
Abstract
Carcinoembryonic antigen-related cell adhesion (CEACAM) molecules belong to a family of membrane glycoproteins that mediate intercellular interactions influencing cellular growth, immune cell activation, apoptosis, and tumor suppression. Several family members (CEACAM1, CEACAM5, and CEACAM6) are highly expressed in cancers, and they share a conserved N-terminal domain that serves as an attractive target for cancer immunotherapy. A multi-epitope vaccine candidate against this conserved domain has been developed using immunoinformatics tools. Specifically, several epitopes predicted to interact with MHC class I and II molecules were linked together with appropriate linkers. The tertiary structure of the vaccine is generated by homology and ab initio modeling. Molecular docking of epitopes to MHC structures have revealed that the lowest energy conformations are the epitopes bound to the antigen-binding groove of the MHC molecules. Subsequent molecular dynamics simulation has confirmed the stability of the binding conformations in solution. The predicted vaccine has relatively high antigenicity and low allergenicity, suggesting that it is an ideal candidate for further refinement and development.
Communicated by Ramaswamy H. Sarma
Introduction
The carcinoembryonic antigen-related cell adhesion molecules (CEACAM) family includes 12 members that generally have one (sometimes two) Immunoglobulin (Ig)-like variable (V)-set domain, but they differ in the number of Ig-like constant C2-set domains, as well as the membrane anchorage (Beauchemin et al., Citation1999). Four members (CEACAM5-8) are associated with the membrane through a glycosylphosphatidylinositol (GPI) linkage, whereas seven members (CEACAM1, 3, 4, 18-21) are anchored to the cellular membrane via bona fide transmembrane domains. Only one member of the CEACAM family, CEACAM16, is a secreted protein with no membrane anchorage (Beauchemin & Arabzadeh, Citation2013).
Functionally, the expression of CEACAM molecules starts early in human embryonic and fetal development (weeks 9-14) (Eades-Perner et al., Citation1994; Nap et al., Citation1988) and is significantly elevated in colorectal (Jothy et al., Citation1993), gastric (Kodera et al., Citation1993), lung (Singer et al., Citation2010), pancreatic (Gebauer et al., Citation2014), and skin (Khatib et al., Citation2011) carcinoma, and thus they belong to a larger family of ‘carcinoembryonic antigens (CEA)’ (Gold & Freedman, Citation1965). In normal adult tissues, CEA is localized in the stomach, tongue, esophagus, cervix, sweat glands and prostate, as well as in columnar epithelial and goblet cells of the colon (Hammarstrom, Citation1999). Research in the past five decades has established that CEACAM molecules are involved in diverse functions in cell adhesion and signaling, and play important roles in cancer progression, inflammation, angiogenesis, and metastasis (Beauchemin et al., 2013; Gray-Owen & Blumberg, Citation2006; Muenzner et al., Citation2005; Sadarangani et al., Citation2011; Tchoupa et al., Citation2014). Three CEACAM proteins (CEACAM1, CEACAM5, and CEACAM6) are considered as valid clinical biomarkers and recently emerged as attractive therapeutic targets for cancer immunotherapy (Dankner et al., Citation2017; Horst & Wagener, Citation2004; Kuespert et al., Citation2006). Indeed, one member of the CEACAM family, CEACAM5, was ranked 13th out of 75 representative cancer antigens based on a suite of pre-defined and pre-weighted criteria (Cheever et al., Citation2009). Since CEACAM molecules share the conserved Ig-like V domain at the N terminus, a cancer vaccine that targets this region potentially has a universal antitumor effect on all cancers that overexpress CEACAM proteins.
The conventional approach to vaccine development typically involves time-consuming and expensive experimental studies, along with ethical concerns. As such, there is a growing interest in utilizing bioinformatics and immunoinformatics tools for vaccine design (Atapour et al., Citation2020; Dorosti et al., Citation2019; Hajighahramani et al., Citation2019; Khalili et al., Citation2015; Mirza et al., Citation2016; Pandey et al., Citation2018; Sabetian et al., Citation2019), which is further empowered by the recent development of synthetic genomics (Bambini & Rappuoli, Citation2009). Immunoinformatics is an emerging field that interfaces computer science and experimental immunology, aiming to use computational methods and resources for the understanding of immunological information (Korber et al., Citation2006; Tong & Ren, Citation2009). One of its primary goals is to develop algorithms to predict potential B- and T-cell epitopes, which reduces the time and cost required for laboratory analysis of antigens. The application of these in silico techniques for epitope mapping has accelerated the development of vaccines (Li Pira et al., Citation2010; Zhao et al., Citation2013).
In this study, a multi-epitope vaccine candidate has been designed targeting the conserved V-set domain of CEACAM molecules (). Several CD4+ and CD8+ epitopes are predicted by a set of immunoinformatics tools, which are then linked together by appropriate linkers for the enhancement of epitope presentation and separation. To the best of our knowledge, this is the first vaccine candidate targeting the conserved N terminal domain of CEACAM molecules that are overexpressed in a variety of cancers, which deserves further refinement and development.
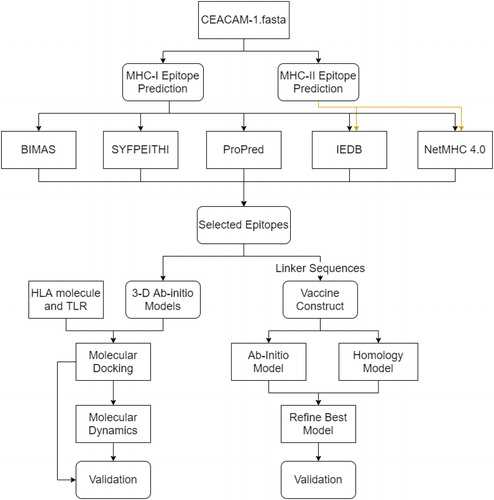
Figure 1. Research strategy workflow. Flowchart of the methods for the prediction and validation of the CEACAM vaccine candidate. Boxes with sharp edges denote methods or tools, whereas boxes with rounded edges represent the data cumulated from tools. The validation for molecular docking/molecular dynamics simulation includes RMSD, RMSF, radius of gyration, snapshots during simulations and binding free energies. The validation for molecular modeling includes Ramachandran plots.
Materials and methods
Sequence and structure retrieval of CEACAM molecules
The protein sequences of CEACAM1 (P13688), CEACAM5 (P06731) and CEACAM6 (P40199) were retrieved from the National Center for Biotechnology Information (NCBI) protein databases. The crystal structure of CEACAM1 Ig-like V-set domain (PDB ID 5DZL) was retrieved from the RCSB PDB database (Berman et al., Citation2000).
Multiple sequence alignment
Multiple sequence alignment of the N-terminal domain of CEACAM1, CEACAM5, and CEACAM6 was performed using the T-Coffee (Notredame et al., Citation2000) multiple sequence aligner hosted by the EBI. T-Coffee uses its progressive alignment algorithm to perform the alignment and is set to produce an alignment output in the ClustalW format.
Prediction of cytotoxic T-lymphocyte (CTL) and helper T-lymphocyte (HTL) epitopes
Five epitope prediction methods including IEDB (Moutaftsi et al., Citation2006), NetMHC 4.0 (Andreatta & Nielsen, Citation2016; Nielsen et al., Citation2003), BIMAS server (Parker et al., Citation1994), SYFPEITHI server (Rammensee et al., Citation1999), and ProPred server (Singh & Raghava, Citation2001) were harnessed to determine potential CTL and HTL epitopes within the CEACAM1 Ig-like V-set domain (see parameters and thresholds of the servers in Supplementary Table S1). These methods predict the peptide epitopes from the antigen of interest based on different algorithms. Specifically, the IEDB server takes in the antigen sequence and runs a sequence alignment method based on artificial neural networks (ANNs) to predict 8-11 amino acid long peptide epitopes. The length was restricted to 9 peptides per epitope for MHC class I and up to 25mers for MHC class II molecules. The NetMHC 4.0 server uses a preset training of 81 MHC alleles to produce novel 9-mer epitopic representations for MHC class I molecules and up to 15mer sequences for MHC class II molecules. The BIMAS server ranks the 9-mer sequences based on independent binding of individual peptide side-chains. The ProPred server uses quantitative matrices that identify promiscuous binding regions useful in selecting vaccine candidates.
Table 1. Top MHC-I-bound epitopes identified from the antigen (* represents different alleles).
Vaccine design and modeling of the 3 D structure of the vaccine
The predicted epitopes were arranged in the order of the antigen, CEACAM1 Ig-like V-set domain, sequence. The epitopes were linked to form a single vaccine candidate with AAY and GPGPG motifs as the linkers to fill gapped sequences between MHC class I and MHC class II epitopes respectively to enhance epitope presentation and separation.
The 3 D structure of the vaccine was predicted by both homology modeling and ab initio modeling. Modeller within the UCSF’s Chimera tool (Pettersen et al., Citation2004) was used to find the three-dimensional homology structure of the vaccine candidate using the original antigen structure (PDB ID 5DZL) as the template. The vaccine structure was also predicted using PEP-FOLD 3.0 server (Shen et al., Citation2014), which predicts structure based on structural alphabet (SA) letters describing the conformations of groups of four consecutive residues. PEP-FOLD 3.0 couples the predicted series of the SA letters with a greedy algorithm and a coarse-grained force field to predict a final 3 D structure.
The Rampage server (Biasini et al., Citation2014) was utilized to assess the quality of the predicted structures by showing the number of residues falling in the favorable and unfavorable regions based on the phi and psi angles of rotation in a molecule.
Physiochemical property, antigenicity, and allergenicity analysis of the vaccine
The analysis of the vaccine construct was done on the ProtParam server (Wilkins et al., Citation1999) which predicts the pI, solubility, Molecular Weight, Half-Life (in-vitro) and Grand Average of Hydropathicity Values (GRAVY) for the input protein sequence.
The antigenicity of the predicted vaccine’s peptide sequence was verified using the ANTIGENpro tool from UC Irvine's Scratch Protein Prediction server (Magnan et al., Citation2010). The peptide sequence of the vaccine structure was entered into the tool and an antigen probability score was produced.
The allergenicity of the candidate vaccine was verified using the AllerTop v. 2.0 web server (Dimitrov et al., Citation2013) from the Medical University of Sofia's Drug Design Group. This tool accepts the vaccine sequence as input and runs a sequence structure mining algorithm on a dataset of known allergens and non-allergens. The tool then determines which member of its database most resembled the vaccine sequence and reports if that protein, as well as the input vaccine sequence, is an allergen or not.
Interferon γ epitopes have been used in the field of immunology to induce the innate as well as the adaptive immune system to elicit anti-tumor pathways (Castro et al., Citation2018). Using the IFNepitope server (Dhanda et al., Citation2013), possible IFN γ epitopes were predicted from the vaccine construct by overlapping regions of protein and predicting their potency and ability to induce IFN γ. Predictions were calculated using a Support Vector Machine (SVM) predictor based on the IEDB’s helper T cell database.
Immune simulation
C-ImmSim 10.1 Server (http://150.146.2.1/C-IMMSIM) was used to interpret the host immune response to the antigen (i.e. the vaccine construct). C-ImmSim is an agent-based computational immune response simulator that utilizes position-specific score matrix (PSSM) and machine learning methods for predicting epitope and immune interactions, respectively (Rapin et al., Citation2010). The parameters in the server were set based on the predominant HLA alleles of predicted epitopes ( and ). The host HLA selection parameter for MHC class I was set on A1010, A2402, and B0702 and for DR MHC class II was sat on DBR1_1101. In accordance with the 3-dose schedule of another cancer vaccine, HPV vaccine, recommended by CDC, the second dose should be given 1-2 months after the first dose, and the third dose should be given 6 months after the first dose. All simulation parameters were set at default with time steps set at 1, 126, 546 (each time step is 8 h and time step 1 is injection at time = 0). Therefore, the intervals between the first and the second dose, as well as between the second and the third dose were 6 and 20 weeks respectively.
Table 2. Top MHC-II-bound epitopes identified from the antigen (* represents different alleles).
Epitope structure modeling
The epitope structures were modeled by the PEPFOLD 3.0 server (Shen et al., Citation2014), which runs a de novo prediction algorithm with the query sequence against a coarse-grained force field to predict the three-dimensional model of the query and validate using hidden Markov models for existing sequences.
Vaccine-HLA docking
The ClusPro server (Kozakov et al., Citation2013; Citation2017; Vajda et al., Citation2017) was employed to analyze the docked binding affinity scores between a given ligand and receptor molecules. The server predicts the energy of the structures based on five different stability forces, namely, Van der Waals forces, electrostatic forces, Decoys as Reference States (DARS) energy algorithm, attractive and repulsive forces. The predicted structures are Fast Fourier Transformed to produce the top optimal cluster with the lowest energy at their centers.
Using the ClusPro server, the epitopes were docked to the Human Leukocyte Antigen (HLA) allele structures available in PDB ( and ). The allele structures for docking were prepared by Chimera. Specifically, the heteroatoms (atoms other than carbon) from the HLA structures were removed and the beta-microglobulin supporting structure was retained due to their function of providing stability to the HLA alleles in the host. Hydrogen atoms were added to the structures using the Tools AddH menu option to create a pre-docking structure. Then several clusters were analyzed to verify the location and binding of the epitope to each HLA allele structure.
The toll-like receptor 4 (TLR4) was used to dock with the epitopes. A 3-dimensional PDB model (PDB ID: 4G8A) of the TLR 4 was used to achieve this. The PDB structure was first cleaned by removing any solvent and non-standard molecules. Then, the variable extracellular domain of the TLR4 (chain B of 4G8A) was extracted and used to dock with the 3-D structures of epitopes.
Vaccine validation using ligand interaction study and molecular dynamics simulation
The Ligplot Tool (Wallace et al., Citation1995) was employed to visualize the interactions and assess the stability of a protein-ligand structure based on H-bonds and hydrophobic contacts. This tool predicts the stability of each of the docked structures.
The molecular dynamics (MD) simulations were performed using Gromacs version 2019.1 (Abraham et al., Citation2015) with the gromos43a1 force field file and plotted using ggplot2. MD simulations were run for each of six different HLA allele structures complexed with their original ligands or with the predicted epitopes (see commands in Supplementary Table S1). Use one of the HLA structures 5XOV as an example. MD simulations were run first using the original 5XOV structure containing the HLA-A*24:02 protein, the beta-2-microglobulin supporting structure, and the original ligand the HIV-1 Nef138-10 peptide. After establishing a baseline, simulation was run on the model, which contains the HLA-A*24:02 protein and the beta-2-microglobulin supporting structure docked with the predicted epitope IYPNASLLI. Simulations were run in the same way over 40 ns by first solvating the molecules in a cube of TIP3P water. Then necessary K + or NA- counterions were added to balance the charge of the system to net zero. Once neutral the model underwent energy minimization using the steepest descent minimization algorithm, to ensure that there were no steric clashes or incorrect geometry. After minimization the simulation solvent and ions were equilibrated, first for temperature by heating the system to 300 K during a 100 ps constant volume simulation with a 2 fs time step. Then the system was equilibrated for pressure at 1 atm during a 100 ps simulation with a 2 fs time step. Both the system’s temperature and pressure were regulated using the Berendsen algorithm. Finally, the simulation production parameters were set, specifying a total simulation runtime of 40 nanoseconds, while the temperature and pressure were held constant at 300 K and 1 atm using the v-rescale temperature and Parrinello − Rahman pressure coupling method.
Once MD simulations were run on each allele epitope and allele original ligand bound structure a variety of methods were used to validate the binding interaction the complexes. RMSD and RMSF information was extracted using the gmx_mpi rms and gmx_mpi rmsf commands and plotted with ggplot2 in R. While further binding interaction verifications were performed on the simulations of the allele epitope bound complexes. PDB structure snapshots of the complexes at timepoints of 1, 20, and 40 ns were obtained from the simulations using the Gromacs command gmx_mpi trjconv -dump 1 -pbc nojump and visualized with UCSC Chimera. The Radius of Gyration (Rg) for each of the simulations was calculated with the gmx_mpi gyrate command and plotted with ggplot2 in R. In addition, the free binding energy of each of the simulations was calculated using the g_mmpbsa tool. G_mmpbsa was run using the g_mmpbsa -pdie 2 -pbsa -decomp command along with the required input files and a production file instructing the program to run calculations of both polar and apolar environments without using the WCA model. The accompanying MmPbSaStat.py python script was then used to calculate the free binding energy from the polar and apolar xvg files generated by g_mmpbsa, which was then plotted with ggplot2 in R.
Results
Sequence retrieval and analysis of CEACAM1, CEACAM5, and CEACAM6 proteins
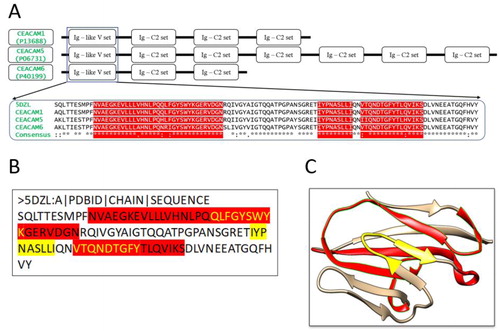
To design an immunogenic multi-epitope vaccine against cancer-related CEACAM molecules such as CEACAM1, CEACAM5, and CEACAM6, a target region that is highly conserved among these molecules needed to be identified. To this aim, the sequences of CEACAM1, CEACAM5, and CEACAM6 were retrieved from the National Center for Biotechnology Information (NCBI) database. Multiple sequence alignment reveals that the N-terminal domain of these molecules is highly conserved with the similarity >91% (). Hence, the domain serves as a perfect region for vaccine design. For the sake of simplicity, this N-terminal domain is referred to as the antigen hereinafter.
Figure 2. Multiple Sequence Alignment of CEACAM molecules and the structures of the conserved N-terminal domain. (a) Domains of CEACAM molecules. The motifs of CEACAM1, 5 and 6 are shown schematically. The sequence of the N-terminal domain, Ig-like V set, of CEACAM1 (P13688), CEACAM5 (P06731) and CEACAM6 (P40199) are aligned, together with the sequence retrieved from the 3 D structure of the CEACAM1 N-terminal domain (PDB: 5DZL Chain A). The predicted MHC class I and II restricted epitopes are highlighted. (b) The protein sequence of the antigen (Chain A in 5DZL). The yellow highlighted regions are the MHC I restricted epitopes obtained from the epitope prediction servers. The red highlighted region represents the MHC II restricted epitopes. The overlapping regions have been colored using yellow text. (c) Three-dimensional structure of the antigen (5DZL, Chain A). The MHC I and II restricted epitopes in the antigen structure are colored with the same coloring scheme as (b).
The ProtParam server (Wilkins et al., Citation1999) was used for the physiochemical analysis of the CEACAM sequences. For instance, the CEACAM1 sequence contains 526 amino acid residues. Its molecular weight is 57560.38 Da and the theoretical pI for the protein is 5.65. It was found that the theoretical half-life of the molecule in mammalian cells is 30 h and the GRAVY (Grand Average of Hydropathy) value is −0.382, which classifies the protein as mildly hydrophilic. This observation is consistent with the fact that CEACAMs attach to the cell membrane and the N-terminal domain of the molecules protrudes into the extracellular matrix (Beauchemin et al., Citation1999; Beauchemin & Arabzadeh, Citation2013).
CTL and HTL epitope prediction
CTLs and HTLs are two subsets of T lymphocytes with CD8+ and CD4+ glycoproteins respectively. CD8+ CTLs interact with the antigen-presenting MHC class I molecules, whereas
CD4+ HTLs interact with the antigen-presenting MHC class II molecules. We predicted antigenic epitopes that can interact with CTLs and HTLs using multiple immunoinformatics tools. and present the consensus epitopes identified by different tools, which shall increase confidence in using these epitopes for designing the vaccine. Note that the MHC alleles were selected based on the ranking of the overall scores provided by the IEDB server and subsequently confirmed by other immunoinformatics tools ( and ). Several alleles have PDB structures available (), which can be used for molecular docking and molecular dynamics simulation studies.
As a result, 7 CTL epitopes were repeatedly predicted by five different methods () and 7 HTL epitopes were detected by three tools (). Several CTL and HTL epitopes are overlapping (), indicating that these regions are highly detectable by different HLA alleles. Further analysis of the population coverage of HLA alleles showed that HLA-A*24:02 and HLA-A*01:01 are found in 40.40% and 52.80% of the global population, respectively (Table S2, supplementary material), indicating that a vaccine candidate based on these epitopes may be effective for a large human population. Note that the corresponding population data for HLA-DRB (MHC class II) alleles are not available.
Construction of a multi-epitope vaccine and physiochemical properties assessment
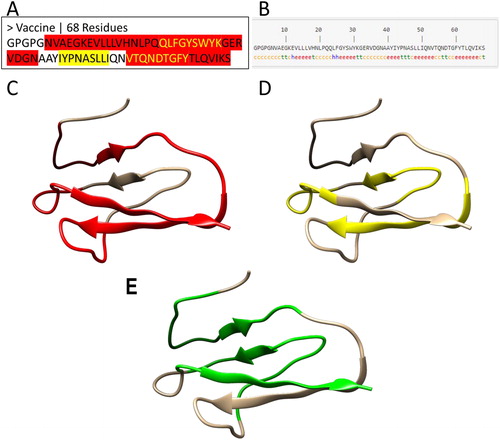
To construct a multi-epitope vaccine, the predicted CTL and HTL epitopes were combined with linkers that play a principal role in the functional and structural features of a protein vaccine (Beauchemin & Arabzadeh, Citation2013). A tandem fusion of these epitopes without proper linkers may result in the generation of a dysfunctional protein with unknown characteristics. In previous studies, AAY has been used as a linker between CTL epitopes for enhancement of epitope presentation whereas GPGPG has been used to link the HTL epitopes (Hajighahramani et al., Citation2017). These two linkers were used in designing of the vaccine (). One of the fragments, TQNDTGFYTLQVIK, is predicted as both a CTL and a HTL epitope ( and ), suggesting that this fragment is recognized by CD8+ and CD4+ cells.
Figure 3. 1-D, 2-D and 3-D structure of a vaccine candidate. (a) The protein sequence of the vaccine candidate. The MHC I restricted epitopes are highlighted in yellow, while MHC II restricted epitopes are highlighted in red. The yellow text in red regions represents the overlapping epitopes. The MHC I and II epitopes are joined by linker sequences GPGPG and AAY. (b) The second structure of the vaccine candidate. The secondary structure of the vaccine was predicted by the SOPMA server (Geourjon & Deleage, Citation1995). The letters ‘h’, ‘c’ and ‘e’ stand for α-helix, random coil and extended β-strand respectively. (c–f) The 3-D structure of the vaccine candidate predicted by a homology modeling method with red color for MHC II epitopes (c), yellow for MHC II epitopes (d), and green for interferon gamma epitopes (e) predicted by IFNepitope server (Dhanda et al., Citation2013).
The presence of IFN-γ produced by CD4+ T cells at the site of infection is important to manage neutrophil recruitment and CXC chemokine production (McLoughlin et al., Citation2008). To confirm that the vaccine candidate can induce IFN-γ, the IFNepitope server was used (Dhanda et al., Citation2013) and it was found that the vaccine candidate contains multiple IFN-γ inducer epitopes (Table S3, supplementary material).
B-cell epitope mapping, antigenicity, and allergenicity prediction of designed vaccine
B-lymphocytes are the key player in humoral immunity by antibody production. They are also one of the main types of antigen-presenting cells. To identify B-cell epitopes in the vaccine candidate, the BepiPred Linear Epitope Prediction 2.0 web tool (Jespersen et al., Citation2017) from IEDB was employed. As a result, 7 peptide regions of variable lengths were predicted to be targeted by B cells. Of the 7 predicted regions, 4 were too short (<10) and therefore removed from further consideration. The remaining 3 regions NVAEGKEVLLL, GPGTQNDTGFYTL, and PNASLLIQNVTQNDTGFYTL were selected not only because they are longer (>10 residues), but also because these regions have a smoothed B-cell epitope likelihood score above the threshold of 0.5 (Jespersen et al., Citation2017). The binding regions were predicted based on the number and concentration of beta-turns in the sequence calculated according to the Chou and Fasman algorithm (Chou & Fasman, Citation1979). Successful identification of B-cell epitopes in the peptide vaccine indicates that it may have the ability to enhance humoral immunity as well as cell-mediated immunity.
To examine whether the designed vaccine is immunogenic in nature, its antigenicity was determined by using the ANTIGENpro server (Magnan et al., Citation2010). It was found that the vaccine has an antigenicity probability of 0.63. Note that the predicted probability of antigenicity score is between 0 and 1, where a higher value indicates a greater likelihood that the input sequence is antigenic. The antigenicity score obtained for this vaccine highlights its antigenic nature and this value exceeds the desired antigenicity value threshold of a 0.6 (Jain et al., Citation2019).
The designed vaccine was further examined to determine if it is a potential allergen. The AllerTOP online server (Castro et al., Citation2018) was used to determine its allergenicity. It was found that the vaccine sequence is likely to be a non-allergen as it is most closely related to a nonallergic sequence in its database (UniProt ID: Q8WVR3). This result indicates that the designed vaccine is nonallergic in nature and probably safe for human use.
To characterize the immunogenicity and immune response profile of the designed vaccine, in silico immune simulations were conducted using the C-ImmSim server (Rapin et al., Citation2010). The levels of antibodies are not elevated in the primary response. The secondary and tertiary responses are characterized by marked increases in levels of IgM, IgG + IgM and IgG1 + IgG2 and B-cell populations (). This profile indicates the development of immune memory and subsequent clearance of the antigen. A similar pattern is also seen in TH (helper) and Tc (cytotoxic) cell populations with corresponding memory development (, D, supplementary material). These results suggest that the designed vaccine likely induce immune reactions as evidenced by a marked increase in the generation of secondary responses.
Vaccine structure prediction, refinement, and assessment
The secondary structure of the vaccine candidate was predicted by the SOPMA server (Geourjon & Deleage, Citation1995), in which most of the structure is covered by β sheets (‘e’) and coils (‘c’) (). To model the 3-dimensional structure of the vaccine candidate, predictions were performed using both homology modeling and ab initio modeling methods. The antigen structure 5DZL was used as the template because the vaccine sequence has 57.3% identity with the antigen sequence (). Consistent with the predicted secondary structure (), the 3-D model is characterized by multiple β sheets and coils, in which the CTL epitopes (), HTL epitopes () and IFN-γ epitopes () are highlighted.
The predicted 3-D models were refined by GalaxyRefine (Ko et al., Citation2012; Shin et al., Citation2014). Table S4 (supplementary material) presents the top five refined models based on the original homology model. Ramachandran plots were used to illustrate the effect of the refinement. Before the refinement, the homology modeling model showed 93.9% of the residues in the favored regions (). After the refinement, the numbers were increased to 100% (, Table S4, supplementary material), indicating that the quality of the structures was greatly improved after the refinement. Similar improvement has been seen for original ab initio model (Table S5, supplementary material).
Figure 4. Ramachandran plots before and after refinement. Ramachandran plots of the vaccine candidate structure predicted by homology modeling method (a) and after refinement by GalaxyRefine refinement (b).
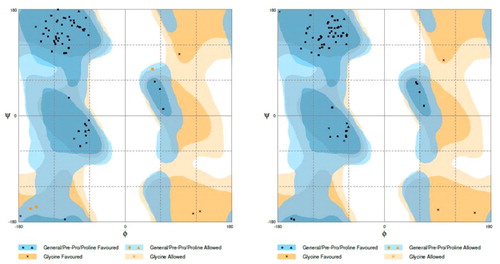
A detailed examination of other parameters such as MolProbity, clash and poor rotamer (Table S4, supplementary material) revealed that the Model 3 of the homology modeling structures has the best quality with the lowest MolProbity score, clash score, no poor rotamer and 100% of residues occurring in the favored regions in the Ramachandran plot (Table S4, supplementary material). Similarly, the Model 2 of the ab initio modeling structures has the best quality with the lowest MolProbity score, clash score, no poor rotamer and 95.3% of residues occurring in the favored regions in the Ramachandran plot (Table S4, supplementary material). The superposition of these two structures showed a high similarity with the RMSD value of 4.45 Å (), which further enhances confidence in the 3-D structure of the vaccine. As such, the Model 3 structure was selected for molecular docking studies.
Molecular docking of epitopes with HLA structures
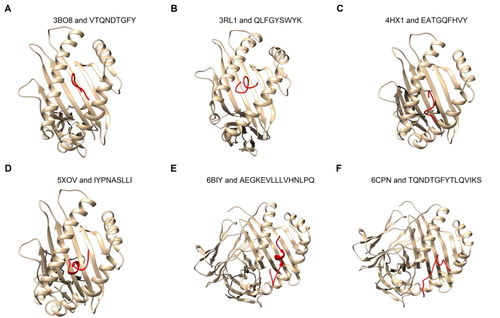
To understand if the epitopes properly interact with MHC molecules, the interactions of these molecules with the original ligands contained in their PDB structures was first examined. For the six HLA complex structures available in PDB, all ligands are bound to the antigen-binding groove of MHC molecules (). These results suggest that the epitopes should appear in the same location for proper interactions with MHC molecules.
The docking of the epitopes to the corresponding HLA allele structures was performed on the ClusPro Server and 39 docked epitope-HLA models were generated. Among them, only the models with the lowest energy score were selected. Detailed analysis of these models showed that all epitopes are bound to the antigen-binding groove (). The lowest energy scores of epitope-HLA models are comparable (supplementary material, Table S6), with the minimal score being −684.1 kCal/mol, indicating that the epitopes interact favorably with the HLA structures.
Figure 5. Docking of epitopes to HLA structures. The epitopes (red) were docked to six HLA 3 D structures available in PDB using Boston University’s ClusPro server. The six complex structures found in PDB include four MHC Class I molecules (a–d, see ) and two MHC Class II molecules (e and f, see ). The epitopes were modeled by PEP-Fold 3.0 that uses ab initio modeling.
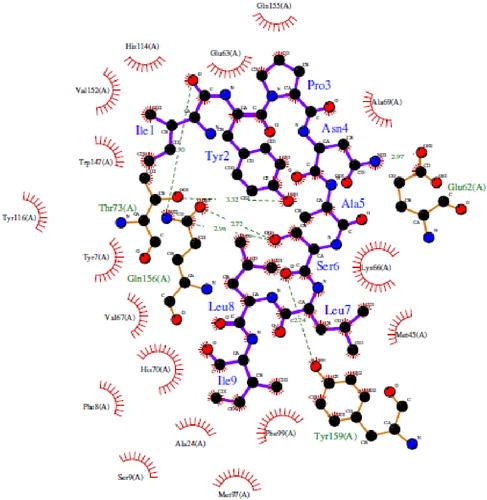
As an illustration, a detailed examination of HLA-epitope interactions was performed on HLA-A*24:02 (5XOV) and the corresponding epitope ‘IYPNASLLI’ using the Ligplot Tool (Wallace et al., Citation1995). It was found that the epitope has numerous hydrogen bonds and hydrophobic interactions with the HLA molecule. Specifically, the epitope region has 6 H-bond interactions where all H-bond length varies from 2.5 − 3.5 Å and multiple hydrophobic interactions between various amino acid residues from the allele (). Notably, this particular epitope was predicted to interact with HLA-A*24:02 by several immunoinformatics tools (). In other words, the docking data confirm the consensus predictions of the immunoinformatics methods (), illustrating the spatial feasibility of interactions between the epitope regions of the designed vaccine and designated MHC molecules.
Figure 6. Interactions between HLA-A*24:02 allele structure and its epitope IYPNASLLI. The residues in blue color represent the epitope from the vaccine, while the green colored residues represent part of the HLA receptor. The bonds represented with green dashed lines show the hydrogen bond and the relative distance is represented in Angstroms. The comb-like residues are the hydrophobic patches found on the receptor as well as the residue atoms of the vaccine.
Toll-like receptors (TLRs) are key components of the innate immune system, recognizing a variety of microbial products (Medzhitov, Citation2001). MoreoverTLRs play an important role in tumor progression (Shcheblyakov et al., Citation2010). It has been reported that the ТLR4 agonists lipopolysaccharide (LPS) from Gram-negative bacteria possess high antitumor activity, when administered intra-tumorally (Okamoto et al., Citation2006). Moreover, it was found that LPS binds to the central domain of TLR4 protein (Ain et al., Citation2020). To understand if TLR4 can recognize the six epitopes in the vaccine, the epitopes were docked to the central domain of TLR4 (the B chain of 4G8A) using ClusPro. Top-ranked docking structures have epitopes located on the central domain of TLR4 (, Table S7; supplementary material), indicating that TLR4 interacts with the epitopes similar to LPS. These results suggest a potential role of innate immunity against the cancer vaccine.
Molecular dynamics simulation for vaccine-HLA complex
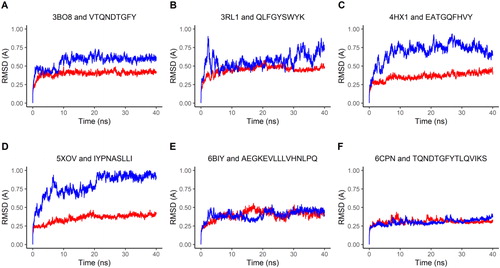
To further study the stability of the epitope-bound HLA structures, molecular dynamics (MD) simulations were performed in GROMACS (Abraham et al., Citation2015) to compare the stability of the epitope-HLA complexes to that of the HLA complexes bound with their original ligands. The stability was measured by RMSD and RMSF values. RMSD computed along a trajectory is the RMSD averaged over atoms as a function of time, while RMSF computed along a trajectory is the RMSF averaged over time as a function of individual atoms. The RMSD of each complex was plotted over time in nanoseconds. The backbone RMSD value of the original ligand-bound complex averaged to 0.2 − 0.4 Å (red lines in ), while the backbone RMSD value of the epitope bound complex averaged to 0.4 − 0.8 Å (blue lines in ). Note that for both the epitope-docked and original ligand-docked simulations, the RMSD values level out over time, which indicates that the epitope-bound complexes are stable in solution.
Figure 7. Molecular Dynamics represented through RMSD Plots. The red-colored lines plot RMSD values of the HLA molecule from six complex structures bound with the original peptide ligand found in PDB, while the blue-colored lines plot the RMSD values of the HLA molecule in the complex bound with the predicted epitopes. The six complex structures found in PDB include four MHC Class I antigens (a–d, see ) and two MHC Class II antigens (e and f, see ).
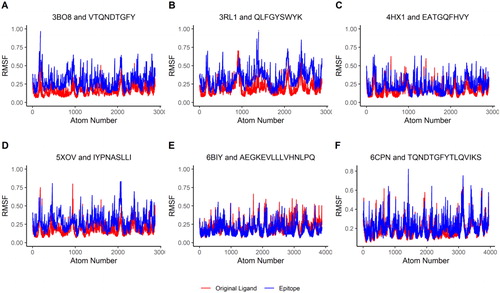
In addition, the RMSF values of both the HLA molecule complexed with the original ligand (red lines in ) and the HLA molecule complexed with the epitopes (blue lines in ) were plotted. The results showed a lack of significant RMSF fluctuations between the two complexes. This lack of significant variation suggests that the epitope-HLA complexes have similar stability as the original HLA complexes and that the binding of the epitopes seems not destabilizing to the HLA molecules.
Figure 8. Molecular Dynamics represented through RMSF Plots. The red-colored lines plot RMSF values of the HLA molecules from six complex structures bound with the original peptide ligands found in PDB, while the blue-colored lines plot RMSF values of the HLA molecules in the complexes bound with the predicted epitopes. The six complex structures contain four MHC Class I antigens (a–d, see ) and two MHC Class II antigens (e and f, see ).
PDB structure snapshots of the vaccine peptide complex MD simulations at 1, 20, and 40 ns showed the peptide’s continued interaction and localization to the known ligand binding domain (). This result indicates that the epitopes remain stable at the docked site. The Radius of Gyration (Rg) of a complex is a measure of the compactness and can be used to indicate complex stability. Rg was calculated for all 6 epitope bound structures, 3bo8, 3rl1, 4hx1, 5xov, 6biy, and 6cpn at each time step in the MD simulation –f), supplementary material). The average Rg for each of the simulations was 2.320, 2.325, 2.159, 2.176, 2.392, and 2.363 nm, respectively. The timestep plots showed that the Rg values fluctuate closely to the average Rg values indicating that the overall shape of the protein complex is stable after binding with the epitopes (Yadav et al., Citation2018). The g_mmpbsa tool was used to calculate the free binding energy of the six MD simulations at each simulation timestep (–f), supplementary material). The average free binding energies for each simulation were also calculated as −16.658 ± 54.984, −449.296 ± 49.972, −184.390 ± 38.207, −28.661 ± 33.628, 307.072 ± 61.670, and −160.319 ± 77.177 kJ/mol, respectively. The negative free binding energy values observed indicate that five of the six predicted epitopes are strongly bound with their HLA receptor alleles making them promising candidates in a cancer vaccine (Ahmad et al., Citation2017).
Discussion
The goal of this project was to design a vaccine construct targeting the highly conserved N-terminal domain of the cancer related CEACAMs using a suite of immunoinformatics and molecular modeling tools. This vaccine, if successful, potentially has a universal antitumor effect on all cancers that overexpress CEACAM proteins.
To this goal, six epitopes have been identified from the domain using immunoinformatics tools, which are predicted to interact with MHC class I alleles in CTLs and MHC class II alleles in HTLs. The predicted CTL and HTL epitopes have the lengths of 9-mer () and 15-mer () respectively. These specific lengths of epitopes were selected based on prior studies on the typical distribution of peptides presented by MHC I and II molecules. Based on the previous study (Bettencourt et al., Citation2020), MHC-I peptides have a narrow distribution with a predominant peak at 9 mer. The selection of 9-mer epitopes () has exactly this length. By contrast, MHC-II peptides show a wide distribution from 12 mer to 18 mer, which is consistent with 11-19 mer from another study (Barra et al., Citation2018). The selection of 15-mer epitopes () is the center of the length distribution.
The selection of the alleles was based on the ranking of the overall scores from the IEDB server. The allele with the highest score is HLA-A*24:02 (). Interestingly, HLA-A*24:02-restricted CTLs recognize antigens of leukemia (Tawara et al., Citation2017), lung cancer (Yamada et al., Citation2003), and stomach cancer (Murahashi et al., 2016), indicating that HLA-A*24:02 is one of the HLA alleles that are critical for cancer antigen recognition. Since CEACAM molecules, especially CEACAM6, are overexpressed in the aforementioned cancers (Hammarstrom, Citation1999), it is highly likely that HLA-A*24:02 also recognize epitopes from CEACAMs, as predicted in this study ().
These epitopes were then linked by AAY and GPGPG motifs for proper separation and presentation of the epitopes to the host immune system. AAY and GPGPG linkers are purposely selected and used in the vaccine construct. AAY is used to link together CTL (MHC I) epitopes to enhance epitope presentation. That is, the vaccine is cleaved by the proteasomal and lysosomal degradation systems after the AAY motif in cytoplasm. Then the generated C-terminal of vaccine binds to the transporter associated with antigen processing (TAP) protein complex, which delivers the epitopes to the endoplasmic reticulum (ER), where they bind to nascent MHC I molecules (Bergmann et al., Citation1996). Thus, binding of epitopes to the TAP transporter, with the help of the AAY linker, is vital for presenting them to MHC I molecules. On the other hand, the GPGPG linker was used because the vaccine contains HTL (MHC II) epitopes and the linker can stimulate HTL response and conserves conformational dependent immunogenicity of the epitopes (Livingston et al., Citation2002).
The interactions between epitopes and HLA structures were shown by molecular docking and molecular dynamics simulation. It was found that the epitopes can bind to the antigen-binding groove of the MHC molecules and this binding is stable over time in solution. These computational results suggest that the designed vaccine candidate has great potential to become an effective vaccine. Although the candidate has a predicted antigenicity value (0.63) that is higher than the threshold (0.6), follow-up experiments are needed to test if the vaccine is immunogenic in humans. One way to enhance the immunogenicity of the vaccine candidate is to use suitable adjuvants (see below).
One of the most important drawbacks of the subunit vaccine is their relatively low immunogenicity compared to whole cell inactivated or live attenuated vaccines (Khalaj-Hedayati et al., Citation2020). To solve this problem, adjuvants are often used in the design of a subunit vaccine. Adjuvants have been widely deployed to further increase the effectiveness of vaccines. They increase vaccine effectiveness in several ways from increasing immune system response to aiding in vaccine transport and increasing the duration of its exposure (Temizoz et al., Citation2016). Vaccine delivery-based adjuvants operate on the principle of creating a deposit of the antigenic compound at the vaccination site that slowly released over time prolonging immune response (Guy, Citation2007). The water in oil emulsion adjuvant, Montanide ISA-51, has shown promise activating T-cell response in cancer vaccines clinical trials that correlate positively with increased patient survival (Chianese-Bullock et al., 2005;; Neninger Vinageras et al., 2008) . Any clinical trial attempting to further test or derive a cancer vaccine from this work is recommended to consider including an appropriate immunity-enhancing adjuvant such as Montanide ISA-51.
Several CEACAMs such as CEACAM5 and CEACAM6 are highly expressed in primary and metastatic cancers in breast, pancreas, colon, and lung (Blumenthal et al., Citation2007), which make them potential targets for cancer vaccines. However, CEACAMs are self-antigens that make it difficult to break immune tolerance when part of them is used in a vaccine. Several approaches have been proposed and tested to break the immune tolerance of cancer self-antigens (Makkouk & Weiner, Citation2015). Additional experiments are required for further development and refinement of the vaccine candidate to break immune tolerance prior to clinical trials.
Conclusions
CEACAMs belong to a family of membrane glycoproteins. Several family members such as CEACAM1, 5 and 6 are highly expressed in a variety of cancers, and thereby they serve as targets for cancer vaccines. Here, a peptide-based vaccine candidate is developed targeting the conserved N-terminal domain of these molecules utilizing a suite of immunoinformatic and molecular modeling tools against cancer-associated CEACAMs. The vaccine candidate contains epitopes predicted to bind to MHC class I and II molecules and has high antigenicity and low allergenicity. The epitopes can bind to the antigen-binding groove of MHC molecules and such binding is stable over time in solution. More experiments are needed to test the immunogenicity of the candidate in humans. Suitable adjuvants can be used for further enhancement of the vaccine. Appropriate strategies to break immune tolerance should be considered prior to clinical trials.
Authors’ contributions
AG and AJR designed and performed the experiments. AG and AJR and FC drafted and edited the manuscript. All authors read and approved the final manuscript.
Supplementary_Tables...xlsx
Download MS Excel (83 KB)Supplementary_Figures...docx
Download MS Word (5.8 MB)Disclosure statement
The authors declare that they have no conflicts of interest.
Additional information
Funding
Related Research Data
References
- Abraham, M. J., Murtola, T., Schulz, R., Páll, S., Smith, J. C., Hess, B., & Lindahl, E. (2015). GROMACS: High performance molecular simulations through multi-level parallelism from laptops to supercomputers. SoftwareX, 1/2, 19–25. https://doi.org/https://doi.org/10.1016/j.softx.2015.06.001
- Ahmad, S., Raza, S., Uddin, R., & Azam, S. S. (2017). Binding mode analysis, dynamic simulation and binding free energy calculations of the MurF ligase from Acinetobacter baumannii. Journal of Molecular Graphics & Modelling, 77, 72–85. https://doi.org/https://doi.org/10.1016/j.jmgm.2017.07.024
- Ain, Q. U., Batool, M., & Choi, S. (2020). TLR4-targeting therapeutics: Structural basis and computer-aided drug discovery approaches. Molecules, 25(3), 627. https://doi.org/https://doi.org/10.3390/molecules25030627
- Andreatta, M., & Nielsen, M. (2016). Gapped sequence alignment using artificial neural networks: Application to the MHC class I system. Bioinformatics (Oxford, England)), 32(4), 511–517. https://doi.org/https://doi.org/10.1093/bioinformatics/btv639
- Atapour, A., Negahdaripour, M., Ghasemi, Y., Razmjuee, D., Savardashtaki, A., Mousavi, S. M., Hashemi, S. A., Aliabadi, A., & Nezafat, N. (2020). In silico designing a candidate vaccine against breast cancer. International Journal of Peptide Research and Therapeutics, 26(1), 369–380. https://doi.org/https://doi.org/10.1007/s10989-019-09843-1
- Bambini, S., & Rappuoli, R. (2009). The use of genomics in microbial vaccine development. Drug Discovery Today, 14(5/6), 252–260. https://doi.org/https://doi.org/10.1016/j.drudis.2008.12.007
- Barra, C., Alvarez, B., Paul, S., Sette, A., Peters, B., Andreatta, M., Buus, S., & Nielsen, M. (2018). Footprints of antigen processing boost MHC class II natural ligand predictions. Genome Medicine, 10(1), 84https://doi.org/https://doi.org/10.1186/s13073-018-0594-6
- Beauchemin, N., & Arabzadeh, A. (2013). Carcinoembryonic antigen-related cell adhesion molecules (CEACAMs) in cancer progression and metastasis. Cancer and Metastasis Reviews, 32(3/4), 643–671. https://doi.org/https://doi.org/10.1007/s10555-013-9444-6
- Beauchemin, N., Draber, P., Dveksler, G., Gold, P., Gray-Owen, S., & Grunert, F. (1999). Redefined nomenclature for members of the carcinoembryonic antigen family. Experimental Cell Research, 122, 467–481.
- Bergmann, C. C., Yao, Q., Ho, C. K., & Buckwold, S. L. (1996). Flanking residues alter antigenicity and immunogenicity of multi-unitCTL epitopes. Journal of Immunology, 157(8), 3242.
- Berman, H. M., Westbrook, J., Feng, Z., Gilliland, G., Bhat, T. N., Weissig, H., Shindyalov, I. N., & Bourne, P. E. (2000). The Protein Data Bank. Nucleic Acids Research, 28(1), 235–242. https://doi.org/https://doi.org/10.1093/nar/28.1.235
- Bettencourt, P., Müller, J., Nicastri, A., Cantillon, D., Madhavan, M., Charles, P. D., Fotso, C. B., Wittenberg, R., Bull, N., Pinpathomrat, N., Waddell, S. J., Stylianou, E., Hill, A. V. S., Ternette, N., & McShane, H. (2020). Identification of antigens presented by MHC for vaccines against tuberculosis. Npj Vaccines, 5, 2https://doi.org/https://doi.org/10.1038/s41541-019-0148-y
- Biasini, M., Bienert, S., Waterhouse, A., Arnold, K., Studer, G., Schmidt, T., Kiefer, F., Gallo Cassarino, T., Bertoni, M., Bordoli, L., & Schwede, T. (2014). SWISS-MODEL: Modelling protein tertiary and quaternary structure using evolutionary information. Nucleic Acids Research, 42(Web Server issue), W252–W258. https://doi.org/https://doi.org/10.1093/nar/gku340
- Blumenthal, R. D., Leon, E., Hansen, H. J., & Goldenberg, D. M. (2007). Expression patterns of CEACAM5 and CEACAM6 in primary and metastatic cancers. BMC Cancer, 7, 2. https://doi.org/https://doi.org/10.1186/1471-2407-7-2
- Castro, F., Cardoso, A. P., Goncalves, R. M., Serre, K., & Oliveira, M. J. (2018). Interferon-gamma at the crossroads of tumor immune surveillance or evasion. Frontiers in Immunology, 9, 847. https://doi.org/https://doi.org/10.3389/fimmu.2018.00847
- Cheever, M. A., Allison, J. P., Ferris, A. S., Finn, O. J., Hastings, B. M., Hecht, T. T., Mellman, I., Prindiville, S. A., Viner, J. L., Weiner, L. M., & Matrisian, L. M. (2009). The prioritization of cancer antigens: A National Cancer Institute pilot project for the acceleration of translational research. Clinical Cancer Research, 15(17), 5323–5337. https://doi.org/https://doi.org/10.1158/1078-0432.CCR-09-0737
- Chianese-Bullock, K. A., Pressley, J., Garbee, C., Hibbitts, S., Murphy, C., Yamshchikov, G., Petroni, G. R., Bissonette, E. A., Neese, P. Y., Grosh, W. W., Merrill, P., Fink, R., Woodson, E. M. H., Wiernasz, C. J., Patterson, J. W., & Slingluff, C. L. (2005). MAGE-A1-, MAGE-A10-, and gp100-derived peptides are immunogenic when combined with granulocyte-macrophage colony-stimulating factor and montanide ISA-51 adjuvant and administered as part of a multipeptide vaccine for melanoma. Journal of Immunology (Baltimore, MD: 1950)), 174(5), 3080–3086. https://doi.org/https://doi.org/10.4049/jimmunol.174.5.3080
- Chou, P. Y., & Fasman, G. D. (1979). Prediction of beta-turns. Biophysical Journal , 26(3), 367–383. https://doi.org/https://doi.org/10.1016/S0006-3495(79)85259-5
- Dankner, M., Gray-Owen, S., Huang, Y.-H., Blumberg, R. S., & Beauchemin, N. (2017). CEACAM1 as a multi-purpose target for cancer immunotherapy. Oncoimmunology, 6(7), e1328336https://doi.org/https://doi.org/10.1080/2162402X.2017.1328336
- Dhanda, S. K., Vir, P., & Raghava, G. P. (2013). Designing of interferon-gamma inducing MHC class-II binders. Biology Direct, 8, 30https://doi.org/https://doi.org/10.1186/1745-6150-8-30
- Dimitrov, I., Flower, D. R., & Doytchinova, I. (2013). AllerTOP–a server for in silico prediction of allergens. BMC Bioinformatics, 14(Suppl 6), S4. https://doi.org/https://doi.org/10.1186/1471-2105-14-S6-S4
- Dorosti, H., Eslami, M., Negahdaripour, M., Ghoshoon, M. B., Gholami, A., Heidari, R., Dehshahri, A., Erfani, N., Nezafat, N., & Ghasemi, Y. (2019). Vaccinomics approach for developing multi-epitope peptide pneumococcal vaccine. Journal of Biomolecular Structure Dynamics, 37(13), 3524–3535. https://doi.org/https://doi.org/10.1080/07391102.2018.1519460
- Eades-Perner, A.-M., van der Putten, H., Hirth, A., Thompson, J., Neumaier, M., von Kleist, S., & Zimmermann, W. (1994). Mice transgenic for the human carcinoembryonic antigen gene maintain its spatiotemporal expression pattern. Cancer Research, 54(15), 4169–4176.
- Gebauer, F., Wicklein, D., Horst, J., Sundermann, P., Maar, H., Streichert, T., Tachezy, M., Izbicki, J. R., Bockhorn, M., & Schumacher, U. (2014). Carcinoembryonic antigen-related cell adhesion molecules (CEACAM) 1, 5 and 6 as biomarkers in pancreatic cancer. PLoS One, 9(11), e113023. https://doi.org/https://doi.org/10.1371/journal.pone.0113023
- Geourjon, C., & Deleage, G. (1995). SOPMA: Significant improvements in protein secondary structure prediction by consensus prediction from multiple alignments. Computer Applications in the Biosciences : Cabios, 11(6), 681–684. https://doi.org/https://doi.org/10.1093/bioinformatics/11.6.681
- Gold, P., & Freedman, S. O. (1965). Specific carcinoembryonic antigens of the human digestive system. The Journal of Experimental Medicine, 122(3), 467–481. https://doi.org/https://doi.org/10.1084/jem.122.3.467
- Gray-Owen, S. D., & Blumberg, R. S. (2006). CEACAM1: Contact-dependent control of immunity. Nature Reviews. Immunology, 6(6), 433–446. https://doi.org/https://doi.org/10.1038/nri1864
- Guy, B. (2007). The perfect mix: Recent progress in adjuvant research. Nature Reviews Microbiology, 5(7), 396–397. https://doi.org/https://doi.org/10.1038/nrmicro1681
- Hajighahramani, N., Eslami, M., Negahdaripour, M., Ghoshoon, M. B., Dehshahri, A., Erfani, N., Heidari, R., Gholami, A., Nezafat, N., & Ghasemi, Y. (2019). Computational design of a chimeric epitope-based vaccine to protect against Staphylococcus aureus infections. Molecular and Cellular Probes, 46, 101414. https://doi.org/https://doi.org/10.1016/j.mcp.2019.06.004
- Hajighahramani, N., Nezafat, N., Eslami, M., Negahdaripour, M., Rahmatabadi, S. S., & Ghasemi, Y. (2017). Immunoinformatics analysis and in silico designing of a novel multi-epitope peptide vaccine against Staphylococcus aureus. Infection, Genetics and Evolution : journal of Molecular Epidemiology and Evolutionary Genetics in Infectious Diseases, 48, 83–94. https://doi.org/https://doi.org/10.1016/j.meegid.2016.12.010
- Hammarstrom, S. (1999). The carcinoembryonic antigen (CEA) family: Structures, suggested functions and expression in normal and malignant tissues. Seminars in Cancer Biology., 9, 67–81.
- Horst, A. K., & Wagener, C. (2004). CEA-related CAMs. Handbook of Experimental Pharmacology., 165, 283–341.
- Jain, R., Singh, S., Verma, S. K., & Jain, A. (2019). Genome-wide prediction of potential vaccine candidates for Campylobacter jejuni using reverse vaccinology. Interdisciplinary Science , 11(3), 337–347. https://doi.org/https://doi.org/10.1007/s12539-017-0260-5
- Jespersen, M. C., Peters, B., Nielsen, M., & Marcatili, P. (2017). BepiPred-2.0: Improving sequence-based B-cell epitope prediction using conformational epitopes. Nucleic Acids Research, 45(W1), W24–W29. https://doi.org/https://doi.org/10.1093/nar/gkx346
- Jothy, S., Yuan, S. Y., & Shirota, K. (1993). Transcription of carcinoembryonic antigen in normal colon and colon carcinoma. In situ hybridization study and implication for a new in vivo functional model. The American Journal of Pathology, 143(1), 250–257.
- Khalaj-Hedayati, A., Chua, C. L. L., Smooker, P., & Lee, K. W. (2020). Nanoparticles in influenza subunit vaccine development: Immunogenicity enhancement. Influenza and Other Respiratory Viruses, 14(1), 92–101. https://doi.org/https://doi.org/10.1111/irv.12697
- Khalili, S., Rahbar, M. R., Dezfulian, M. H., & Jahangiri, A. (2015). In silico analyses of Wilms׳ tumor protein to designing a novel multi-epitope DNA vaccine against cancer. Journal of Theoretical Biology, 379, 66–78. https://doi.org/https://doi.org/10.1016/j.jtbi.2015.04.026
- Khatib, N., Pe'er, J., Ortenberg, R., Schachter, J., Frenkel, S., Markel, G., & Amer, R. (2011). Carcinoembryonic antigen cell adhesion molecule-1 (CEACAM1) in posterior uveal melanoma: Correlation with clinical and histological survival markers. Investigative Ophthalmology & Visual Science, 52(13), 9368–9372. https://doi.org/https://doi.org/10.1167/iovs.10-6006
- Ko, J., Park, H., Heo, L., & Seok, C. (2012). GalaxyWEB server for protein structure prediction and refinement. Nucleic Acids Research, 40(Web Server issue), W294–W297. https://doi.org/https://doi.org/10.1093/nar/gks493
- Kodera, Y., Isobc, K., Yamauchi, M., Satta, T., Hascgawa, T., Oikawa, S., Kondoh, K., Akiyama, S., Itoh, K., Nakashima, I., & Taakagi, H. (1993). Expression of carcinoembryonic antigen (CEA) and nonspecific crossreacting antigen (NCA) in gastrointestinal cancer; the correlation with degree of differentiation. British Journal of Cancer, 68(1), 130–136. https://doi.org/https://doi.org/10.1038/bjc.1993.300
- Korber, B., LaBute, M., & Yusim, K. (2006). Immunoinformatics comes of age. PLoS Computational Biology, 2(6), e71https://doi.org/https://doi.org/10.1371/journal.pcbi.0020071
- Kozakov, D., Beglov, D., Bohnuud, T., Mottarella, S., Xia, B., Hall, D. R., & Vajda, S. (2013). How good is automated protein docking?. Proteins, 81(12), 2159–2166. https://doi.org/https://doi.org/10.1002/prot.24403
- Kozakov, D., Hall, D. R., Xia, B., Porter, K. A., Padhorny, D., Yueh, C., Beglov, D., & Vajda, S. (2017). The ClusPro web server for protein-protein docking. Nature Protocols, 12(2), 255–278. https://doi.org/https://doi.org/10.1038/nprot.2016.169
- Kuespert, K., Pils, S., & Hauck, C. R. (2006). CEACAMs: Their role in physiology and pathophysiology. Current Opinion in Cell Biology, 9, 616–626.
- Li Pira, G., Ivaldi, F., Moretti, P., & Manca, F. (2010). High throughput T epitope mapping and vaccine development. Journal of Biomedicine and Biotechnology. , 2010, 1–12. https://doi.org/https://doi.org/10.1155/2010/325720
- Livingston, B., Crimi, C., Newman, M., Higashimoto, Y., Appella, E., Sidney, J., & Sette, A. (2002). A Rational Strategy to Design Multiepitope Immunogens Based on Multiple Th Lymphocyte Epitopes. The Journal of Immunology, 168(11), 5499–5506. https://doi.org/https://doi.org/10.4049/jimmunol.168.11.5499
- Magnan, C. N., Zeller, M., Kayala, M. A., Vigil, A., Randall, A., Felgner, P. L., & Baldi, P. (2010). High-throughput prediction of protein antigenicity using protein microarray data. Bioinformatics (Oxford, England)), 26(23), 2936–2943. https://doi.org/https://doi.org/10.1093/bioinformatics/btq551
- Makkouk, A., & Weiner, G. J. (2015). Cancer immunotherapy and breaking immune tolerance: New approaches to an old challenge. Cancer Research, 75(1), 5–10. https://doi.org/https://doi.org/10.1158/0008-5472.CAN-14-2538
- McLoughlin, R. M., Lee, J. C., Kasper, D. L., & Tzianabos, A. O. (2008). IFN-gamma regulated chemokine production determines the outcome of Staphylococcus aureus infection. Journal of Immunology (Baltimore, Md. : 1950)), 181(2), 1323–1332. https://doi.org/https://doi.org/10.4049/jimmunol.181.2.1323
- Medzhitov, R. (2001). Toll-like receptors and innate immunity. Nature Reviews. Immunology, 1(2), 135–145. https://doi.org/https://doi.org/10.1038/35100529
- Mirza, M. U., Rafique, S., Ali, A., Munir, M., Ikram, N., Manan, A., Salo-Ahen, O. M. H., & Idrees, M. (2016). Towards peptide vaccines against Zika virus: Immunoinformatics combined with molecular dynamics simulations to predict antigenic epitopes of Zika viral proteins. Scientific Reports, 6, 37313https://doi.org/https://doi.org/10.1038/srep37313
- Moutaftsi, M., Peters, B., Pasquetto, V., Tscharke, D. C., Sidney, J., Bui, H. H., Grey, H., & Sette, A. (2006). A consensus epitope prediction approach identifies the breadth of murine T(CD8+)-cell responses to vaccinia virus . Nature Biotechnology, 24(7), 817–819. https://doi.org/https://doi.org/10.1038/nbt1215
- Muenzner, P., Rohde, M., Kneitz, S., & Hauck, C. R. (2005). CEACAM engagement by human pathogens enhances cell adhesion and counteracts bacteria-induced detachment of epithelial cells. The Journal of Cell Biology, 170(5), 825–836. https://doi.org/https://doi.org/10.1083/jcb.200412151
- Murahashi, M., Hijikata, Y., Yamada, K., Tanaka, Y., Kishimoto, J., Inoue, H., Marumoto, T., Takahashi, A., Okazaki, T., Takeda, K., Hirakawa, M., Fujii, H., Okano, S., Morita, M., Baba, E., Mizumoto, K., Maehara, Y., Tanaka, M., Akashi, K., … Tani, K. (2016). Phase I clinical trial of a five-peptide cancer vaccine combined with cyclophosphamide in advanced solid tumors. Clinical Immunology (Orlando, FL.).), 166-167, 48–58. https://doi.org/https://doi.org/10.1016/j.clim.2016.03.015
- Nap, M., Mollgard, K., Burtin, P., & Fleuren, G. J. (1988). Immunohistochemistry of carcino-embryonic antigen in the embryo, fetus, and adult. Tumour Biology : The Journal of the International Society for Oncodevelopmental Biology and Medicine, 9(2/3), 145–153. https://doi.org/https://doi.org/10.1159/000217555
- Neninger Vinageras, E., de la Torre, A., Osorio Rodríguez, M., Catalá Ferrer, M., Bravo, I., Mendoza del Pino, M., Abreu Abreu, D., Acosta Brooks, S., Rives, R., del Castillo Carrillo, C., González Dueñas, M., Viada, C., García Verdecia, B., Crombet Ramos, T., González Marinello, G., & Lage Dávila, A. (2008). Phase II randomized controlled trial of an epidermal growth factor vaccine in advanced non-small-cell lung cancer. Journal of Clinical Oncology : official Journal of the American Society of Clinical Oncology, 26(9), 1452–1458. https://doi.org/https://doi.org/10.1200/JCO.2007.11.5980
- Nielsen, M., Lundegaard, C., Worning, P., Lauemoller, S. L., Lamberth, K., Buus, S., Brunak, S., & Lund, O. (2003). Reliable prediction of T-cell epitopes using neural networks with novel sequence representations . Protein Science : a Publication of the Protein Society, 12(5), 1007–1017. https://doi.org/https://doi.org/10.1110/ps.0239403
- Notredame, C., Higgins, D. G., & Heringa, J. (2000). T-Coffee: A novel method for fast and accurate multiple sequence alignment. Journal of Molecular Biology, 302(1), 205–217. https://doi.org/https://doi.org/10.1006/jmbi.2000.4042
- Okamoto, M., Oshikawa, T., Tano, T., Ahmed, S. U., Kan, S., Sasai, A., Akashi, S., Miyake, K., Moriya, Y., Ryoma, Y., Saito, M., & Sato, M. (2006). Mechanism of anticancer host response induced by OK-432, a Streptococcal Preparation, mediated by phagocytosis and toll-like receptor 4 signaling. Journal of Immunotherapy, 29, 79–86.
- Pandey, R. K., Bhatt, T. K., & Prajapati, V. K. (2018). Novel immunoinformatics approaches to design multi-epitope subunit vaccine for Malaria by investigating Anopheles Salivary protein. Scientific Reports, 8(1), 1125https://doi.org/https://doi.org/10.1038/s41598-018-19456-1
- Parker, K. C., Bednarek, M. A., & Coligan, J. E. (1994). Scheme for ranking potential HLA-A2 binding peptides based on independent binding of individual peptide side-chains. Journal of Immunology (Baltimore, MD. : 1950), 152(1), 163–175.
- Pettersen, E. F., Goddard, T. D., Huang, C. C., Couch, G. S., Greenblatt, D. M., Meng, E. C., & Ferrin, T. E. (2004). UCSF Chimera-a visualization system for exploratory research and analysis. Journal of Computing Chemistry, 25(13), 1605–1612. https://doi.org/https://doi.org/10.1002/jcc.20084
- Rammensee, H.-G., Bachmann, J., Emmerich, N. N., Bachor, O. A., & Stevanovic, S. (1999). SYFPEITHI: Database for MHC ligands and peptide motifs. Immunogenetics, 50(3-4), 213–219. https://doi.org/https://doi.org/10.1007/s002510050595
- Rapin, N., Lund, O., Bernaschi, M., & Castiglione, F. (2010). Computational immunology meets bioinformatics: The use of prediction tools for molecular binding in the simulation of the immune system. PLoS One, 5(4), e9862https://doi.org/https://doi.org/10.1371/journal.pone.0009862
- Sabetian, S., Nezafat, N., Dorosti, H., Zarei, M., & Ghasemi, Y. (2019). Exploring dengue proteome to design an effective epitope-based vaccine against dengue virus. Journal of Biomolecular Structure & Dynamics, 37(10), 2546–2563. https://doi.org/https://doi.org/10.1080/07391102.2018.1491890
- Sadarangani, M., Pollard, A. J., & Gray-Owen, S. D. (2011). Opa proteins and CEACAMs: Pathways of immune engagement for pathogenic Neisseria. FEMS Microbiology Review, 35(3), 498–514. https://doi.org/https://doi.org/10.1111/j.1574-6976.2010.00260.x
- Shcheblyakov, D. V., Logunov, D. Y., Tukhvatulin, A. I., Shmarov, M. M., Naroditsky, B. S., & Gintsburg, A. L. (2010). Toll-like receptors (TLRs): the Role in tumor progression. Acta Naturae, 2(3), 21–29.
- Shen, Y., Maupetit, J., Derreumaux, P., & Tuffery, P. (2014). Improved PEP-FOLD approach for peptide and miniprotein structure prediction. Journal of Chemical Theory and Computation, 10(10), 4745–4758. https://doi.org/https://doi.org/10.1021/ct500592m
- Shin, W.-H., Lee, G. R., Heo, L., Lee, H., & Seok, C. (2014). Prediction of protein structure and interaction by GALAXY protein modeling programs. Bio Design, 2, 1–11.
- Singer, B. B., Scheffrahn, I., Kammerer, R., Suttorp, N., Ergun, S., & Slevogt, H. (2010). Deregulation of the CEACAM expression pattern causes undifferentiated cell growth in human lung adenocarcinoma cells. PLoS One, 5(1), e8747https://doi.org/https://doi.org/10.1371/journal.pone.0008747
- Singh, H., & Raghava, G. (2001). ProPred: Prediction of HLA-DR binding sites. Bioinformatics (Oxford, England)), 17(12), 1236–1237. https://doi.org/https://doi.org/10.1093/bioinformatics/17.12.1236
- Tawara, I., Kageyama, S., Miyahara, Y., Fujiwara, H., Nishida, T., Akatsuka, Y., Ikeda, H., Tanimoto, K., Terakura, S., Murata, M., Inaguma, Y., Masuya, M., Inoue, N., Kidokoro, T., Okamoto, S., Tomura, D., Chono, H., Nukaya, I., Mineno, J., … Shiku, H. (2017). Safety and persistence of WT1-specific T-cell receptor gene-transduced lymphocytes in patients with AML and MDS. Blood, 130(18), 1985–1994. https://doi.org/https://doi.org/10.1182/blood-2017-06-791202
- Tchoupa, A. K., Schuhmacher, T., & Hauck, C. R. (2014). Signaling by epithelial members of the CEACAM family - mucosal docking sites for pathogenic bacteria. Cell Communication and Signaling, 12, 27https://doi.org/https://doi.org/10.1186/1478-811X-12-27
- Temizoz, B., Kuroda, E., & Ishii, K. J. (2016). Vaccine adjuvants as potential cancer immunotherapeutics. International Immunology, 28(7), 329–338. https://doi.org/https://doi.org/10.1093/intimm/dxw015
- Tong, J. C., & Ren, E. C. (2009). Immunoinformatics: Current trends and future directions. Drug Discovery Today, 14(13/14), 684–689. https://doi.org/https://doi.org/10.1016/j.drudis.2009.04.001
- Vajda, S., Yueh, C., Beglov, D., Bohnuud, T., Mottarella, S. E., Xia, B., Hall, D. R., & Kozakov, D. (2017). New additions to the ClusPro server motivated by CAPRI. Proteins, 85(3), 435–444. https://doi.org/https://doi.org/10.1002/prot.25219
- Wallace, A. C., Laskowski, R. A., & Thornton, J. M. (1995). LIGPLOT: A program to generate schematic diagrams of protein-ligand interactions. Protein Engineering, 8(2), 127–134. https://doi.org/https://doi.org/10.1093/protein/8.2.127
- Wilkins, M. R., Gasteiger, E., Bairoch, A., Sanchez, J. C., Williams, K. L., Appel, R. D., & Hochstrasser, D. F. (1999). Protein identification and analysis tools in the ExPASy server. Methods in Molecular Biology (Clifton, N.J.).), 112, 531–552. https://doi.org/https://doi.org/10.1385/1-59259-584-7:531
- Yadav, D. K., Kumar, S., Misra, S., Yadav, L., & Teli, M. (2018). Molecular Insights into the Interaction of RONS and Thieno [3, 2-c] pyran analogs with SIRT6/COX-2: A molecular dynamics study. Scientific Reports, 8(1), 1–16. https://doi.org/https://doi.org/10.1038/s41598-018-22972-9
- Yamada, A., Kawano, K., Koga, M., Takamori, S., Nakagawa, M., & Itoh, K. (2003). Gene and peptide analysis of newly defined lung cancer antigens recognized by HLA-A2402-restricted tumor-specific cytotoxic T lymphocytes. Cancer Research, 63, 2829–2835.
- Zhao, L., Zhang, M., & Cong, H. (2013). Advances in the study of HLA-restricted epitope vaccines. Human Vaccines & Immunotherapeutics, 9(12), 2566–2577. https://doi.org/https://doi.org/10.4161/hv.26088