
Abstract
The inosine triphosphate pyrophosphatase (ITPA) protein is responsible for removing noncanonical purine nucleoside triphosphates from intracellular nucleotide pools. Absence of ITPA results in genomic instability and increased levels of inosine in DNA and RNA. The proline to threonine substitution at position 32 (P32T) affects roughly 15% of the global population and can modulate treatment outcomes for cancer, lupus, and hepatitis C patients. The substitution of arginine with cysteine at position 178 (R178C) is extremely uncommon and has only been reported in a small cohort of early infantile encephalopathy patients suggesting that a functional ITPA protein is required for life in humans. Here we present molecular dynamic simulations that describe the structure and dynamics of the wild-type ITPA homodimer and two of its clinically relevant mutants, P32T and R178C. The simulation results indicate that both the P32T and R178C mutations alter the structure and dynamic properties of the protein and provide a possible explanation of the experimentally observed effect of the mutations on ITPA activity. Specifically, the mutations increased the overall flexibility of the protein and changed the dominant collective motions of the top lobe as well as the helix 2 of the lower lobe. Moreover, we have identified key active-site residues that are classified as essential or intermediate for inosine triphosphate (ITP) hydrolyzing activity based on their hydrogen bond occupancy. Here we also present biochemical data indicating that the R178C mutant has very low ITP hydrolyzing activity.
Communicated by Ramaswamy H. Sarma
1. Introduction
The ITPA protein plays a protective role in the cell by removing noncanonical purine containing nucleoside triphosphates from the intracellular nucleoside triphosphate pools (Burgis & Cunningham, Citation2006; Chung, Park, Lee, & Jang, Citation2002; Galperin, Moroz, Wilson, & Murzin, Citation2006). Nucleotides containing noncanonical purines, such as hypoxanthine and xanthine, can arise in cells via normal de novo purine metabolism, but also by oxidative damage. ITPA specifically acts on both ribose and deoxyribose nucleoside triphosphates ((d)NTPs) containing noncanonical purines by performing a pyrophosphohydrolysis reaction (Lin et al., Citation2001; Vanderheiden, Citation1970). The major role of ITPA is thought to be the conversion of (deoxy)inosine triphosphate ((d)ITP) to (deoxy)inosine monophosphate ((d)IMP) with release of pyrophosphate. Conversion of (d)ITP excludes noncanonical purine containing (d)NTPs from (d)NTP pools, preventing their incorporation into DNA or RNA molecules, and improper binding by nucleotide binding proteins (Burgis, Brucker, & Cunningham, Citation2003; Pang et al., Citation2012). ITPA dysfunction can cause increased levels of inosine in nucleic acids, sensitivity to noncanonical purines, elevated mutation rates, double-strand breaks in DNA, delayed cell cycle progression, and apoptosis (Abolhassani et al., Citation2010; Burgis et al., Citation2003; Noskov et al., Citation1996; Pang et al., Citation2012).
In 2015, Kevelam et al. demonstrated that arginine to cysteine substitution at position 178 (R178C (c.532C > T)) produced an ITPA deficient phenotype which resulted in a lethal early infantile encephalopathy (Kevelam et al., Citation2015). This position has previously been identified as essential for enzyme activity based on a site-directed mutational analysis of the substrate-binding site (Gall et al., Citation2013). ITPA also plays a role in drug metabolism and lower levels of ITPA activity can modulate drug metabolism for some treatment regimens. The P32T (c94C > A, rs1127354) mutant has been shown to play a role in both thiopurine and ribavirin metabolism (Bierau, Lindhout, & Bakker, Citation2007). Herting et al. have performed biochemical studies on P32T mutant and demonstrated that this mutant has reduced catalytic activity and stability (Herting, Barber, Zappala, Cunningham, & Burgis, Citation2010). Low levels of ITPA activity, such as found in P32T individuals, are associated with poorer outcomes for patients undergoing thiopurine therapy due to increased intracellular concentrations of the active form of the drug (Marinaki et al., Citation2004; Shipkova et al., Citation2011). On the other hand, P32T polymorphism is linked to improved outcomes for hepatitis C patients undergoing ribavirin therapy (Fellay et al., Citation2010) and, more recently, HIV patients undergoing tenofovir therapy (Peltenburg et al., Citation2018).
The crystal structures of ITPA have shown that the protein forms a homodimer in which the monomers consist of two lobes composed of beta sheet and alpha helical segments (Porta, Kolar, Kozmin, Pavlov, & Borgstahl, Citation2006; Stenmark et al., Citation2007). Stepchenkova et al. have used size-exclusion chromatography to examine the dimerization of the ITPA P32T mutant and found that this mutant is a dimer in solution, same as wild-type (Stepchenkova et al., Citation2009). Later, it was proposed from the crystal structure determination that the P32T has the wild-type structure, but has overall greater structural disorder, and flexibility (Simone et al., Citation2013).
Thus far, the ITPA protein has mostly been subject to experimental studies. Most experimental studies have focused on the more common polymorphism, c.94C > A (p.Pro32Thr). Molecular modeling methods, and especially molecular dynamics simulations, have become a very useful tool in the study of biological molecules, providing atomic detail and high temporal resolution about their structure and motion (Houndonougbo, Kuczera, & Jas, Citation2005, Citation2008; Mahadevan, Xu, Siahaan, & Kuczera, Citation2002; Nagarajan, Alkayed, Kaul, & Barnes, Citation2020; Wan et al., Citation2020; Yu et al., Citation2020). In addition, the results of computer simulations are valuable and complement experimental data. In particular, single 100–300 ns long molecular dynamics simulation of model built mutant structures using experimentally determined wild type coordinates has been widely employed to study the effect of mutations on protein structure and dynamics and the results are reported to be in good agreement with experiment (Boopathi & Kolandaivel, Citation2017; Fukuyoshi et al., Citation2016; Narang, Shuaib, Goyal, & Goyal, Citation2018; Tugba & Emil, Citation2015). For ITPA, computational studies can supplement the existing experiments for P32T and moreover give new insight into the structure and dynamics of wild-type and other mutants (Das et al., Citation2019; Saldaña-Rivera, Bello, & Méndez-Luna, Citation2019; Zhang et al., Citation2020).
In the current study, we present results of molecular dynamics simulations in aqueous solution for the wild-type ITPA dimer as well as the P32T and R178C clinical ITPA mutants. Although the simulation of the monomer will reduce the computational time, the monomer as such is not considered a functional unit of the ITPA protein. Moreover, given the interactions between monomers in the homodimer simulation, we expect that the dynamics of the monomer alone will be different from that of the homodimer. For all of these reasons, we have only considered the homodimer protein. We focus on the effect of the mutations on the structure, dynamics, and interaction properties of ITPA, which is used to provide a possible structural explanation of the observed reduction of the enzymatic activity in the mutants. To this end, we analyze root-mean square deviation, radius of gyration, secondary structure, distance fluctuations, eigenvector projection analysis, and the hydrogen bonding patterns. Our simulations showed that the wild-type homodimer and the two clinically relevant mutants mostly retain their secondary structures in aqueous solution. We have found that both mutations produced a reorientation of α-helix 2 in the lower lobe and increase the overall protein flexibility. Additionally, the results of the MD simulations correlate well with the enzyme activity measurements presented in this study and in Gall et al. (Citation2013), Herting et al. (Citation2010) and Kevelam et al. (Citation2015). We have also observed based on the first principal component analysis that the top lobes of the homodimer move in opposite direction in the wild-type as well as in the mutants. Importantly, the key hydrogen bond interactions and residues in the active site for enzyme activity were identified based on the hydrogen bond occupancy analysis of the MD trajectories. We also present activity measurements showing that the R178C mutant has very low ITPA catalytic activity.
2. Materials and methods
2.1. Molecular dynamics simulations
The initial configuration of the wild-type dimer was taken from the crystal structure of human ITPA (Stenmark et al., Citation2007), Protein Data Bank file 2CAR. The mutants’ structures were built from the file 2CAR by constructing the coordinates of the side chains of the mutated residues. The GROMACS program version 5.1.2 (Van der Spoel et al., Citation2005), running on a Linux cluster, and the CHARMM version 36 all-atom force field (Best et al., Citation2012) was employed to generate the simulation systems and to perform MD simulations. Hydrogen atoms needed in the all-atom model were added and the proteins were placed in rectangular cells which were overlaid with TIP3P (Jorgensen, Chandrasekhar, Madura, Impey, & Klein, Citation1983) water molecules. Solvent molecules were randomly replaced by sodium and chlorine counterions to neutralize the system at biologically realistic conditions (at pH 7.0 and concentration of 0.15 M). The protonation state of titratable residues is set to the normal protonation state corresponding to pH 7.0. The histidine residues were modeled as neutral. Additionally, we have calculated pKa of the residues using the ProteinPrepare tool in PlayMolecule web application (Martínez-Rosell, Giorgino, & De Fabritiis, Citation2017) and the obtained values are consistent with the protonation state assignments. The minimum solute-box distance is set to 1 nm. The cell dimensions are 6.5 × 6.4 × 5.5 nm for the wild-type and 8.5 × 8.4 × 7.5 nm for the mutants. The system comprises 54 Na+ and 48 Cl– for the wild-type, 56 Na+ and 48 Cl– for the R178C, as well as 54 Na+ and 48 Cl– for the P32T.
Each cell was energy minimized with the steepest descent algorithm and was equilibrated with the protein fixed in the NVT ensemble for 25 ps using a 1 fs time step and a target temperature of 310 K. Further 10 ns equilibration was applied in the NPT ensemble at 310 K using a 2 fs time step with the whole protein-solvent system being allowed to move. Finally, 1 μs NPT simulation of the unconstrained proteins was performed. The leapfrog algorithm was employed for integration of equations of motion, the Nose–Hoover thermostat (Hoover, Citation1985; Nose, Citation1984) was used to maintain constant temperature, and the Parrinello–Rahman coupling algorithm (Parrinello & Rahman, Citation1981) was applied to keep the pressure at 1 bar. The periodic boundary conditions were employed. All the bonds involving hydrogen atoms were fixed using the LINCS constraints algorithm (Hess, Citation2008) in order to integrate the equations of motion with a time step of 2 fs (Barth, Kuczera, Leimkuhler, & Skeel, Citation1995). The water molecules were kept rigid with the Settle algorithm (Miyamoto & Kollman, Citation1992). The van der Waals interactions were truncated at 1.2 nm and a switching function was employed between 1.0 and 1.2 nm. The particle-mesh Ewald (PME) scheme (Sagui & Darden, Citation1999) with a cutoff of 1.2 nm was used to calculate the long-range electrostatic interactions.
The mutants’ initial structures were built using the program CHARMM (Brooks et al., Citation1983) version 40 and the GROMACS program version 5.1.2 (Van der Spoel et al., Citation2005) was employed to generate the simulation systems and to perform both equilibrations and the trajectories generation. The CHARMM version 36 all-atom force field (Best et al., Citation2012) was used. The generation of the trajectories for each simulation took an average of 9 h/ns, running in parallel on one node of a 2.80 GHz dual Quad-Core Intel Xeon Linux cluster.
In each of the simulations, frames were saved every 10 ps. The root-mean-square deviations (RMSDs) with respect to the starting structures and the root-mean-square fluctuations (RMSFs) were computed after removing the effects of the overall translation and rotation. Hydrogen bonds patterns, radius of gyration, secondary structure elements, as well as distance fluctuation were computed and the principal component within the trajectories were also analyzed. The analyses of the trajectories were performed using the GROMACS package version 5.1.2 (Van der Spoel et al., Citation2005).
2.2. Plasmid construction
The pET28a-based mutant plasmid was constructed similar to Gall et al. (Citation2013) using the pET28a-ITPA plasmid as a template (Burgis & Cunningham, Citation2006).
2.3. Purification of refolded His6-tagged recombinant wild-type ITPA and R178C ITPA
Wild-type ITPA and R178C ITPA were overexpressed as described previously (Gall et al., Citation2013) and pelleted cells were stored at −80 °C. Cell pellets were resuspended in 8 M urea, 20 mM phosphate pH 7.4, 0.5 M NaCl (Buffer A) then sonicated as in Gall et al. (Citation2013). Cleared lysates were loaded onto a 1 mL Ni2+ charged HisTrap HP affinity chromatography column and washed with 10–15 column volumes of Buffer A using a BioLogic LP System (www.bio-rad.com). Recombinant protein was eluted using Buffer A containing 500 mM imidazole and eluent was analyzed using SDS-PAGE (Sambrook, Fritsch, & Maniatis, Citation1989). Fractions containing purified target protein were selected for dialysis on ice against 20 mM Tris–HCl pH 7.4, 100 mM NaCl, 10 mM MgCl2 and 1 mM 1,4-dithiothreitol (Buffer B) containing 8 M urea in which the concentration of urea was gradually reduced using a constant flow dialysis system that pumped in Buffer B at a rate of 2 mL/min for about 32 h. Absence of urea was determined using a urea assay kit (www.abnova.com). The final protein product was stored in 50% glycerol at −20 °C. After 48 h at −20 °C, precipitate was removed by centrifugation and protein concentration was quantified using a NanoDrop 2000 (www.thermofisher.com).
2.4. Specific activity measurement
Pyrophosphohydrolase activity with ITP was measured for refolded R178C and wild-type enzymes prepared as above and similar to the standard method (Gall et al., Citation2013). De-ionized H2O was used throughout the assay and to prepare all solutions and buffers. ITP was purchased from Sigma-Aldrich (www.sigmaaldrich.com). Reactions were performed in 100 µl volumes containing 20 mM Tris-HCl at pH 7.4, 100 mM NaCl, 10 mM MgCl2, 100 µM ITP, and 0.2 pmol standard wild-type, 0.4 pmol refolded wild-type and 100 pmol refolded R178C. After 10 min preincubated at 37 °C, the enzyme was added. After 10 min at 37 °C reactions were stopped by adding 100 µl of 2% sodium dodecyl sulfate solution. The suspension was mixed and centrifuged. An aliquot of the supernatant was removed, and used for IMP quantification. HPLC was performed similar to Iyama, Abolhassani, Tsuchimoto, Nonaka, and Nakabeppu (Citation2010). Reaction products were separated on a Nucleogen 60-7 DEAE column (www.mn-net.com) using a ThermoFisher UltiMate 3000 HPLC system at a flow rate of 0.6 ml/min and buffer containing 75 mM sodium phosphate, pH 6.4, 5% acetonitirile and 0.4 mM EDTA. IMP was quantified by UV absorption at 248 nm using an UltiMate 3000 VWD-3400RS UV detector. IMP produced was calculated using an IMP standard curve and specific activity was calculated. Two-tailed Student’s t-tests were used to determine statistical significance.
3. Results
3.1. Overall structure
The root-mean-square deviations (RMSD) of backbone atoms and the radius of gyration (Rg) were calculated to assess the convergence to equilibrium and the structural stability of the simulations. The RMSDs () tend to fluctuate around constant values after an initial steady rise period of about 20 ns for the wild-type and R178C and 40 ns for the P32T, indicating that the simulations converge towards equilibrium structures. The RMSD average values are 0.23 for the wild-type, 0.31 for R178C, and 0.35 nm for P32T. The difference between the average RMSD values is small, in the 0.08–0.12 nm range. It can however be observed that the RMSD for the mutants tends to be slightly higher than for the wild-type during the simulations with the largest observed for P32T, suggesting possible structural difference upon mutations. This structural difference can also be significant for the R178C as an abrupt increase in in the RMSD of this mutant is visible around 600 ns.
Figure 1. Instantaneous backbone RMSDs from the starting structure (a) and radius of gyration (b) for the wild-type (WT) and the mutants.
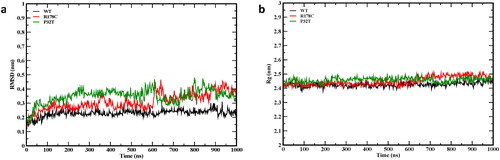
The Rg characterizes protein structures compactness. From , we observed that the Rg of the wild-type and the mutants fluctuate around stable values during the simulations confirming the convergence of the simulations.
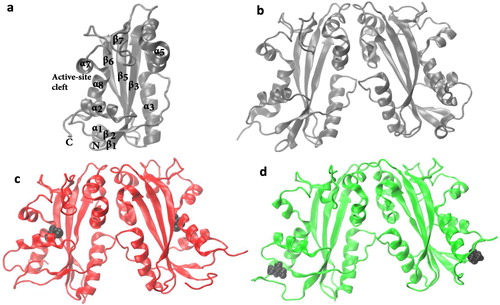
In order to understand the effect of mutations on the secondary structure of ITPA, we have visualized the final structures of the 1 μs simulations () and have examined the variation with time of the secondary structure elements in the trajectories (Figure S1). The secondary structure elements in the crystal structure () are mostly observed in the final simulation structures of the wild-type () and the mutants (), except the loss of the N-terminal helix (α1 in ) in the final structures of the mutants. The secondary structures of the wild-type and the mutants tend to remain stable throughout the simulation and have comparable secondary structure components for the most part (Figure S1), although noticeable differences can be observed in the regions of helices 1. The structure of the N-terminal helix 1 fluctuates between the α and 3-helix in the wild-type while the structure of this α-helix is lost in the mutants, confirming the results of the visualization of the final simulation snapshots. Moreover, these results rule out the possibility of structural and dynamical artifacts due to the simulation process. We have also compared the wild-type and mutants’ structures by aligning the final structures of the MD simulations (Figure S2). The alignment was carried out using the Multiseq tool in VMD and the structures were colored using the structural similarity Qres (Roberts, Eargle, Wright, & Luthey-Schulten, Citation2006). Blue indicates high structural similarity and red indicates low structural similarity. The structure alignments show that the mutant structures are similar to that of the wild-type, with RMSD value of 0.27 for the WT-R178C pair and 0.24 nm for the WT-P32T pair. Significant differences can be observed in the regions of N- and -C termini, α-helix 2, and the loop after beta strand 6. In addition, the structure alignments show that the mutations cause a remarkable shift of the α-helix 2, compared to that of wild-type (Figure S2).
Figure 2. (a) The secondary structural element of the ITPA monomer. All the alpha helices and the beta sheets are labeled except those with residues less than four. Final simulation structures of the ITPA dimer (b) wild-type, (c) R178C, and (d) P32T. The point mutation residues (P32 and R178) are shown in black van der Waals representations.
3.2. Effect of R178C and P32T mutations on ITPA dynamics
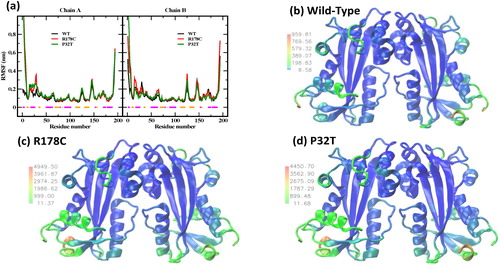
To assess the effect of mutations on the dynamic behavior of ITPA, the root mean-square fluctuations (RMSFs) of backbone atoms within the equilibrated trajectories were calculated (). We have converted the RMSF values into B-factor values (B-factor = 8π2×(RMSF)2/3) that were used to represent the structure of the wild-type and mutants () on color scale for better visualization. As shown in , the wild-type protein exhibits high atom RMSFs (values of more than 0.19 nm) in the regions of N- and C-termini (residues 1–4 and 191–194), α- helices 2 (residues 15–18 and 29–31), and the loops before and after beta strand 6 (residues 125–126 and 145–148). Other than the N- and -C termini, the two most flexible residues are in the loops after helices 2 (residue 30) and beta strand 6 (residue 146). It should be noted that helix 7 has relatively large fluctuations (). The RMSF profile of the mutants is for the most part similar to that obtained for the wild-type (). However, we observed in the mutants an increase of the flexibility in most of the regions described above, with the R178C mutant showing the highest flexibility value (see the maximum values of the labels and the legend in ). In addition, the B-factor average values are 197 for the wild-type, 443 for P32T, and 450 nm2 for R178C. This suggests that the overall flexibility in the mutant proteins is higher than in the wild-type protein and the R178C mutant is more flexible than the P32T mutant.
Figure 3. (a) Distribution along the polypeptide of backbone atom RMSFs around trajectory average structures for the wild-type and the mutants. The horizontal lines represent the X-ray crystal structure α-helices (magenta) and β-strands (orange). Graphical representations of the wild-type (b), R178C (c), and P32T (d) colored using the B-factor values. The labels beside the color bar show the range of the values of the B-factor in nm2.
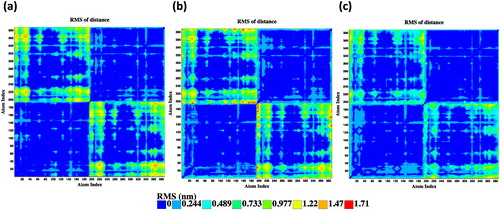
We have analyzed the distance fluctuations between the Cα atoms to further evaluate internal rigid regions (lowest distance fluctuation values), flexible regions moving together (coordinated movements, representing relatively low distance fluctuation values), and regions moving in opposite direction (less coordinated movements, representing highest distance fluctuation values). The matrices of the distance fluctuations for the wild-type and mutants are shown in . In general, each monomer of the wild-type and mutants exhibit rigid-body movements (blue blocks in ). However, regions of loops and helix 2 moving in coordination (green strips) are noticeable in each monomer of the three systems. In the mutants, these mobilities are observed for the whole helix 2 (17–28 residues) and also in the N-and-C-termini. Further, less coordinated movement of intermonomer regions (orange to red color in the top left and low right squares) are observed in the wild-type and mutants. Nevertheless, significant increase in the intermonomer movements is visible in the R178C (red dashes in ). These results agree with our RMSF analysis that the R178C mutation is associated with a larger increase in structural flexibility than the P32T mutation.
Figure 4. Distance fluctuation maps between the Cα atoms within the equilibrated trajectories of the wild-type (a), R178C (b), and P32T (c).
To further investigate the effects of mutations on the structure of ITPA protein, RMSD conformation cluster analysis were performed using the GROMOS algorithm (Daura et al., Citation1999) in GROMACS program. The algorithm calculates RMSD between all pairs of structures in a trajectory as well as the structure with the highest number of other structures for which the RMSD is within a chosen cutoff (neighbor conformations) and is identified as the center of the first cluster. The structures of the first cluster are removed from the pool and the process is repeated until the pool of structures is empty. Clustering calculations with different cutoff were performed and the cutoff of 0.2 nm was found to capture more than 90% of the trajectories in fewer number of clusters. The clustering analysis has identified 6 clusters for the wild-type, 15 clusters for R178C, and 11 clusters for P32T that cover more than 90% of the total populations (Table S1). The alignments of the middle structures of the clusters are shown in Figure S3. It can be observed that the structures of the wild-type and the mutants diverge in the N- and C-termini, and in the loops after helix 2 and strand 6. In addition, structural differences can be also observed in the helix 7 for the wild-type and in the helix 2 for both mutants, with the structures of R178C showing most divergence in helix 2. The clustering data (Table S1) indicate that the trajectories of the mutants give a larger number of significant clusters and less structures in the most populated clusters than the trajectory of the wild-type, suggesting that both mutations led to the increase of structural flexibility of the ITPA protein. In addition, the R178C mutant has the highest number of clusters and the lowest percentage of cluster members in the most populated cluster (cluster 1) which supports our RMSF and distance fluctuations analysis that the R178C structures are more flexible than the P32T structures.
3.3. Essential dynamics
We have performed essential dynamics (ED) analysis of the simulation trajectories to describe the dominant subdomain motions of ITPA and the effects of mutations on these motions. We have used the GROMACS program to calculate the mass-weighted covariance matrix of position fluctuations of the backbone atoms from the starting structures. The calculations were done after removing the effects of the overall translations and rotations of the protein (Amadei, Linssen, & Berendsen, Citation1993). In previous work (Houndonougbo et al., Citation2005, Citation2008), it was found that fewer eigenvectors with the largest eigenvalues represent most of the collective motions and the trajectory fluctuations. Thus, the collective motions important for protein functions are separated from the local fluctuations. The eigenvalues describe the extent of correlated motions about each eigenvector. The principal component analysis (PCA) consists of projecting the trajectories of the molecular dynamics simulations on the eigenvectors (principal components). The result of the covariance matrix calculations indicates that the contribution of the first component (PC1) to the variance of the motion is 19.2% for the wild-type, 27.6% for R178C, and 24.4% for P32T. These values indicate that PC1 has larger contribution to the overall motion in the mutants than in the wild-type. Information about the overall structural flexibility can be obtained from the PCA by calculating values of the trace of the covariance matrix for which larger value means larger flexibility. The values of the trace of the covariance matrix are 7.7735, 17.432, and 17.1425 nm2 for the wild-type, R178C, and P32T, respectively. These values are in agreement with the RMSF analyses for the overall flexibility.
The 2 D projections onto the first two eigenvectors (principal components PC1 and PC2) in Figures S4 and S5 illustrate the motion of the wild-type and the mutants in phase space. In these projections, a conformational region or subspace is represented by a distribution of dots within a compact cluster. It is clear that the structures of the mutants are generally more flexible than the wild-type which sampled only one conformation region, while the mutants visited several conformation regions. The simulation of the wild-type continuously samples conformations that are very close to the starting structure (Figure S4(a)). On the other hand, the R178C mutant sampled a single state with conformations fairly close to the initial structure (crystal structure) during the first 600 ns of simulation but, then, transitioned to a different state with different structures (Figure S4(b)). Additionally, In Figure S5, the P32T mutant samples a state with conformations close to that of the crystal structure, but the trajectory also visited different states (red and green in Figure S5). These differences in the two mutants are characterized by the loss of the secondary structure at the N- and C-termini and the movement of the lower lobe subunits including helix 2. However, the reduced presence of the blue color in the representations in Figure S4(b) compared to Figure S5 indicates that the degree of structural deviation in the R178C is higher than in the P32T. For the R178C, the transition around 600 ns to different state which is characterized by the loss of N- and C-termini structures and the movements of the lower lobe subunits explains the cause of the sudden rise in the RMSD around 600 ns.
We have calculated the inner products between the eigenvectors of the mutants with the eigenvectors for the wild-type to explore the similarity between the structural subspaces. The inner product is positive for correlated subspaces, negative for anti-correlated subspaces, and is close to zero for poor overlapping subspaces (Martínez-Archundia, Correa-Basurto, Montaño, & Rosas-Trigueros, Citation2019). For these calculations, we have used the first 30 eigenvectors which capture more than 80% of the overall motion. The resulting overlap matrices are displayed in Figure S6. Both mutant proteins share some similarities with the wild-type protein, with overlapping values up to 0.657 for R178C and 0.573 for P32T. Concerning the first two PCs of the wild-type, only PC2 is anti-correlated (dark blue square) with PC5 of R178C, whereas, PC1 exhibits anti-correlation with PC3 and PC6 of P32T and PC2 is correlated (red square) with PC7 in P32T. These results suggest that the mutations lead to a significant change of the motion of the protein. Moreover, Figure S6 shows that PC8 in wild-type is correlated to PC13 of R178C, while, PC5 and PC7 of the wild-type display anti-correlation with PC10 and PC11 in P32T, respectively. The results of the inner product analysis seem to be consistent with the different subspace exploration behaviors we observed in Figures S4 and S5, i.e. the P32T mutant visited more conformation subspaces than the R178C mutant. These data may also explain the largest average RMSD value noticed earlier for P32T.
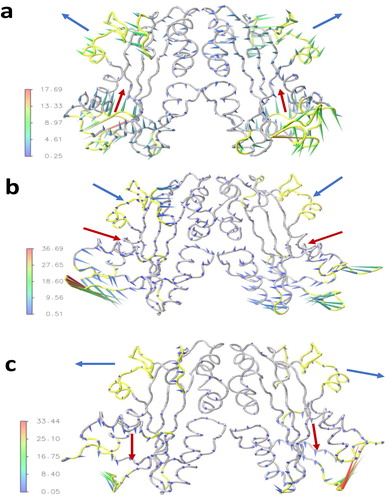
Porcupine plots are used to display the direction and magnitude of the eigenvectors of the first principal components for the wild-type and the mutants. The dominant collective motions of the wild-type protein observed in the first principal component (color scale arrows from green to red in ) are in the N- and C-termini (residues 1–9 and 188–194), in the α-helices 2 and 7 (residues 15–17 and 169–175), in the loops after α-helix 2 (residues 30–33) and before α-helix 7 (157–168) as well as the loops before and after beta strand 6 (residues 125–127 and 144–149), consistent with the high flexibility areas of the RMSF analysis (see for the different secondary elements). The trajectory projection on the first eigenvector of the wild-type dimer simulation () shows the top lobe subunits of one monomer moving outward of the active site in opposite direction of the other monomer, while the two α-helices in the lower lobe (helix 2) predominantly move upward in the active site in the same direction. Interestingly, such motion between monomers would not be observable with a single monomer simulation. The first principal component for mutants is illustrated in . The most prominent motions are in the N- and C-termini for the R178C and in the N-terminal for P32T. The mobility amplitude of the dominant motions is larger in the mutants as compared to the wild-type and the largest amplitude is observed in the R178C (see color scale in ) which indicate that R178C is more unstable than P32T. This result was also supported by the RMSF data. We note that the amplitude of the motions of α-helix 7 of the top lobe and the α-helix 2 of the lower lobe are considerably reduced in both mutants. The mutation at position 178 has changed the direction of the prominent motions observed in the wild-type, such as the top lobe motions are reversed and the helix 2 of the lower lobe moves laterally (see arrows in ). The mutation at the position 32 preserves in general the direction of the motion of the top lobe, but has reversed the motion of the helix 2 of the lower lobe (see arrows in ).
Figure 5. Porcupine plot to visualize the first principle eigenvector of the wild-type (a), R178C (b), and P32T (c). The arrows attached to the Cα atoms show the direction of motion and the length of arrows shows the magnitude of the eigenvector. The regions of dominant motions in the wild-type are highlighted in yellow color. The blue arrows indicate the direction of motion of the top lobe subunits and the red arrows show the direction of motion of the α-helix 2 of the lower lobe.
3.4. Effect of R178C and P32T mutations on the hydrogen bond interactions
In order to evaluate the effect of mutation on the hydrogen bond interactions of ITPA, the total number of hydrogen bonds formed along the simulation trajectories was calculated (Figure S7). The number of intramolecular hydrogen bonds of the wild-type is noticeably greater over the course of the simulation as compared to the mutants with the average of 281, 261, and 265 hydrogen bonds for the wild-type, R178C, and P32T, respectively. Thus, the hydrogen bond interactions were weakened by R178C and P32T mutations. R178C has less intramolecular hydrogen bond formations in the simulations, suggesting that the hydrogen bond interactions of ITPA protein are more altered by R178C mutation than the P32T mutation, consistent with the RMSF, ED analyses.
To further assess the effect of mutation on the interactions of key residues in the active site of ITPA and the mutated residues, we have analyzed the hydrogen bonding interactions of Asn16, Lys19, Asp41, Glu44, Lys56, Asp72, Asp152, Lys172, His177, and mutated residues at positions 178 and 32. The calculated hydrogen bond occupancy, which is defined as the fraction of frames where a hydrogen bond is formed between two residues, are reported in Table S2 for the occupancies greater than 30%. It can be observed that these key residues in the active site of ITPA, including the mutated residues in this study, are involved in very stable hydrogen bonding in the wild-type with neighboring residues, supporting that they play an important role in ITPA activity. However, the hydrogen bonding analysis shows that the hydrogen bond interactions of residues 19, 32, 72, 152, and 178 are affected by the mutations. Hence, we focused on the details of these interactions (). The hydrogen bond occupancy of the active-site residues in the wild-type shows that Asp152 and Arg178 are engaged in three stable hydrogen bonds and Lys19 makes five stable hydrogen bonds ( and ). It is worth noting that three out of the four strong hydrogen bond interactions (occupancies greater than 75%) for the wild-type in are associated with the active-site residue 178 which suggests that Arg178 is essential for ITPA activity. In the active site, the R178C mutation leads to the loss of three interactions, Lys172-Asp152, Arg178-Glu22 and Arg178-Glu71, while the P32T mutation caused a loss of the Lys19-Glu71 interaction (). The P32T mutation also resulted in the gain of two stable hydrogen bonds in the active site, Lys19-Asp152 and Arg178-Ser176. These results suggest that the R178C mutation has disrupted the hydrogen bonding in the active site to a greater extent than the P32T mutation (loss of three for R178C compared to no net change for P32T). Hence, the degree of alteration found in this analysis of the hydrogen bonding occupancy of the active-site residues in R178C and P32T mutants is consistent with the relative decrease of ITPA activity for the two mutants, that is, the R178C mutation reduces the ITPA activity to a greater extent than that of P32T mutation. These results also correlate well with the level of increase in overall flexibility and the decrease in total number of hydrogen bonds as presented above. The analysis of the hydrogen bond occupancy for the active site suggests that the Arg178-Glu22, Arg178-Glu71 and Asp152-Lys172 hydrogen bond interactions are important for ITPA activity. In the wild-type, the hydrogen bonds occupancy of Arg178-Glu22, Arg178-Glu71 and Asp152-Lys172 interactions are 77.7, 99.9 and 43.8, respectively. Based on the strength of the interaction, we classify the residues Arg178, Glu22, Glu71 as essential for ITPA activity while the residues, Asp152 and Lys172 are of intermediate importance for ITPA activity. This classification agrees well with a site-directed mutagenesis study which has categorized Arg178 and Glu22 as essential and Asp152 as well as Lys172 as intermediate (Gall et al., Citation2013).
Figure 6. Illustration of the hydrogen bonding interactions of key residues in the active site. The red dotted lines represent the interactions that exist in the wild-type. The green dotted lines represent new interactions. The point mutation residues (R178 and P32) are shown in orange licorice representations.
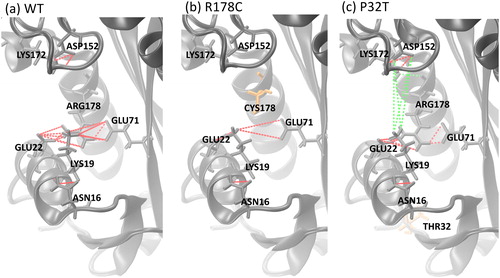
Table 1. Percentage of occupancy of hydrogen bond interactions in the equilibrated simulation trajectories of wild-type, R178C, and P32T.
3.5. R178C ITPA catalytic activity
The specific activity for wild-type ITPA prepared using the standard and urea refolding purification scheme and refolded R187C ITPA was determined. Side-by side specific activity measurements for standard wild-type, refolded wild-type, and refolded R178C ITPA proteins were performed as described in materials and methods. We observed that refolded wild-type protein had approximately four-fold less activity than wild-type prepared from our standard purification scheme (Gall et al., Citation2013). Comparing refolded wild-type to refolded R178C, we observed a nearly two-hundred fold reduction in activity for the R178C mutant (). This is consistent with our previous determination of Arg-178 as essential for enzyme activity (Gall et al., Citation2013). In contrast, the P32T mutant was observed to have a roughly two-fold reduction for the rate of catalysis and specificity constant (Herting et al., Citation2010).
Table 2. Specific activity measurements.
4. Discussion
Over the last decade, the pharmacogenetic significance of ITPA status has been well established. Altogether, nine clinically relevant ITPA polymorphisms/mutations have been identified (Burgis, Citation2016; Handley et al., Citation2019; Kaur, Neethukrishna, Kumble, Girisha, & Shukla, Citation2019). Two of these, P32T and R178C, were investigated here. It has been proposed, based on structural and biochemical data, that the P32T is a homodimer in solution and shifts the position of the α-helix 2 (Simone et al., Citation2013; Stenmark et al., Citation2007). The structural features of the R178C mutant have not been reported. Comparing this study to others (Gall et al., Citation2013; Herting et al., Citation2010; Kevelam et al., Citation2015), indicates that the R178C mutant has much less ITP hydrolyzing activity than the P32T protein. To the best of our knowledge, this is the first time that molecular dynamics simulation has been used to better understand how these two mutations affect the structural and dynamic behavior of ITPA and understand the molecular basis for the degree of reduction of the activity of ITPA by R178C and P32T mutations. Moreover, this is the first report we are aware of which investigates biochemical attributes of the R178C mutant protein.
The biochemical data presented here show that the R178C mutant has very low enzyme activity. Because Kevelam et al. (Citation2015) had such a limited number of samples (only one R178C patient), and assays were performed with whole cell lysates, it is important to replicate their findings in vitro using modern molecular biology/biochemistry techniques. Considering that a cysteine residue is being introduced into the native structure, it is possible that this could result in improper disulfide bond formation and an altered structure. The difficulties we encountered during preliminary purification of the R178C ITPA using the standard protein purification scheme were overcome by addition of 8 M urea, however, solubility was not altered by the addition of 2-mercaptoethanol (unpublished data). This supports that the mutant protein has a native-like fold, with reduced stability, and no improper disulfide bond formation.
Molecular dynamics simulations offer useful insights into protein structure and dynamics that often play important roles in ligand binding (Durrant & McCammon, Citation2011; Rosales-Hernández & Correa-Basurto, Citation2015). Our MD simulations have been produced over a reasonably long time period of equilibrated trajectories, which allows us to gain insight into the structural basis of the reduction of stability of ITPA R178C and P32T mutants. We have found by analyzing the RMSD, the Rg, the variation with time of the secondary structure elements, as well as the molecular view and alignments of the final simulation structures that the wild-type and both mutated proteins maintained in general their secondary structure elements during the simulations. Considering the P32T mutant, the simulation results agree with previously published structural data, namely that the P32T alteration retains the dimeric structure of wild-type in solution and causes the rearrangement of α-helix 2 (Simone et al., Citation2013; Stenmark et al., Citation2007) (, S1 and S2). In addition, this study has shown that the R178C in solution also retains the secondary structure of the wild-type (, S1 and S2).
The RMSF, RMSD cluster and PCA analyses suggest that both the R178C and P32T mutants exhibit larger overall flexibility than that of the wild-type, with the R178C undergoing the largest overall flexibility. Additionally, we have found in the wild-type that the α-helices 2 and 7 exhibit significant fluctuations (). This is consistent with experimental results, where residues of these helices close in upon substrate binding (Porta et al., Citation2006; Stenmark et al., Citation2007) which implies a degree of flexibility of the subunits. α-helix 2 includes active-site residue 19 and a change in the flexibility of this helix may affect enzymatic activity. Interestingly, both mutations increase the flexibility of the helix 2. The analysis of the distance fluctuations revealed that each monomer of the wild-type and the mutants move in rigid-body manner. Nevertheless, the motions between monomers are less coordinated in the wild-type as well as in the mutants and substantial increase of these motions are observed in the R178C protein. Additionally, the analysis of principal components overlap suggests a major change in ITPA motion due to the R178C mutation. Porcupine plots based on the first principal component analysis show that both mutations significantly reduced the motions in α-helices 2 and 7. Note that α-helix 7 includes active-site residue 172. The dominant collective motions in the mutants are located in the N-and-C-termini and the R178C displays the largest amplitude. Hence, these analyses support the experimental finding that the catalytic activity is more reduced in R178C than in P32T.
Additional information concerning the effect of mutations on molecular interactions was gained by analyzing the hydrogen bonding interactions in the wild-type and mutants. The R178C and P32T mutations have impacted the total number of intramolecular hydrogen bonds formed in the simulation trajectories (Figure S7) which may affect substrate binding and in turn lead to a reduction of enzyme activity. In our simulations, the results of the total hydrogen bond analysis are consistent with the conclusion of the fluctuations analysis in that the R178C mutant could experience more diminution of the enzymatic activity than the P32T protein. Moreover, the hydrogen bond occupancy analysis found that the R178C mutation has suppressed the active-site interactions of Lys172 with Asp152 as well as residue 178 with Glu22, and Glu71, while the P32T mutation leads to the loss of the interaction of Lys19 with Glu71 in the active site ( and ). We have categorized, based on hydrogen bond occupancy, the residues with reduced interactions as essential (more than 75% occupancy) and as intermediate (less than 75% occupancy) for ITPA activity. We have thus classified Arg178, Glu22, and Glu71 as essential and Asp152 and Lys172 as intermediate in importance for ITPA activity. Interestingly, this classification is in good agreement with the classification for Arg178, Glu22, Asp152 and Lys172 in Gall et al. (Citation2013). Note that residue Glu71 had not been investigated in Gall et al. (Citation2013). Hence, the hydrogen bonding occupancy analysis of the residues in the active site indicates that the two mutations have modified the electrostatic interactions between donor and acceptor atoms which may explain the loss of enzymatic activity of the two mutants with the larger reduction in the R178C mutant. This result agrees with the total number of intramolecular hydrogen bonds analysis.
In conclusion, we have used extensive MD simulations to investigate the structural and dynamics features of ITPA dimer and two of its clinical mutants, namely R178C and P32T. Maintenance of wild-type secondary elements and the shift of helix 2 were observed in the simulations of the mutants. We have found that both mutations change most of the properties investigated and R178C shows the largest change, which could explain the experimental report that the R178C mutation decreases more the catalytic activity than the P32T mutation. The MD simulations reveal that R178C and P32T mutations alter the direction and the amplitude of the motions of the upper lobe as well as the lower lobe helix 2. Additionally, we have classified key hydrogen interactions found in the active-site. These data underscore the contribution of protein stability to the development of clinical symptoms for affected patients. Our study will be useful for developing therapeutic aptamers to improve in vivo stabilization of ITPA proteins in affected patients.
Supplementary-J-Biomol-Struct-Dyn_YH_NEB_revised_2.pdf
Download PDF (47.9 MB)Acknowledgment
Y.H. thanks Prof. K. Kuczera for his constructive suggestions.
Disclosure statement
No potential conflict of interest was reported by the author(s).
ORCID
Yao Houndonougbo [oricid logo] http://orcid.org/0000-0003-0434-3521
Nicholas Burgis [orcid logo] http://orcid.org/0000-0002-8279-7193
Additional information
Funding
Related Research Data
References
- Abolhassani, N., Iyama, T., Tsuchimoto, D., Sakumi, K., Ohno, M., Behmanesh, M., & Nakabeppu, Y. (2010). NUDT16 and ITPA play a dual protective role in maintaining chromosome stability and cell growth by eliminating dIDP/IDP and dITP/ITP from nucleotide pools in mammals. Nucleic Acids Research, 38(9), 2891–2903. doi:10.1093/nar/gkp1250
- Amadei, A., Linssen, A. B. M., & Berendsen, H. J. C. (1993). Essential dynamics of proteins. Proteins: Structure, Function, and Genetics, 17(4), 412–425. doi:10.1002/prot.340170408
- Barth, E., Kuczera, K., Leimkuhler, B., & Skeel, R. (1995). Algorithms for constrained molecular dynamics. Journal of Computational Chemistry, 16(10), 1192–1209. doi:10.1002/jcc.540161003
- Best, R. B., Zhu, X., Shim, J., Lopes, P. E. M., Mittal, J., Feig, M., & MacKerell, A. D., Jr. (2012). Optimization of the additive CHARMM all-atom protein force field targeting improved sampling of the backbone ϕ, ψ and side-chain χ1 and χ2 dihedral angles. Journal of Chemical Theory and Computation, 8(9), 3257–3273. doi:10.1021/ct300400x
- Bierau, J., Lindhout, M., & Bakker, J. A. (2007). Pharmacogenetic significance of inosine triphosphatase. Pharmacogenomics, 8(9), 1221–1228. doi:10.2217/14622416.8.9.1221
- Boopathi, S., & Kolandaivel, P. (2017). Effect of mutation on A beta(1-42)-Heme complex in aggregation mechanism: Alzheimer’s disease. Journal of Molecular Graphics and Modelling, 76, 224–233. doi:10.1016/j.jmgm.2017.06.016
- Brooks, B. R., Bruccoleri, R., Olafson, B., States, D., Swaminathan, S., & Karplus, M. (1983). CHARMM: A program for macromolecular energy, minimization and dynamics calculations. Journal of Computational Chemistry, 4(2), 187–217. doi:10.1002/jcc.540040211
- Burgis, N. E. (2016). A disease spectrum for ITPA variation: Advances in biochemical and clinical research. Journal of Biomedical Science, 23(1), 13. doi:10.1186/s12929-016-0291-y
- Burgis, N. E., Brucker, J. J., & Cunningham, R. P. (2003). Repair system for noncanonical purines in Escherichia coli. Journal of Bacteriology, 185(10), 3101–3110. doi:10.1128/jb.185.10.3101-3110.2003
- Burgis, N. E., & Cunningham, R. P. (2006). Substrate specificity of RdgB protein, a deoxyribonucleoside triphosphate pyrophosphohydrolase. Journal of Biological Chemistry, 282(6), 3531–3538. doi:10.1074/jbc.M608708200
- Chung, J., Park, H., Lee, J., & Jang, Y. (2002). Identification of the dITP- and XTP-hydrolyzing protein from Escherichia coli. Journal of Biochemistry and Molecular Biology, 35, 403–408. doi:10.5483/BMBRep.2002.35.4.403
- Das, S., Santra, S., Rohman, M. A., Ray, M., Jana, M., & Singha Roy, A. (2019). An insight into the binding of 6-hydroxyflavone with hen egg white lysozyme: A combined approach of multi-spectroscopic and computational studies. Journal of Biomolecular Structure and Dynamics, 37(15), 4019–4034. doi:10.1080/07391102.2018.1535451
- Daura, X., Gademann, K., Jaun, B., Seebach, D., Gunsteren, W. F. V., & Mark, A. E. (1999). Peptide folding: When simulation meets experiment. Angewandte Chemie International Edition, 38(1–2), 236–240. doi:10.1002/(SICI)1521-3773(19990115)38:1/2<236::AID-ANIE236>3.0.CO;2-M
- Durrant, J. D., & McCammon, J. A. (2011). Molecular dynamics simulations and drug discovery. BMC Biology, 9(1), 1–9. doi:10.1186/1741-7007-9-71
- Fellay, J., Thompson, A. J., Ge, D., Gumbs, C. E., Urban, T. J., Shianna, K. V., … Goldstein, D. B. (2010). ITPA gene variants protect against anaemia in patients treated for chronic hepatitis C. Nature, 464(7287), 405–408. doi:10.1038/nature08825
- Fukuyoshi, S., Kometani, M., Watanabe, Y., Hiratsuka, M., Yamaotsu, N., Hirono, S., … Oda, A. (2016). Molecular dynamics simulations to investigate the influences of amino acid mutations on protein three-dimensional structures of cytochrome P450 2D6.1, 2, 10, 14A, 51, and 62. PLoS One, 11(4), e0152946. doi:10.1371/journal.pone.0152946
- Gall, A. D., Gall, A., Moore, A. C., Aune, M. K., Heid, S., Mori, A., & Burgis, N. E. (2013). Analysis of human ITPase nucleobase specificity by site-directed mutagenesis. Biochimie, 95(9), 1711–1721. doi:10.1016/j.biochi.2013.05.016
- Galperin, M., Moroz, O., Wilson, K., & Murzin, A. (2006). House cleaning, a part of good housekeeping. Molecular Microbiology, 59(1), 5–19. doi:10.1111/j.1365-2958.2005.04950.x
- Handley, M. T., Reddy, K., Wills, J., Rosser, E., Kamath, A., Halachev, M., … FitzPatrick, D. R. (2019). ITPase deficiency causes a Martsolf-like syndrome with a lethal infantile dilated cardiomyopathy. PLoS Genetics, 15(3), e1007605. doi:10.1371/journal.pgen.1007605
- Herting, G., Barber, K., Zappala, M. R., Cunningham, R. P., & Burgis, N. E. (2010). Quantitative in vitro and in vivo characterization of the human P32T mutant ITPase. Biochimica et Biophysica Acta, 1802(2), 269–274. doi:10.1016/j.bbadis.2009.11.002
- Hess, B. (2008). P-LINCS: A parallel linear constraint solver for molecular simulation. Journal of Chemical Theory and Computation, 4(1), 116–122. doi:10.1021/ct700200b
- Hoover, W. G. (1985). Canonical dynamics: Equilibrium phase-space distributions. Physical Review A, 31(3), 1695–1697. doi:10.1103/PhysRevA.31.1695
- Houndonougbo, Y., Kuczera, K., & Jas, G. S. (2005). Structure and dynamics of phospholamban in solution and in membrane bilayer: Computer simulations. Biochemistry, 44(6), 1780–1792. doi:10.1021/bi0488404
- Houndonougbo, Y., Kuczera, K., & Jas, G. S. (2008). Effects of CMAP and electrostatic cutoffs on the dynamics of an integral membrane protein: The phospholamban study. Journal of Biomolecular Structure and Dynamics, 26(1), 17–34. doi:10.1080/07391102.2008.10507220
- Iyama, T., Abolhassani, N., Tsuchimoto, D., Nonaka, M., & Nakabeppu, Y. (2010). NUDT16 is a (deoxy)inosine diphosphatase, and its deficiency induces accumulation of single-strand breaks in nuclear DNA and growth arrest. Nucleic Acids Research, 38(14), 4834–4843. doi:10.1093/nar/gkq249
- Jorgensen, W. L., Chandrasekhar, J., Madura, J. D., Impey, R. W., & Klein, M. L. (1983). Comparison of simple potential functions for simulating liquid water. Journal of Chemical Physics, 79(2), 926–935. doi:10.1063/1.445869
- Kaur, P., Neethukrishna, K., Kumble, A., Girisha, K. M., & Shukla, A. (2019). Identification of a novel homozygous variant confirms ITPA as a developmental and epileptic encephalopathy gene. American Journal of Medical Genetics Part A, 179(5), 857–861. doi:10.1002/ajmg.a.61103
- Kevelam, S. H., Bierau, J., Salvarinova, R., Agrawal, S., Honzik, T., Visser, D., … van der Knaap, M. S. (2015). Recessive ITPA mutations cause an early infantile encephalopathy. Annals of Neurology, 78(4), 649–658. doi:10.1002/ana.24496
- Lin, S., McLennan, A., Ying, K., Wang, Z., Gu, S., Jin, H., … Mao, Y. (2001). Cloning, expression, and characterization of a human inosine triphosphate pyrophosphatase encoded by the ITPA gene. Journal of Biological Chemistry, 276(22), 18695–18701. doi:10.1074/jbc.M011084200
- Mahadevan, J., Xu, C., Siahaan, T., & Kuczera, K. (2002). Molecular dynamics simulations of conformational behavior of linear RGD peptidomimetics and cyclic prodrugs in aqueous and octane solutions. Journal of Biomolecular Structure and Dynamics, 19(5), 775–788. doi:10.1080/07391102.2002.10506784
- Marinaki, A., Duley, J., Arenas, M., Ansari, A., Sumi, S., Lewis, C., … Sanderson, J. (2004). Mutation in the ITPA gene predicts intolerance to azathioprine. Nucleosides Nucleotides and Nucleic Acids, 23(8–9), 1393–1397. doi:10.1081/NCN-200027639
- Martínez-Archundia, M., Correa-Basurto, J., Montaño, S., & Rosas-Trigueros, J. L. (2019). Studying the collective motions of the adenosine A2A receptor as a result of ligand binding using principal component analysis. Journal of Biomolecular Structure and Dynamics, 37(18), 4685–4700. doi:10.1080/07391102.2018.1564700
- Martínez-Rosell, G., Giorgino, T., & De Fabritiis, G. (2017). PlayMolecule ProteinPrepare: A web application for protein preparation for molecular dynamics simulations. Journal of Chemical Information and Modeling, 57(7), 1511–1516. doi:10.1021/acs.jcim.7b00190
- Miyamoto, S., & Kollman, P. A. (1992). Settle: An analytical version of the SHAKE and RATTLE algorithm for rigid water models. Journal of Computational Chemistry, 13(8), 952–962. doi:10.1002/jcc.540130805
- Nagarajan, S., Alkayed, N. J., Kaul, S., & Barnes, A. P. (2020). Effect of thermostable mutations on the neurotensin receptor 1 (NTSR1) activation state. Journal of Biomolecular Structure and Dynamics, 38(2), 340–353. doi:10.1080/07391102.2019.1573705
- Narang, S. S., Shuaib, S., Goyal, D., & Goyal, B. (2018). Assessing the effect of D59P mutation in the DE loop region in amyloid aggregation propensity of 2-microglobulin: A molecular dynamics simulation study. Journal of Cellular Biochemistry, 119(1), 782–792. doi:10.1002/jcb.26241
- Nose, S. (1984). A molecular dynamics method for simulation in the canonical ensemble. Molecular Physics, 52, 255–268.
- Noskov, V., Staak, K., Shcherbakova, P., Kozmin, S., Negishi, K., Ono, B., … Pavlov, Y. (1996). HAM1, the gene controlling 6-N-hydroxylaminopurine sensitivity and mutagenesis in the yeast Saccharomyces cerevisiae. Yeast, 12(1), 17–29. doi:10.1002/(SICI)1097-0061(199601)12:1<17::AID-YEA875>3.0.CO;2-I
- Pang, B., McFaline, J. L., Burgis, N. E., Dong, M., Taghizadeh, K., Sullivan, M. R., … Dedon, P. C. (2012). Defects in purine nucleotide metabolism lead to substantial incorporation of xanthine and hypoxanthine into DNA and RNA. Proceedings of the National Academy of Sciences of the United States of America, 109(7), 2319–2324. doi:10.1073/pnas.1118455109
- Parrinello, M., & Rahman, A. (1981). Polymorphic transitions in single crystals: A new molecular dynamics method. Journal of Applied Physics, 52(12), 7182–7190. doi:10.1063/1.328693
- Peltenburg, N. C., Bierau, J., Bakker, J. A., Schippers, J. A., Lowe, S. H., Paulussen, A. D. C., … Verbon, A. (2018). Erythrocyte Inosine triphosphatase activity: A potential biomarker for adverse events during combination antiretroviral therapy for HIV. PLoS One, 13(1), e0191069. doi:10.1371/journal.pone.0191069
- Porta, J., Kolar, C., Kozmin, S. G., Pavlov, Y. I., & Borgstahl, G. E. O. (2006). Structure of the orthorhombic form of human inosine triphosphate pyrophosphatase. Acta Crystallographica Section F: Structural Biology and Crystallization Communications, 62(11), 1076–1081.
- Roberts, E., Eargle, J., Wright, D., & Luthey-Schulten, Z. (2006). MultiSeq: Unifying sequence and structure data for evolutionary analysis. BMC Bioinformatics, 7, 382. doi:10.1186/1471-2105-7-382
- Rosales-Hernández, M. C., & Correa-Basurto, J. (2015). The importance of employing computational resources for the automation of drug discovery. Expert Opinion on Drug Discovery, 10(3), 213–219. doi:10.1517/17460441.2015.1005071
- Sagui, C., & Darden, T. A. (1999). Molecular dynamics simulations of biomolecules: Long-range electrostatic effects. Annual Review of Biophysics and Biomolecular Structure, 28(1), 155–179. doi:10.1146/annurev.biophys.28.1.155
- Saldaña-Rivera, L., Bello, M., & Méndez-Luna, D. (2019). Structural insight into the binding mechanism of ATP to EGFR and L858R, and T790M and L858R/T790 mutants. Journal of Biomolecular Structure and Dynamics, 37(17), 4671–4684. doi:10.1080/07391102.2018.1558112
- Sambrook, J., Fritsch, E.F., & Maniatis, T. (Eds.). (1989). Molecular cloning: A laboratory manual (2nd ed.).Cold Spring Harbor, NY: Cold Spring Harbor Laboratory Press.
- Shipkova, M., Franz, J., Abe, M., Klett, C., Wieland, E., & Andus, T. (2011). Association between adverse effects under azathioprine therapy and inosine triphosphate pyrophosphatase activity in patients with chronic inflammatory bowel disease. Therapeutic Drug Monitoring, 33(3), 321–328. doi:10.1097/FTD.0b013e31821a7c34
- Simone, P. D., Struble, L. R., Kellezi, A., Brown, C. A., Grabow, C. E., Khutsishvili, I., … Borgstahl, G. E. O. (2013). The human ITPA polymorphic variant P32T is destabilized by the unpacking of the hydrophobic core. Journal of Structural Biology, 182(3), 197–208. doi:10.1016/j.jsb.2013.03.007
- Stenmark, P., Kursula, P., Flodin, S., Gräslund, S., Landry, R., Nordlund, P., & Schüler, H. (2007). Crystal structure of human inosine triphosphatase – Substrate binding and implication of the inosine triphosphatase deficiency mutation P32T. Journal of Biological Chemistry, 282(5), 3182–3187. doi:10.1074/jbc.M609838200
- Stepchenkova, E. I., Tarakhovskaya, E. R., Spitler, K., Frahm, C., Menezes, M. R., Simone, P. D., … Pavlov, Y. I. (2009). Functional study of the P32T ITPA variant associated with drug sensitivity in humans. Journal of Molecular Biology, 392(3), 602–613. doi:10.1016/j.jmb.2009.07.051
- Tugba, G. K., & Emil, A. (2015). Structural, dynamical, and energetical consequences of Rett syndrome mutation R133C in MeCP2. Computational and Mathematical Methods in Medicine, 2015, 746157.
- Van der Spoel, D., Lindahl, E., Hess, B., Groenhof, G., Mark, A., & Berendsen, H. (2005). GROMACS: Fast, flexible, and free. Journal of Computational Chemistry, 26(16), 1701–1718. doi:10.1002/jcc.20291
- Vanderheiden, B. S. (1970). Human erythrocyte “ITPase”: An ITP pyrophosphohydrolase. Biochimica et Biophysica Acta, 215(3), 555–558. doi:10.1016/0304-4165(70)90109-1
- Wan, Y., Guan, S., Qian, M., Huang, H., Han, F., Wang, S., & Zhang, H. (2020). Structural basis of fullerene derivatives as novel potent inhibitors of protein acetylcholinesterase without catalytic active site interaction: Insight into the inhibitory mechanism through molecular modeling studies. Journal of Biomolecular Structure and Dynamics, 38(2), 410–425. doi:10.1080/07391102.2019.1576543
- Yu, Z., Liu, Y., Zhu, J., Han, J., Tian, X., Han, W., & Zhao, L. (2020). Insights from molecular dynamics simulations and steered molecular dynamics simulations to exploit new trends of the interaction between HIF-1α and p300. Journal of Biomolecular Structure and Dynamics, 38(1), 1–12. doi:10.1080/07391102.2019.1580616
- Zhang, H., He, X., Ni, D., Mou, L., Chen, X., & Lu, S. (2020). How does the novel T315L mutation of breakpoint cluster region-abelson (BCR-ABL) kinase confer resistance to ponatinib: A comparative molecular dynamics simulation study. Journal of Biomolecular Structure and Dynamics, 38(1), 89–100. doi:10.1080/07391102.2019.1567390