study, three-dimensional quantitative structure-activity relationship (3D-QSAR) models of PPARα receptor (CoMFA-1, q2 = 0.636, r2 = 0.953; CoMSIA-1, q2 = 0.779, r2 = 0.999) and PPARδ receptor (CoMFA-2, q2 = 0.624, r2 = 0.906; CoMSIA-2, q2 = 0.627, r2 = 0.959) were successfully constructed using 35 triazolone ring derivatives. Contour map analysis revealed that the electrostatic and hydrophobic fields played vital roles in the bioactivity of dual antagonists. Molecular docking studies suggested that the hydrogen bonding, electrostatic and hydrophobic interactions all influenced the binding of receptor-ligand complex. Based on the information obtained above, we designed a series of compounds. The docking results were mutually validated with 3D-QSAR results. Three-dimensional-QSAR and absorption, distribution, metabolism, excretion and toxicity (ADMET) predictions indicated that 19 newly designed compounds possessed excellent biological activity and physicochemical properties. In summary, this research could provide theoretical guidance for the structural optimization of novel PPARα and δ dual antagonists.
Communicated by Ramaswamy H. Sarma
GRAPHICAL ABSTRACT
In this study, the CoMFA/CoMSIA, molecular docking studies and molecule dynamics simulations were performed to reveal the SAR of triazolone ring derivatives with PPARα/δ dual antagonistic activities. On the basis of QSAR models and bioisostere replacement, new compounds were designed and their ADMET properties were predicted.
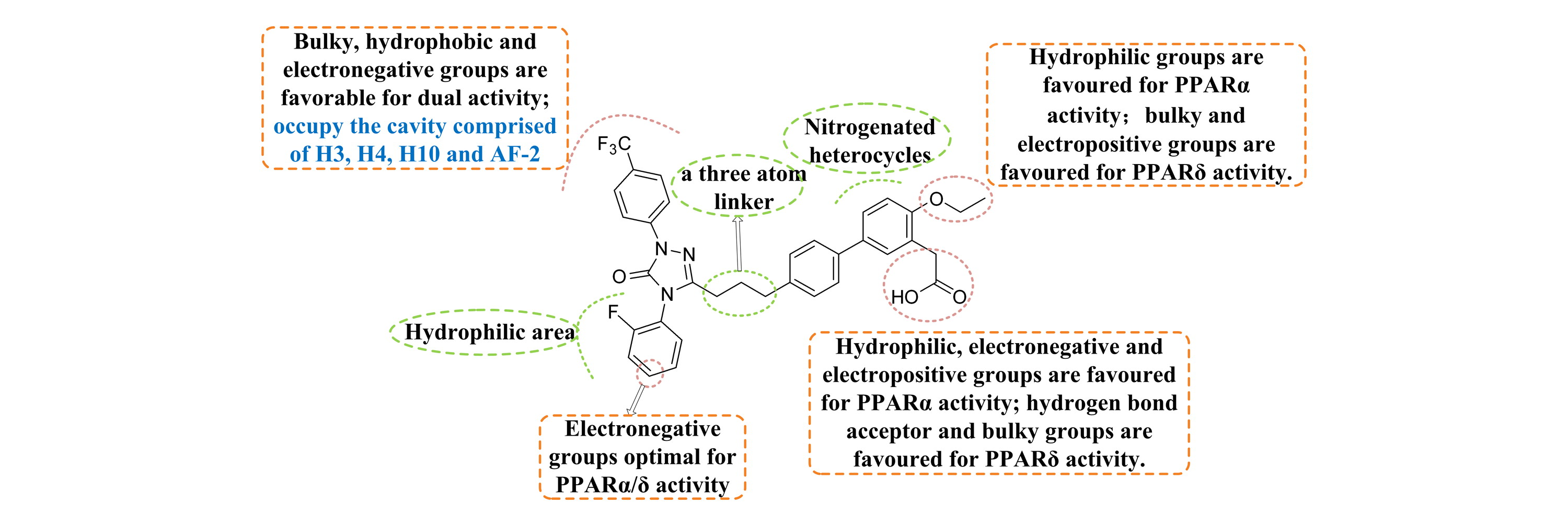
CoMFA and CoMSIA analysis were performed to investigate the SAR of triazolone ring derivatives with PPARα/δ dual antagonistic activities.
Molecular docking simulation was conducted to provide us much more insights into the active site where ligands bind.
According to the docking results and contour maps derived from the 3D-QSAR model, we carried out structural optimization and designed several new compounds to improve the predicted biological activity.
The docking results were mutually validated with 3D-QSAR results.
3D-QSAR and ADMET prediction indicated that 19 newly designed compounds had good physicochemical properties, weak side effects and low toxicity.
Highlights
Disclosure statement
No potential conflict of interest was reported by the authors.