
 ?Mathematical formulae have been encoded as MathML and are displayed in this HTML version using MathJax in order to improve their display. Uncheck the box to turn MathJax off. This feature requires Javascript. Click on a formula to zoom.
?Mathematical formulae have been encoded as MathML and are displayed in this HTML version using MathJax in order to improve their display. Uncheck the box to turn MathJax off. This feature requires Javascript. Click on a formula to zoom.Abstract
During biological events, the water molecules associated with the protein are re-oriented to adapt to the new conditions, inducing changes in the system’s free energy. The characterization of water structure and thermodynamics may facilitate the prediction of certain biological events, such as the binding of a ligand and the membrane-associated parts of a protein. In this computational study, we calculated the hydration thermodynamics of cytosolic phospholipase A2 group IV (GIVA cPLA2) to study the hydration properties of the protein’s surface and binding pocket. Hydrophobicity scales and the Grid Inhomogeneous Solvation Theory (GIST) tool were employed for the calculations. The hydrophobic areas of the protein’s surface were predicted more accurately with the GIST method rather than with the hydrophobicity scales. Based on this, a model of the protein-membrane complex was constructed. In addition, the calculation revealed the highly hydrated binding pocket that further contribute to our understanding of the ligands’ binding.
Communicated by Ramaswamy H. Sarma
Introduction
Phospholipases A2 (PLA2s) is a super-family of enzymes that cleaves a fatty acid from the sn-2 position of phospholipids (Dennis, Cao, Hsu, Magrioti, & Kokotos, Citation2011). The products of this enzymatic reaction are metabolized through several biological paths (e.g. COX, LOX), promoting the synthesis of inflammatory mediators. The exact conditions that promote the upregulation of these enzymes and the interactions between the isoforms are still under investigation. The selective inhibition of PLA2s has long been a promising approach for the elucidation of their contribution to the mechanism that leads to inflammation and the discovery of an effective treatment. However, both the high homology of the enzymes belonging to the same group and the similarities of their catalytic mechanism, make this target challenging.
In this work, we focused on group IVA of cytosolic phospholipase A2 (GIVA cPLA2) (Leslie, Citation2015). This enzyme is the most well studied among the cytosolic PLA2s and its Ca2+-dependence for activation and substrate preference for arachidonic acid (Dennis et al., Citation2011) characterizes it. The enzyme is located in the cytosol. There is a translocation of the enzyme close to the cell membrane upon its activation by two calcium ions, which bind in the sub-domain of the enzyme. The translocation is necessary for the enzyme to reach the substrate and therefore its association with the cell membrane is a crucial step for its catalytic activity. Hydrogen/deuterium exchange mass spectrometry (DXMS) has been employed in the past for the description of the GIVA cPLA2 orientation on the membrane and the interactions formed in the interfacial binding region (Mouchlis, Bucher, McCammon, & Dennis, Citation2015). However, this type of analysis is not available for every membrane-associated enzyme and theoretical calculations are usually employed in order to predict the parts of proteins that interact with the cell membrane.
Known GIVA cPLA2 inhibitors are characterized by a diversity of the chemical groups they belong to, as it has been described in a number of reviews (Kokotou, Limnios, Nikolaou, Psarra, & Kokotos, Citation2017; Nikolaou, Kokotou, Vasilakaki, & Kokotos, Citation2019). More specifically, pyrrolidine-based inhibitors (Seno et al., Citation2000) and indole-based inhibitors (Lee et al., Citation2007; McKew et al., Citation2006) are potent inhibitors of GIVA cPLA2 and some of them advanced to clinical trials (http://ClinicalTrials Identifier: NCT00396955 (Citation2006) and NCT02795832 (Citation2016)). In addition, novel inhibitors belonging to indole series have been designed and studied by Tomoo et al. (Citation2014) and demonstrate some promising results. Activated carbonyl-based indol-1-yl-propan-2-ones inhibitors (Bovens et al., Citation2010; Ludwig, Bovens, Brauch, Elfringhoff, & Lehr, Citation2006) have also been systematically studied by Lehr and co-workers (Bovens et al., Citation2010), while Kokotos and co-workers have developed a series of 2-oxoamides derivatives (Kokotos et al., Citation2004; Six et al., Citation2007) thiazolyl ketones (Kokotos et al., Citation2014) and 2-oxoesters (Kokotou, Galiatsatou, et al., Citation2017), which demonstrate potent and selective inhibition against PLA2s. However, the development of optimized lead compounds for the inhibition of GIVA cPLA2 action is still an active field of research.
The only available crystallographic structure of GIVA cPLA2 is of the apo form of the enzyme (Dessen et al., Citation1999). Due to the lack of any co-crystalized X-ray ligand structure, the binding mode of its inhibitors is unknown, which make the design of new inhibitors challenging. Additionally, the lid region is closed in the apo form and the binding mode of few inhibitors has been defined using DXMS, a method that does not provide details about the conformation of the ligand (Burke et al., Citation2009). Therefore, only computational simulations (Burke et al., Citation2008) may describe the structural rearrangement of the binding pocket upon ligand’s insertion.
Both events, the GIVA cPLA2’s binding on the cell membrane and the formation of the ligand-protein complex, are partly driven by hydration effects. Here, we have addressed both events by computationally calculating the hydration properties of the protein’s surface and binding region. We aim to understand the role of hydrophilic and hydrophobic forces lead to the assembly of the protein-membrane complex and the hydration properties of the binding pocket.
Hydrophobicity scales have been employed in the past to assign hydrophobicity values to amino acids and predict the hydrophobic domains of proteins. Various hydrophobicity scales have been published (Cornette et al., Citation1987; Eisenberg, Schwarz, Komaromy, & Wall, Citation1984; Engelman, Steitz, & Goldman, Citation1986; Hopp & Woods, Citation1983; Janin, Citation1979; Kyte & Doolittle, Citation1982; Rose, Geselowitz, Lesser, Lee, & Zehfus, Citation1985), but the complete range of an amino acid behavior is difficult to be described by one parameter, and therefore the results are discordant. Lately, progress has been made to improve the accuracy of such scales by using modern computational methods (Nicolau, Paszek, Fulga, & Nicolau, Citation2014; Schauperl, Podewitz, Waldner, & Liedl, Citation2016). Increasing the resolution by replacing the amino acid values by atomic values is one parameter that has been introduced in the development of these scales. In addition, the environment of each amino acid, which includes other amino acids and their effect on the surrounding water molecules, has also been considered.
In this work, we assigned hydrophobicity values on the amino acids based on the calculation of the thermodynamic properties of the surrounding water molecules. We implemented this using the GIST (Nguyen, Cruz, Gilson, & Kurtzman, Citation2014; Nguyen, Gilson, & Young, Citation2011; Nguyen, Kurtzman, & Gilson, Citation2012; Raman & MacKerell, Citation2013) tool in AmberTools, an application of the inhomogeneous fluid solvation theory (IFST) on biomolecules (Green, Citation1952; Huggins, Citation2012; Lazaridis, Citation1998; Li & Lazaridis, Citation2006). GIST divides a defined solvent area in a 3D grid, which consists of small grid boxes. It discretizes the equations of IFST to approximate the localized thermodynamic properties of these small grid boxes. The calculation of the localized thermodynamic properties, including entropy and enthalpy, is an important feature of GIST.
Herein, we report the application of hydrophobicity scales and a method based on GIST analysis to map the hydrophobic areas of GIVA cPLA2 surface in order to predict the hydrophobic areas of the protein’s surface that may interact with the membrane. We also describe the thermodynamic properties of the solvent areas in the active site of GIVA cPLA2 that may affect ligands’ binding.
Computational methods
The only available crystallographic structure of GIVA cPLA2 (PDB ID: 1CJY) (Dessen et al., Citation1999) was used for the calculations. The missing amino acids of the apo structure were added in Chimera (Pettersen et al., Citation2004), using Modeller 9.17 (Sali & Blundell, Citation1993). The charmm-gui on-line tool was used for building the lipid bilayer of 1-palmitoyl-2-oleoyl-sn-glycero-3-phosphocholine (POPC) and placing the protein on the lipids surface (), according to a previously published model created on the basis of DXMS data (Burke et al., Citation2009).
Figure 1. GIVA cPLA2 on a lipid bilayer of POPC generated with the charmm-gui tool.
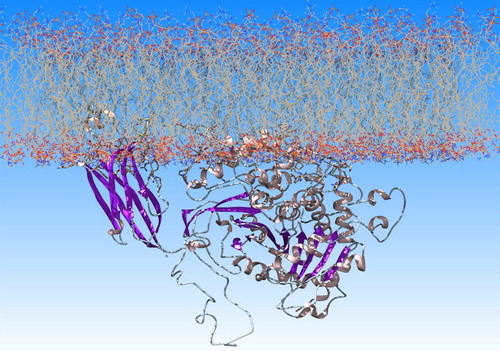
All atoms unrestrained MD simulations in explicitly solvent were performed with AMBER (Case et al., Citation2016). The one system we simulated was the apo GIVA cPLA2 in solvent (named ‘the apo system’ in this paper) and the other was the GIVA cPLA2-membrane complex in solvent (named ‘the membrane system’). The crystal water molecules and the two calcium ions were retained, because calcium ions keep the waters ordered and create interactions with the membrane, which contribute to the formation of the complex. In addition, a water molecule close to the catalytic dyad has been resolved in the X-ray structure. This water may be involved in the catalytic mechanism and therefore it was important for our calculations to keep it. tLEaP module and lipid14 (Dickson et al., Citation2014) and ff14SB (Maier et al., Citation2015) force fields were used to produce the parameter files. Systems were solvated in tLEaP with approximately 37,000 (the apo system) and 62,000 (the membrane system) water molecules. The TIP3P water model was used to describe the properties of the solvent. A truncated octahedral water box with 12.0 Å periodic boundary conditions and a nonbonded cutoff distance of 8 Å were used for the apo system. The solvation of the membrane system was accomplished with the AddToBox program and SetBox command in tLEaP to create a periodic system of {133.769 134.537 178.282} dimensions in Å. The nonbonded cutoff distance was set at 10 Å. Both systems were neutralized by adding 28 in the apo and 36 in the membrane system Na+ ions using the Joung and Cheatham ion parameters (Joung & Cheatham, Citation2008).
Consequently, we performed minimization with SANDER (Salomon-Ferrer, Case, & Walker, Citation2012). For the apo system, a four-step energy minimization workflow including the steepest descent algorithm followed by the conjugate gradient method was set. During the first step, the solute was kept fixed with a harmonic force constant of 500 kcal·mol−1·Å−1. We applied two additional minimization steps during of which the strength of the restraint was gradually reduced. For the membrane system, a four-step energy minimization workflow was also set: 10,000 steps of the steepest descent algorithm and 5,000 steps of conjugate gradient with the solute constrained and 40,000 steps of the steepest descent algorithm and 10,000 steps of conjugate gradient unconstrained. The solute was kept fixed with a harmonic force constant of 500 kcal·mol−1·Å−1. Minimization is necessary to reduce any bad contacts and allow the solvent and sidechains to reach an optimal orientation.
The systems were heated from 0 to 310 K in three steps under constant volume and with a reduced restraint of 10 kcal·mol−1·Å−1. The SHAKE algorithm was applied and the Langevin thermostat with a collision frequency of γ = 2.0 ps−1 for the apo system and 1.0 ps−1 for the membrane system was used. Equilibration was performed in the isothermal isobaric (NPT) ensemble at 310 K and 1 atm pressure. For the apo system, the equilibration was performed in two steps: one with the solute restrained and only the water molecules allowed to move and one with the entire system unrestrained. The total time of equilibration was 200 ps and the Berendsen barostat was utilized for pressure control with isotropic position scaling (ntp = 1). For the membrane system, equilibration was performed for 1.2 ns in four sequential steps, keeping the protein and lipid atoms fixed and 5 ns keeping only the protein fixed. The Monte Carlo barostat was utilized for pressure control with anisotropic pressure scaling (ntp = 2). The long-range electrostatic interactions were accounted for using the particle mesh Ewald (PME) method (Darden, York, & Pedersen, Citation1993). The pmemd.cuda (Salomon-Ferrer, Götz, Poole, Le Grand, & Walker, Citation2013) engine of AMBER was used for the heating, equilibration and production time. The total time of the unconstrained MD simulation was 300 ns.
The MD trajectories were clustered and the top three clusters were further proceeded for a GIST analysis. We repeated the GIST analysis three times to allow a better sampling of the analysis and increased the accuracy of the results. Clustering was performed using AmberTools. The bottom-up clustering method was used and distance between frames calculated via best-fit coordinate RMSD using residues 182-186, 214, 218, 248-251, 253, 277, 281, 382-384, 387, 390, 403, 407, 535-537, 541, 564, 664 and 666-670, which define the lid region. Each cluster was then prepared and simulated for 20 ns keeping the solute constrained (harmonic force constant of 10 kcal·mol−1·Å−1) and allowing only the waters to move. It is suggested (Ramsey et al., Citation2016) to keep the solute of interest restrained in the lab frame, according to the source particle method used in IST (Lazaridis, Citation1998). 10,000 simulation snapshots were generated from each simulation. The GIST analysis was performed with AmberTools17 and the grid spacing was set to 0.5 Å. The grid dimensions along each coordinate axis were set to 150 Å to cover the whole protein surface. The grid points within 5 Å distance from the solute were counted in for determining the thermodynamic properties of the hydrating water molecules. The gistpp post-processing tool (Ramsey et al., Citation2016) and in-house scripts were used for analyzing the GIST output files. For the calculation of the localized values for the free energy, the ΔG values of the grid points around each atom in a distance of 5 Å were integrated and normalized by the number of water molecules at each grid point.
Results
Hydrophobicity scales and GIST analysis of GIVA cPLA2 surface
Hydrophobicity scales have been used in the past to identify the hydrophobic regions of transmembrane proteins that interact with the membrane (Eisenberg et al., Citation1984). Here, we employed well known hydrophobicity scales as well as a method build on the GIST tool to calculate these regions of GIVA cPLA2, based on amino acids’ and water molecules’ thermodynamic properties, respectively. Then, we compared our results with the experimental data to justify the most accurate computational method for predicting the membrane-associated part of the protein.
The hydrophobic surface of GIVA cPLA2 generated by various hydrophobicity scales is presented in by a colour-coded representation. The results generated by the GIST analysis of the apo system are shown in . presents the orientation of the protein on the membrane according to DXMS data.
Figure 2. Amino acids color-coded (red hydrophobic; blue hydrophilic) based on the values of hydrophobicity scales of: (a) Cornette et al. (Citation1987) (−3.1 to 5.7); (b) Eisenberg et al. (Citation1984) (−2.53 to 1.38); (c) Engelman et al. (Citation1986) (−12.3 to 3.7); (d) Hopp and Woods (Citation1983) (−3.4 to 3.0); (e) Janin (Citation1979) (−1.8 to 0.9); (f) Kyte & Doolittle (Citation1982) (−4.5 kcal/mol to 4.5 kcal/mol); (g) Rose et al. (Citation1985) (0.52 to 0.91); (i) Schauperl et al. (Citation2016) (−15 to −5) and (k) the ΔG values of the surrounding water molecules (cut-off 4 Å) calculated using GIST tool (−4kcal/mol to 1 kcal/mol).

Figure 3. Orientation of GIVA cPLA2 in a lipid bilayer according to DXMS data. The description of the hydrophobic areas (red) of the protein surface was produced by the GIST tool. These areas penetrate the membrane and this is in accordance with the DXMS data.
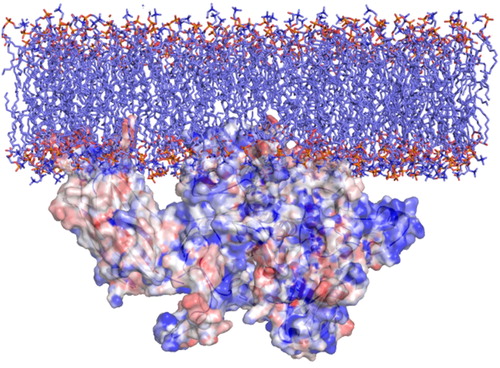
According to DXMS data, the areas of the enzyme which penetrate the cell membrane are addressed by residues 35 to 39 and 96 to 98, belonging to C2 domain, including the residues around the entrance of the tunnel which leads to the catalytic dyad. Based on our GIST calculations, these areas were predicted to be hydrophobic (colored in red, in ) and so expected to penetrate the membrane and interact with the aliphatic parts of it.
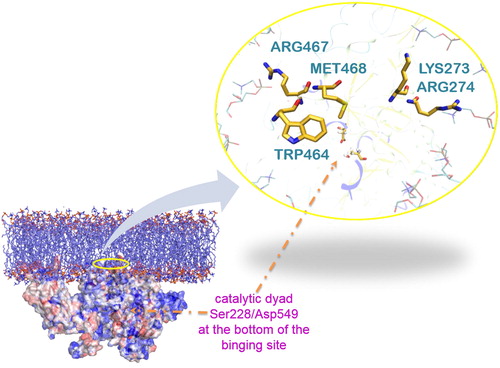
To understand further these results, we studied the interactions formed between the protein and the lipids during the MD simulation of the complex. Based on the simulation, residues at the entrance of the channel form interactions with the aliphatic chains and the polar head groups of the lipids membrane (depending on the type of the amino acids), which stabilize the protein-membrane association (). We think that the combination of the GIST results with the MD simulations may be harnessed to identify regions in proteins that are more likely to bind to a membrane than other methods.
Figure 4. A representative snap shot from the MD simulation of the protein-membrane complex. The entrance of the tunnel (yellow residues) is highlighted with a yellow circle. It leads to the catalytic dyad Ser228/Asp549 shown by the orange arrow. Met468 and Trp464 (yellow) interact with the lipophilic part of lipids (cyan) and Arg467, Lys273 and Arg274 (yellow) with the polar head groups, in accordance with the DXMS data (only some parts of the lipids are represented here (cyan ‘stick’) for clarity reasons).
GIST analysis of the solvent areas in the binding pocket of GIVA cPLA2
The binding pocket of GIVA cPLA2 has the shape of a tunnel, which starts at the protein’s surface and ends at the catalytic dyad. The number of water molecules there is limited, mainly because the tunnel is narrow and consists of lipophilic amino acids. We focused our study on the hydration properties of the binding pocket close to the catalytic dyad. As notice above, only one water molecule was resolved close to the catalytic dyad by the X-ray structure.
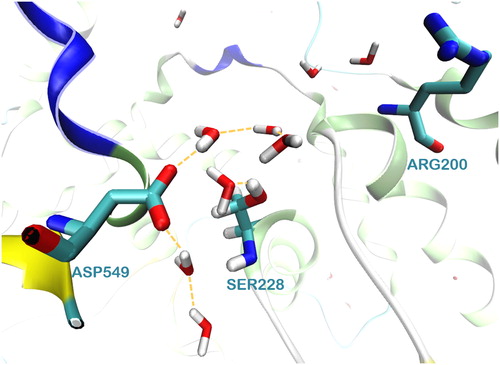
The MD simulations revealed that during the simulation time a well-organized network of waters is formed around the catalytic dyad Ser228/Asp549 ( and Figure S4 (Supplementary material)). It is worth to mention here that the formation of this network was observed in both systems, the apo system and the membrane system. Two more water molecules are located close to Arg200, which according to the catalytic mechanism stabilizes the intermediate product of the reaction.
Figure 5. The network of water molecules around the catalytic dyad Ser228/Asp549 of GIVA cPLA2 formed during a 300 ns MD simulation.
To investigate further the water occupancy of the binding pocket, we calculated the water occupancy around the catalytic serine within 6.5 Å using the watershell command in the AmberTools. In the graphical representation (Supplementary material, S1), the area around the catalytic serine is been occupied by 6 to 7 water molecules for most of the simulation time. These waters make interactions with the catalytic dyad of Ser/Asp and the amino acids Gly551, Asn555, Gly198, Trp393, Thr231, Gly226 and Thr330. The high occupancy map (calculated with GIST) has identified the locations of these hydrophilic centers. Using the ‘closest’ command in AmberTools we obtained the 10 closest residues to the catalytic serine (Supplementary material, S2). The results relieved that the water molecules persist in this area for a considerable simulation time. They do not exchange with the solvent frequently due to the interactions create with the amino acids of the binding pocket.
GIST analysis was applied on the binding pocket of GIVA cPLA2 in the apo system. For the GIST analysis, three clusters, produced by the 300 ns simulation time, were used. ΔG values were calculated based on the GIST analysis:
(1)
(1)
where the
is the sum of the
(water-water enthalpy referenced to the bulk) and
(solute-water enthalpy referenced to the bulk). The
term represents the total entropy. The calculated values of ΔG around the binding pocket are represented as a grid in . The positions of the water molecules are from a representative structure of the most populated cluster.
Figure 6. The values are presented as orange grid, with an energy cutoff of −5 kcal/mol.
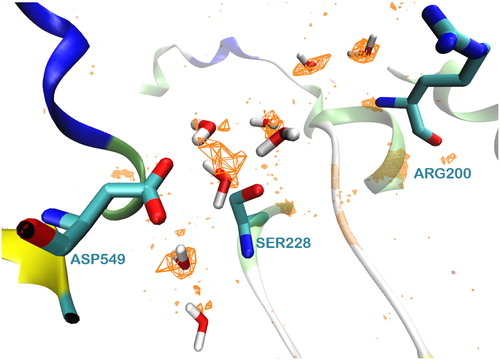
The energy calculations could be used to identify the less favorably bound water molecules. They could be replaced by a part of the ligand, towards a higher binding affinity, as it has been addressed elsewhere (Abel, Young, Farid, Berne, & Friesner, Citation2008; Abel et al., Citation2011). The hydration properties of a binding pocket may be useful in identifying key features that a drug molecule could mimic when binding its target. Therefore, the structure of the ligand should be suitable to replace specific water molecules and mimic their interactions with the key amino acids.
Conclusions
Hydration properties of proteins’ surface play a pivotal role in protein-protein and protein-membrane interactions. Hydration scales have been used in the past in an attempt to predict hydration properties of proteins’ surface, based on the amino acid’s properties. The available hydrophobicity scales assign a fixed value for every type of amino acid, neglecting their individual environments. Hence, these scales fail to describe accurately the energetic contributors of hydration. To accurately describe the hydrophobic character of the individual amino acids, the thermodynamic properties of water molecules around every amino acid have to be investigated including also the influence of local environment e.g. other molecules or amino acids.
In order to account the local difference, we calculated the hydrophobic character per atom of every amino acid using GIST analysis and MD simulations. We apply this method on the surface of the apo structure of GIVA cPLA2 in explicit solvent, in order to identify the hydrophobic points of the surface. We concluded that the prediction of GIVA cPLA2 surface parts, which penetrates the membrane, better meets the previously experimental DXMS data, when made based on the GIST analysis and MD simulations rather than other hydrophobicity scales. The main difference between the methods used by the hydrophobic scales and GIST tool for the calculations is that GIST includes the influence of the surrounding molecules.
GIST uses the discrete sums of the solvation energy and entropy quantities over the voxels of a 3 D grid, in order to estimate a local free energy of solvation for the voxels. The published hydrophobicity scales, referenced in , use a variety of approaches to calculate the amino acids properties. Our results show that by including the solvation energy and entropy terms, assisted by MD simulations, a quantified description of the environment of the amino acids is possible.
Based on the MD simulations of the GIVA cPLA2 – membrane complex, interactions between the lipids and the surface amino acids are formed and stabilize the protein’s orientation in the membrane. These interactions may be used as a target to inhibit enzyme’s activity by inhibiting its attachment to the membrane.
In addition, we analyzed the hydration properties of the binding pocket in an attempt to understand better ligands’ binding. The MD simulation of the apo structure revealed a highly solvated catalytic center. In addition, the tunnel that leads to the catalytic center of the enzyme is narrow and it consist of, mainly, lipophilic amino acids. Given the structure of the natural substrate (membrane phospholipids), we suggest that this highly hydrated area around the catalytic center at the bottom of the tunnel, promote the right placement of the ligand near the catalytic serine via its polar groups. Although the favorable ΔG values (cutoff of −5 kcal/mol) of the waters around this area may suggest that the replacement of these waters is not promoted, we know – based on the catalytic mechanism and experimental data – that this area may be occupied by a ligand upon its insertion (Dessen et al., Citation1999; Kokotou, Galiatsatou, et al., Citation2017). Thus, we suggest that the replacement of certain water molecules should occur upon ligand’s insertion, when a favorable interaction between the ligand and the amino acids is formed.
In conclusion, hydration thermodynamics assist by MD simulations may be used to predict the interactions in the interface between enzyme-membrane complexes. In the case of GIVA cPLA2, the predicted protein-membrane model based on the hydrophobicity properties of its surface, calculated by GIST and MD simulations, was in a good agreement with the DXMS data. Thus, we could suggest this application for the prediction of the interactions in a protein-membrane complex, in cases where no experimental data are available.
Moreover, we analyzed the hydration thermodynamics of the catalytic site of GIVA cPLA2. The area, where the catalytic mechanism takes place, seems to be highly hydrated in contrast with the rest of the binding pocket, which consist of lipophilic amino acids. This event may act as a driving force for the right placement of the natural substrate close to the catalytic serine to promote the hydrolysis. The less favorably bound water molecules may be replaced by a part of a ligand and therefore these results may be used in the design of new GIVA cPLA2 inhibitors.
| Abbreviations | ||
| COX | = | cyclooxygenase |
| DXMS | = | Hydrogen/deuterium exchange mass spectrometry |
| GIVA cPLA2 | = | cPLA2 cytosolic phospholipase A2 group IVA |
| GIST | = | Grid Inhomogeneous Solvation Theory |
| LOX | = | lipoxygenase |
| MD | = | molecular dynamics |
| POPC | = | 1-palmitoyl-2-oleoyl-sn-glycero-3-phosphocholine |
GIVAcPLA2_in_lipids.pdb
Download (19.8 MB)GIVAcPLA2_representantive_GISTstructure.pdb
Download (876 KB)MD_simulation_active_site_apo_str.mp4
Download MP4 Video (53.5 MB)Supporting_info.docx
Download MS Word (631.3 KB)Disclosure statement
No potential conflict of interest was reported by the authors.
Additional information
Funding
References
- Abel, R., Young, T., Farid, R., Berne, B. J., & Friesner, R. A. (2008). Role of the active-site solvent in the thermodynamics of factor Xa ligand binding. Journal of the American Chemical Society, 130(9), 2817–2831. doi:10.1021/ja0771033
- Abel, R., Salam, N. K., Shelley, J., Farid, R., Friesner, R. A., & Sherman, W. (2011). Contribution of explicit solvent effects to the binding affinity of small‐molecule inhibitors in blood coagulation factor serine proteases. Chemmedchem, 6(6), 1049–1066. doi:10.1002/cmdc.201000533
- Bovens, S., Schulze Elfringhoff, A., Kaptur, M., Reinhardt, D., SchäFers, M., & Lehr, M. (2010). 1-(5-Carboxyindol-1-yl) propan-2-one inhibitors of human cytosolic phospholipase A2α: Effect of substituents in position 3 of the indole scaffold on inhibitory potency, metabolic stability, solubility, and bioavailability. Journal of Medicinal Chemistry, 53(23), 8298–8308. doi:10.1021/jm101094p
- Burke, J. E., Babakhani, A., Gorfe, A. A., Kokotos, G., Li, S., Woods, V. L., Jr., … Dennis, E. A. (2009). Location of inhibitors bound to group IVA phospholipase A2 determined by molecular dynamics and deuterium exchange mass spectrometry. Journal of the American Chemical Society, 131(23), 8083–8091. doi:10.1021/ja900098y
- Burke, J. E., Hsu, Y. H., Deems, R. A., Li, S., Woods, V. L., Jr., & Dennis, E. A. (2008). A phospholipid substrate molecule residing in the membrane surface mediates opening of the lid region in group IVA cytosolic phospholipase A2. Journal of Biological Chemistry, 283(45), 31227–31236. doi:10.1074/jbc.M804492200
- Case, D. A., Betz, R. M., Cerutti, D. S., Cheatham, T. E., III, Darden, T. A., Duke, R. E., … Kollman, P. A. (2016). AMBER 2016, San Francisco: University of California.
- Cornette, J. L., Cease, K. B., Margalit, H., Spouge, J. L., Berzofsky, J. A., & DeLisi, C. (1987). Hydrophobicity scales and computational techniques for detecting amphipathic structures in proteins. Journal of Molecular Biology, 195(3), 659–685. doi:10.1016/0022-2836(87)90189-6
- Darden, T., York, D., & Pedersen, L. (1993). Particle mesh Ewald: An N⋅log(N) method for Ewald sums in large systems. Journal of Chemical Physics, 98(12), 10089–10092. doi:10.1063/1.464397
- Dennis, E. A., Cao, J., Hsu, Y. H., Magrioti, V., & Kokotos, G. (2011). Phospholipase A2 enzymes: Physical structure, biological function, disease implication, chemical inhibition, and therapeutic intervention. Chemical Reviews, 111(10), 6130–6185. doi:10.1021/cr200085w
- Dessen, A., Tang, J., Schmidt, H., Stahl, M., Clark, J. D., Seehra, J., & Somers, W. S. (1999). Crystal structure of human cytosolic phospholipase A2 reveals a novel topology and catalytic mechanism. Cell, 97(3), 349–360. doi:10.1016/S0092-8674(00)80744-8
- Dickson, C. J., Madej, B. D., Skjevik, Å. A., Betz, R. M., Teigen, K., Gould, I. R., & Walker, R. C. (2014). Lipid14: The amber lipid force field. Journal of Chemical Theory and Computation, 10(2), 865–879. doi:10.1021/ct4010307
- Eisenberg, D., Schwarz, E., Komaromy, M., & Wall, R. (1984). Analysis of membrane and surface protein sequences with the hydrophobic moment plot. Journal of Molecular Biology, 179(1), 125–142. doi:10.1016/0022-2836(84)90309-7
- Engelman, D. M., Steitz, T. A., & Goldman, A. (1986). Identifying nonpolar transbilayer helices in amino acid sequences of membrane proteins. Annual Review of Biophysics and Biophysical Chemistry, 15(1), 321–353. doi:10.1146/annurev.bb.15.060186.001541
- Green, H. S. (1952). The molecular theory of fluids. Amsterdam: North-Holland Publishing Company.
- Hopp, T. P., & Woods, K. R. (1983). A computer program for predicting protein antigenic determinants. Molecular Immunology, 20(4), 483–489. doi:10.1016/0161-5890(83)90029-9
- Huggins, D. J. (2012). Application of inhomogeneous fluid solvation theory to model the distribution and thermodynamics of water molecules around biomolecules. Physical Chemistry Chemical Physics, 14(43), 15106–15117. doi:10.1039/c2cp42631e
- Janin, J. (1979). Surface and inside volumes in globular proteins. Nature, 277(5696), 491–492. doi:10.1038/277491a0
- Joung, I. S., & Cheatham, T. E. III. (2008). Determination of alkali and halide monovalent ion parameters for use in explicitly solvated biomolecular simulations. The Journal of Physical Chemistry B, 112(30), 9020–9041. doi:10.1021/jp8001614
- Kokotos, G., Feuerherm, A. J., Barbayianni, E., Shah, I., Saether, M., Magrioti, V., … Johansen, B. (2014). Inhibition of group IVA cytosolic phospholipase A2 by thiazolyl ketones in vitro, ex vivo, and in vivo. Journal of Medicinal Chemistry, 57(18), 7523–7535. doi:10.1021/jm500192s
- Kokotos, G., Six, D. A., Loukas, V., Smith, T., Constantinou-Kokotou, V., Hadjipavlou-Litina, D., … Dennis, E. A. (2004). Inhibition of group IVA cytosolic phospholipase A2 by novel 2-oxoamides in vitro, in cells, and in vivo. Journal of Medicinal Chemistry, 47(14), 3615–3628. doi:10.1021/jm030485c
- Kokotou, M., Galiatsatou, G., Magrioti, V., Koutoulogenis, G., Barbayianni, E., Limnios, D., … Kokotos, G. (2017). 2-Oxoesters: A novel class of potent and selective inhibitors of cytosolic group IVA phospholipase A2. Scientific Reports, 7(1), 7025. doi:10.1038/s41598-017-07
- Kokotou, M. G., Limnios, D., Nikolaou, A., Psarra, A., & Kokotos, G. (2017). Inhibitors of phospholipase A2 and their therapeutic potential: An update on patents (2012–2016). Expert Opinion on Therapeutic Patents, 27(2), 217–225. doi:10.1080/13543776.2017.1246540
- Kyte, J., & Doolittle, R. F. (1982). A simple method for displaying the hydropathic character of a protein. Journal of Molecular Biology, 157(1), 105–132. doi:10.1016/0022-2836(82)90515-0
- Lazaridis, T. (1998). Inhomogeneous fluid approach to solvation thermodynamics. 1. Theory. The Journal of Physical Chemistry B, 102(18), 3531–3541. doi:10.1021/jp9723574
- Lee, K. L., Foley, M. A., Chen, L., Behnke, M. L., Lovering, F. E., Kirincich, S. J., … McKew, J. C. (2007). Discovery of Ecopladib, an indole inhibitor of cytosolic phospholipase A2α. Journal of Medicinal Chemistry, 50(6), 1380–1400. 2007). doi:10.1021/jm061131z
- Leslie, C. C. (2015). Cytosolic phospholipase A2: Physiological function and role in disease. Journal of Lipid Research, 56(8), 1386–1402. doi:10.1194/jlr.R057588
- Li, Z., & Lazaridis, T. (2006). Thermodynamics of buried water clusters at a protein − ligand binding interface. The Journal of Physical Chemistry B, 110(3), 1464–1475. doi:10.1021/jp056020a
- Ludwig, J., Bovens, S., Brauch, C., Elfringhoff, A. S., & Lehr, M. (2006). Design and synthesis of 1-Indol-1-yl-propan-2-ones as inhibitors of human cytosolic phospholipase A2a. Journal of Medicinal Chemistry, 49(8), 2611–2620. doi:10.1021/jm051243a
- Maier, J. A., Martinez, C., Kasavajhala, K., Wickstrom, L., Hauser, K. E., & Simmerling, C.(2015). ff14SB: Improving the Accuracy of Protein Side Chain and Backbone Parameters from ff99SB. Journal of Chemical Theory and Computation, 11(8), 3696–3713. doi:10.1021/acs.jctc.5b00255.
- Maier, J. A., Martinez, C., Kasavajhala, K., Wickstrom, L., Hauser, K. E., & Simmerling, C. L. (2015). ff14SB: Improving the accuracy of protein side chain and backbone parameters from ff99SB. Journal of Chemical Theory and Computation, 11(8), 3696–3713. doi:10.1021/acs.jctc.5b00255
- McKew, J. C., Foley, M. A., Thakker, P., Behnke, M. L., Lovering, F. E., Sum, F. W., … Clark, J. D. (2006). Inhibition of cytosolic phospholipase A2α: Hit to lead optimization. Journal of Medicinal Chemistry, 49(1), 135–158. doi:10.1021/jm0507882
- Mouchlis, V. D., Bucher, D., McCammon, J. A., & Dennis, E. A. (2015). Membranes serve as allosteric activators of phospholipase A2, enabling it to extract, bind, and hydrolyze phospholipid substrates. Proceedings of the National Academy of Sciences, 112(6), E516–525. doi:10.1073/pnas.1424651112
- NCT00396955. (2006). http://ClinicalTrials.gov/. Identifier: NCT00396955.
- NCT02795832. (2016). http://ClinicalTrials.gov/. Identifier: NCT02795832.
- Nguyen, C., Gilson, M. K., & Young, T. (2011). Structure and thermodynamics of molecular hydration via grid inhomogeneous solvation theory. arXiv, 1108, 4876.
- Nguyen, C. N., Cruz, A., Gilson, M. K., & Kurtzman, T. (2014). Thermodynamics of water in an enzyme active site: Grid-based hydration analysis of coagulation factor Xa. Journal of Chemical Theory and Computation, 10(7), 2769–2780. doi:10.1021/ct401110x
- Nguyen, C. N., Kurtzman, T., & Gilson, M. K. (2012). Grid inhomogeneous solvation theory: Hydration structure and thermodynamics of the miniature receptor cucurbit[7]uril. Journal of Chemical Physics., 137(4), 044101. doi:10.1063/1.4733951
- Nicolau, D. V., Jr., Paszek, E., Fulga, F., & Nicolau, V. D. (2014). Mapping hydrophobicity on the protein molecular surface at atom-level resolution. PLoS One, 9(12), e114042. doi:10.1371/journal.pone.0114042
- Nikolaou, A., Kokotou, M. G., Vasilakaki, S., & Kokotos, G. (2019). Small-molecule inhibitors as potential therapeutics and as tools to understand the role of phospholipases A2. Biochimica et Biophysica Acta (Bba) - Molecular and Cell Biology of Lipids, 1864(6), 941–956. doi:10.1016/j.bbalip.2018.08.009
- Pettersen, E. F., Goddard, T. D., Huang, C. C., Couch, G. S., Greenblatt, D. M., Meng, E. C., & Ferrin, T. E. (2004). UCSF Chimera–a visualization system for exploratory research and analysis. Journal of Computational Chemistry, 25(13), 1605–1612. doi:10.1002/jcc.20084
- Raman, E. P., & MacKerell, A. D. Jr. (2013). Rapid estimation of hydration thermodynamics of macromolecular regions. Journal of Chemical Physics, 139(5), 055105. doi:10.1063/1.4817344
- Ramsey, S., Nguyen, C., Salomon‐Ferrer, R., Walker, R. C., Gilson, M. K., & Kurtzman, T. (2016). Solvation thermodynamic mapping of molecular surfaces in AmberTools: GIST. Journal of Computational Chemistry, 37(21), 2029–2037. doi:10.1002/jcc.24417
- Rose, G., Geselowitz, A., Lesser, G., Lee, R., & Zehfus, M. (1985). Hydrophobicity of amino acid residues in globular proteins. Science, 229(4716), 834–838. doi:10.1126/science.4023714
- Sali, A., & Blundell, T. L. (1993). Comparative protein modeling by satisfaction of spatial restraints. Journal of Molecular Biology, 234, 779–815. doi:10.1006/jmbi.1993.1626
- Salomon-Ferrer, R., Case, D. A., & Walker, R. C. (2012). An overview of the Amber biomolecular simulation package. Wiley Interdisciplinary Reviews: Computational Molecular Science, 3, 198–210. doi:10.1002/wcms.1121
- Salomon-Ferrer, R., Götz, A. W., Poole, D., Le Grand, S., & Walker, R. C. (2013). Routine microsecond molecular dynamics simulations with amber on gpus. 2. Explicit solvent particle mesh ewald. Journal of Chemical Theory and Computation, 9(9), 3878–3888. doi:10.1021/ct400314y
- Schauperl, M., Podewitz, M., Waldner, B. J., & Liedl, K. R. (2016). Enthalpic and entropic contributions to hydrophobicity. Journal of Chemical Theory and Computation, 12(9), 4600–4610. doi:10.1021/acs.jctc.6b00422
- Seno, K., Okuno, T., Nishi, K., Murakami, Y., Watanabe, F., Matsuura, T., … Ohtani, M. (2000). Pyrrolidine inhibitors of human cytosolic phospholipase A2. Journal of Medicinal Chemistry, 43(6), 1041–1044. doi:10.1016/S0960-894X(01)00003-8
- Six, D. A., Barbayianni, E., Loukas, V., Constantinou-Kokotou, V., Hadjipavlou-Litina, D., Stephens, D., … Kokotos, G. (2007). Structure − activity relationship of 2-oxoamide inhibition of group IVA cytosolic phospholipase A2 and group V secreted phospholipase A2. Journal of Medicinal Chemistry, 50(17), 4222–4235. doi:10.1021/jm0613673
- Tomoo, T., Nakatsuka, T., Katayama, T., Hayashi, Y., Fujieda, Y., Terakawa, M., & Nagahira, K. (2014). Design, synthesis, and biological evaluation of 3‐(1-aryl‐1H‐indol-5-yl)propanoic acids as new indole-based cytosolic phospholipase A2α inhibitors. Journal of Medicinal Chemistry, 57(17), 7244–7262. doi:10.1021/jm500494y