
 ?Mathematical formulae have been encoded as MathML and are displayed in this HTML version using MathJax in order to improve their display. Uncheck the box to turn MathJax off. This feature requires Javascript. Click on a formula to zoom.
?Mathematical formulae have been encoded as MathML and are displayed in this HTML version using MathJax in order to improve their display. Uncheck the box to turn MathJax off. This feature requires Javascript. Click on a formula to zoom.Abstract
Sodium-glucose co-transporter 2 (SGLT-2) is a major transport protein responsible for reabsorption of glucose from the kidney back to the bloodstream. Inhibiting this protein effectively lowers the glucose level of diabetic patients; however, the use of synthetic SGLT-2 inhibitors has been linked to some serious adverse effects. There is a need to identify safer alternatives that are equally or more effective as the current inhibitor drugs. Phytochemicals are known for their efficacy as herbal remedies, but these molecules remain underexplored as source of therapeutic agents. In this study, we performed in silico screening to identify potential SGLT-2 inhibitors from the 21 phytochemicals from Centella asiatica. Docking results identified eleven compounds with estimated binding energies comparable to that of known inhibitors drugs. The stability of the complexes was then elucidated using 100 ns MD simulations. From our dynamic binding free energy calculations using MM/PBSA, asiaticoside, betulinic acid, centellasapogenol, methyl brahmate, and rutin exceeded at least one of the binding energies of the reference compounds, which highlights their strong affinity towards SGLT-2. Among the five, betulinic acid, centellasapogenol, and methyl brahmate maintained their structural stability to the same extent as the references and exhibited better oral bioavailability and excellent drug-like properties. Because of these results, it is recommended to prioritize betulinic acid, centellasapogenol, and methyl brahmate in future in vitro and in vivo studies to verify their potential as inhibitor drugs for diabetes therapies.
Communicated by Ramaswamy H. Sarma
Introduction
Diabetes is a chronic disease characterized by an increased level of glucose in the blood due to the body’s inefficiency to produce or use insulin. It is mostly common to adults with 8.5% prevalence and tends to be diagnosed in older people in their 40–50s (Centers for Disease Control and Prevention, Citation2020). If left uncontrolled, diabetes can lead to several complications including heart attack, kidney disease, lower-limb amputations, and adult-onset blindness. Additionally, 43% of diabetic patients die prematurely before the age of 70 due to complications (Centers for Disease Control and Prevention, 2020; Roglic, Citation2016). In the Philippines, diabetes has one of the highest mortality rates, accounting for 33,295 deaths in 2016, and it is also the 6th leading cause of death in the country (Bersales, Citation2018).
Sodium-glucose co-transporter 2 (SGLT-2) inhibitors are among the newest class of anti-hyperglycemic medications to receive approval from the FDA (Hsia et al., Citation2017). SGLT-2 is the most prevalent sodium-glucose co-transporter subtype which facilitates 90% of glucose reabsorption in the kidney back to the bloodstream (Hsia et al., 2017). It is therefore an ideal target for the treatment of diabetes because of their large contribution to glucose reabsorption. SGLT-2 inhibitors have a unique mechanism that is independent of the level of insulin secretion–they block glucose attachment to the protein in the kidney and disrupt the main function of the sugar transporter thereby preventing glucose reabsorption to the bloodstream. Once SGLT-2 transport of glucose is inhibited, glucosuria or the excretion of glucose through the urine is induced to lower the sugar level of the body.
The binding site of the SGLT-2 protein is sandwiched by a group of residues that form intracellular and extracellular gates. In the intracellular gate, Tyr 263 forms the strongest van der Waals interactions to hold the glucose molecule and prevents the sugar’s exit further into the cavity (Tamura et al., Citation2015). The extracellular gate, composed of Met 73, Tyr 87, and Phe 424, hinders the detachment of glucose (Wright et al., Citation2011). Certain residues such as Ala 294, Gln 428, Gln 69, Glu 68, Glu 88, Ser 91, and Asn 260 were also determined to be of importance in the absorption mechanism (Tamura et al., 2015; Wright et al., 2011). Among these, Gln 428 is established to be the most vital because of its direct interaction with the sugar for absorption and any mutation in this residue hinders sugar absorption (Rizvi et al., Citation2013). Additionally, Glu 88 and Glu 68 provides the strongest electrostatic interactions – a significant contributor to the strong binding affinity observed between the sugar molecule and the protein (Tamura et al., 2015)
There are currently three approved SGLT-2 drugs by the FDA – canagliflozin (CAN), dapagliflozin (DAP), and empagliflozin (EMP) – while some are still undergoing clinal trials (Hsia et al., 2017). However, the use of these drugs are associated to the development of urinary tract infections and genital infection (Singh & Kumar, Citation2018). Aside from these common adverse effects of SGLT-2 inhibitors, the chemically synthesized SGLT-2 inhibitor drugs carry additional health risks. An increased risk of bone fractures and decreased bone mineral density have been linked to the use of canagliflozin, for which the FDA issued a relevant warning (Tentolouris et al., Citation2019). Lower limb amputations and pancreatitis – though exceedingly rare – were also observed from the use of this drug. For dapagliflozin, patients reportedly developed nasopharyngitis, a unique adverse reaction of the drug (Sha et al., Citation2015), while empagliflozin has been correlated with an increased chance of upper respiratory infections which led to a drug label warning from the FDA. The exact mechanism of this adverse effects remains unknown so whether this is caused by SLGT-2 inhibition or not is yet to be verified (Tentolouris et al., 2019). Because of the occurrence of these undesirable side effects, there is a need for novel agents that may offer superior efficacy and safer treatment options.
One alternative source of drug candidates are phytochemicals or bioactive compounds from plants whose vast structural diversity of natural molecules remains largely untapped. 80% of the population from developing countries still relies on herbal extracts because of their proven safety and efficacy in traditional medicine (Veeresham, Citation2012). Natural compounds have also been established in various in vivo and in vitro studies as one of the major sources of anti-diabetic agents (Jugran et al., Citation2020; Kabir et al., Citation2014; Supkamonseni et al., Citation2014). The first SGLT-2 inhibitor discovered, phlorizin, was a natural compound isolated from the bark of an apple tree; however, the work was discontinued because of the compound’s non-selective nature and poor oral bioavailability (Kalra, Citation2014). Following this, numerous studies began characterizing extracts from plants as anti-diabetic inhibitors or activator of various proteins but only few explored the inhibition of the SGLT-2 protein (Choi, Citation2016; Hung et al., Citation2012). Thus, efforts to identify novel SGLT-2 inhibitors derived from plants are still on-going.
In this study, we explored the potential of the phytochemicals from Centella asiatica as an SGLT-2 inhibitor (). Centella asiatica is a small and edible perennial plant with a wide range of folk medicinal applications indigenous to Asian and African countries. Extracts from the plant have been found to exhibit antioxidant (Pittella et al., Citation2009), antifungal, antibacterial (Dash et al., Citation2011), and anti-inflammatory (George & Joseph, Citation2009) properties mostly attributed to its vast number of phytochemicals, specifically triterpenes and flavonoids (Hashim et al., Citation2011). Although Centella asiatica’s main usage in folk medicine is to treat skin lesions and promote wound healing (Gohil et al., Citation2010), some in-vivo studies have probed the use of the plant’s extract as a cure for diabetes. A significant reduction of glucose by up to 69% was observed in induced diabetic rats from the methanolic extract of the plant which was highly comparable to the effects of the known diabetic drug, glibenclamide (Chauhan et al., Citation2010). There was also a protective effect against diabetes which may be due to the plant’s high number of flavonoids working synergistically. Two more recent studies identified the triterpene, asiaticoside (AST), and the flavonoid, rutin (RUT) as the active ingredients responsible for the anti-diabetic activity observed in rats. It was also theorized that asiaticoside enhanced the insulin secretion of pancreatic β-cells that led to the reduced glucose levels (Fitrianda et al., Citation2017). Rutin, on the other hand, was shown to slow down carbohydrate digestion and glucose absorption in the body (Supkamonseni et al., 2014). But the exact pharmacological mechanisms of these effects are not yet clear, thus glycemic control due to SGLT-2 inhibition could be a possibility. The continuous use of Centella asiatica in herbal medicine, and its perceived therapeutic efficacy and versatility has driven the scientific community to accumulate large bodies of evidence to highlight the immense potential of this plant to cure various diseases (Gohil et al., 2010). To the best of our knowledge, an investigation of the inhibitory properties of Centella asiatica’s phytochemicals against SGLT-2 have never been done. This study aims to test if Centella asiatica is a potential source of safer alternative drug molecules capable of inhibiting SGLT-2 to lower the chances of diabetes complications.
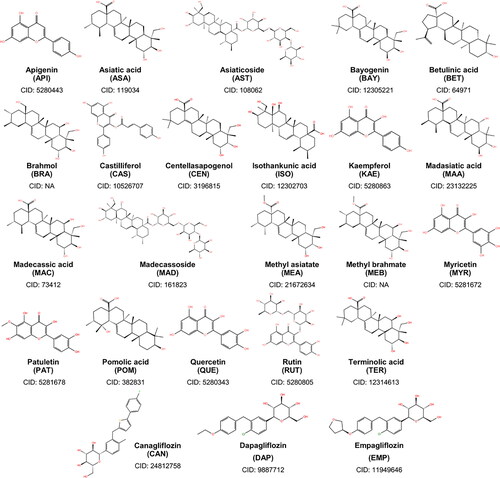
Figure 1. Two-dimensional structures of the 21 phytochemicals of Centella asiatica considered in this study. Also shown are the structures of canagliflozin, dapagliflozin, and empagliflozin – the known SGLT-2 inhibitors.
Materials and methods
The phytochemical structures of triterpenes and flavonoids (, Supplementary Materials) of Centella asiatica were obtained from PubChem (www.pubchem.com) (Kim et al., Citation2016). The structures of brahmol (BRA) and methyl brahmate (MEB) are unavailable in PubChem so their structures were constructed in Maestro (Schrödinger, Citation2020a) and were prepared in Avogadro 1.2 (Hanwell et al., Citation2012) using General AMBER Force Field (GAFF). Steepest descent algorithm with 10,000 iterations was employed to optimize and establish the relaxed conformation. In total, 21 triterpenes and flavonoids were considered in this study. The protein structure of SGLT-2 (PDB ID: 2XQ2) (Watanabe et al., Citation2010) (, Supplementary Materials) was imported from the Research Collaboratory for Structural Bioinformatics Protein Data Bank (www.rcsb.org) (Rose et al., Citation2016).
Table 1. ADMET and drug-likeness results of the ligands and the references.
ADMET and drug-like property evaluation
The QikProp module of Schrödinger (Schrödinger, Citation2020b) was used to evaluate the absorption, distribution, metabolism, elimination, and toxicity (ADMET) properties of all the 21 phytochemicals and the three known SGLT-2 inhibitors. There are 45 descriptors and parameters from the QikProp modules, each with their own range of accepted values. Part of the QikProp descriptors is Lipinski’s Rule of Five (Ro5), a simple yet widely recognized rule of thumb to evaluate the drug’s oral absorption. The rule states that an ideal candidate drug must have less than five H-bond donors and less than ten H-bond acceptors, must weigh less than 500 g/mol, and the calculated octanol-water partition coefficient log P must be less than five. Having more violations from these guidelines increases the likelihood of poor solubility and permeability (Lipinski, Citation2004). Two additional servers were also used to evaluate the compounds’ drug-likeness and toxicity. Molsoft (www.molsoft.com) (Molsoft, Citation2018) provides the overall drug-likeness score and ChemBioSever (www.chemibioserver.com) (Athanasiadis et al., Citation2012) detects the presence of some known toxic moieties.
Molecular docking
Molecular docking was performed in Autodock 4.2 (Morris et al., Citation2009) using Lamarckian Genetic Algorithm (LGA). The ligands were treated as flexible molecules and the receptor was kept rigid. The grid center coordinates for the binding cavity of SGLT-2 were set to 2.111, 37.221, 52.376 in the x, y, z direction, and a grid box size of 40 × 40 × 40 Å with 0.375 Å spacing was used to cover the known active residues within the binding cavity. The location of the active site of SGLT-2 was determined using the known residues that surround the active pocket of the protein (Deng et al., Citation2014; Wright et al., 2011). For exhaustiveness, the number of GA runs was set to 1000, with 1000 poses generated per run. Only the best pose per run was saved. This is to get as close as possible to the correct conformation. The docking score of each pose was estimated using the binding free energy (ΔG, EquationEquation (1)(1)
(1) ), which is the sum of the energies from van der Waals and H-bond interactions, desolvation energy, electrostatic energy, and torsional free energy (Morris et al., 2009). Aside from the 21 phytochemicals, three known SGLT-2 inhibitors are used as control compounds.
(1)
(1)
From the 1000 poses generated, only the binding pose with the most negative docking score was chosen for further analysis. The ligands and references were evaluated using the same scoring function. The docking scores were used to rank the compounds to determine which ones scored higher than the known inhibitors. The free energy of binding of each of the top poses of the 21 ligands was estimated using Molecular Mechanics/Poisson-Boltzmann Surface Area (MM/PBSA) methods. Docking experiments and the MM/PBSA calculation were done in triplicates.
Molecular dynamics simulations
Molecular dynamics (MD) simulations were carried out using Groningen Machine for Chemical Simulations (GROMACS) 2019.3 (Lemkul, Citation2018) on complexes with binding energies stronger than at least one of the references, estimated either by the docking scoring function or MMM/PBSA. The force field parameter utilized for the protein is the November 2018 version of Chemistry at Harvard Macromolecular Mechanics (CHARMM) 36 while the ligands’ force field parameters were generated from the CGenFF (https://cgenff.umaryland.edu/) server of the University of Maryland Baltimore (Vanommeslaeghe et al., Citation2010). A cubic box with periodic boundary conditions was used to contain the system, 10 Å from the protein’s edge. The box was then solvated using three-site transferable water model (TIP3P), to which Na+ ions were added to neutralize the system. Additional 0.15 M Na+ and Cl- ions were added to mimic the physiological conditions. A 5,000-step structure optimization with steepest descent algorithm was performed to minimize the energy of the system, followed by a 100 ps equilibration in the NVT and NPT ensembles using V-rescale thermostat and Berendsen barostat for temperature and pressure coupling. The cutoff for both van der Waals and electrostatic interactions were kept at 1.2 nm, and the Particle Mesh Ewald method was used to calculate the long-range electrostatic interactions. In the production run, the temperature and pressure was kept at 310 K and 1 bar ( and , Supplementary Materials). V-rescale thermostat with a time constant of 0.1 ps was used, while pressure was controlled by the Berendsen barostat using a time constant of 1 ps and a compressibility of 4.5 × 10−5 bar−1. Simulation run was 100 ns and snapshots of the trajectory were saved every 100 ps. All MD simulations were done in triplicates. The stability of the resulting trajectories were then evaluated using the root-mean-square deviation (RMSD), root-mean-square fluctuations (RMSF), hydrogen bond interactions, and MM/PBSA tools of GROMACS. In the H-bond analysis, the cutoff angle and distance were set to 30° and 3.5 nm, respectively.
Figure 2. Reside interactions within 4.0 Å of the top eleven ligands and references with the SGLT-2 protein. References: (A) CAN, (B) DAP, (C) EMP. Candidate ligands: (D) AST, (E) BET, (F) CAS, (G) CEN, (H) ISO, (I) MAA, (J) MAD, (K) MEB, (L) MYR, (M) QUE, (N) RUT. Majority of the interactions were hydrophobic and polar while only a few electrostatic contacts were observed. Three residues (Glu 68, Ile 427, and Tyr 263) were also observed to have interacted with all the candidate ligands and the references.
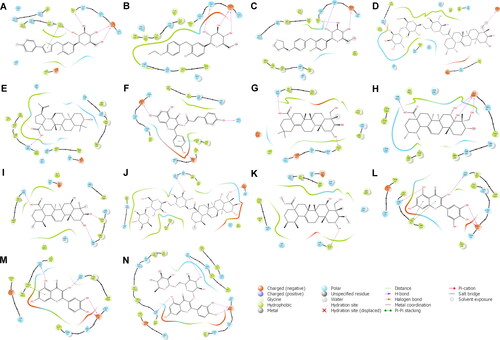
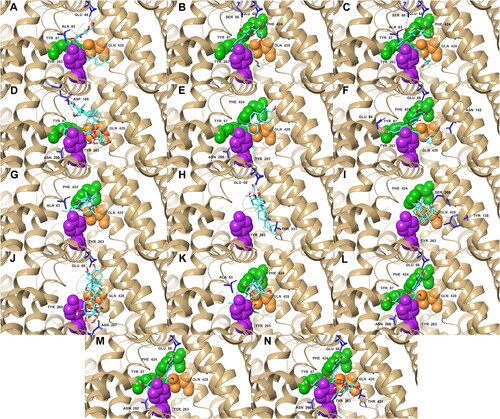
Figure 3. Docking poses of the top five ligands and the references inside the binding pocket of SGLT-2. Also shown are the some of the interacting residues and their corresponding interaction types: hydrogen bonds (pink); aromatic hydrogen bonds (blue); pi-pi stacking (black). References: (A) CAN, (B) DAP, (C) EMP. Candidate ligands: (D) AST, (E) BET, (F) CAS, (G) CEN, (H) ISO, (I) MAA, (J) MAD, (K) MEB, (L) MYR, (M) QUE, (N) RUT. Extracellular (Phe 424, Tyr 87, Met 73) and intracellular (Tyr 263) gate residues are represented with green and purple spheres, respectively. Gln 428, the residue with the most critical role in the absorption mechanism, is represented with orange spheres.
Molecular mechanics/Poisson Boltzmann surface area
The Molecular Mechanics/Poisson Boltzmann method gives a more accurate estimation for the free energy of binding than the scoring function of docking experiments (Genheden & Ryde, Citation2015). It incorporates three energetic terms, including the molecular mechanics energies (bonded and non-bonded interactions), polar solvation energy, and non-polar solvation energy to solve for the change in the free energy.
The g_mmpbsa tool (Kumari et al., Citation2014), a GROMACS compatible package, is used to implement the MM/PBSA calculations, and the python script MmPbSaStat.py provided in the g_mmpbsa package, compiles and estimates the interaction free energies. The tool currently does not include the calculation of the entropic energy and is therefore unable to give the absolute binding energy (Kumari et al., 2014).
The bonded interactions consisting of bond, angle, dihedral, and improper interactions of both the ligand and protein in the bound and unbound forms are assumed to not change so the first term of Equation (2) is zero. Non-bonded interactions include both electrostatic and van der Waals energies, which are based on the molecular mechanics force-field parameters. The last two terms, and
are the energies required to transfer a solute from a vacuum into the solvent. The former is estimated from the Poisson − Boltzmann (PB) equation, while latter makes use of solvent accessible surface area (SASA) model to estimate the repulsive and attractive forces associated with the cavity formation and van der Waals interactions (Kumari et al., 2014). Thus, Equation (2) can be transformed into:
The clustering tool of GROMACS was also used to group together similar conformational states that are within 2.0 Å RMSD of each other in the stable phase of the trajectory. The resulting structures represent the most probable binding conformation of the ligand inside the protein.
Results and discussion
ADMET and drug-likeness of ligands
From the 45 properties and descriptors of QikProp, the three references scored favorably in 41 of these tests (). Methyl asiatate (MEA) obtained the highest consolidated QikProp score of 44, followed by bayogenin (BAY), madasiatic acid (MAA), and methyl brahmate (MEB), all with 43 points. Betulinic acid (BET), MEA, and MEB also had the most negative predicted aqueous solubility (QPlogS) that is within the recommended range, which correlates to high oral bioavailability. Moreover, the partition values (QPlogPC16, QPlogPoct, QPlogPw) of these compounds indicate high lipophilicity resulting to good passive diffusion and high gastrointestinal absorption (Shaikh & Siu, Citation2016). In addition, MEB had a ‘high’ HOA classification and scored 95.84% in PHOA, the highest among all phytochemicals and control compounds. Based on these prediction results, it is possible that MEB is a more potent drug than the three known SGLT-2 (PDB ID: 2XQ2) inhibitors.
Three compounds, asiaticoside (AST), madecassoside (MAD), and rutin (RUT), were rated poorly in QikProp, with an aggregate score of 23, 25, and 30 respectively. They also each incurred three violations of the Ro5 guidelines. These ligands got a ‘low’ classification in HOA and obtained a zero in PHOA. They also did not meet the criteria set on the allowable number of hydrogen bond donor, hydrogen bond acceptor, and molecular weight due to their relatively large structures. In accordance with Lipinski’s Rule of Five, these three compounds will have low chances of good permeability and solubility when taken orally. However, it also important to consider that Ro5 is an observation made only on known orally administered synthetic drugs and the rule does not include natural or semi-natural produced drugs. It has been determined that only 51% of all US FDA-approved drugs are compliant to Lipinski’s Rule, and macrocyclic and macrolide antibiotic drugs (i.e. erythromycin, cyclosporine, etc.), with molecular weight over 700 g/mol, obviously do not comply to these rules (Zhang & Wilkinson, Citation2007). But molecules with more than one violation should not be necessarily removed from further consideration but shall only receive low prioritization in the succeeding steps (Petit et al., Citation2012). RUT and AST have also been shown in in-vivo studies to possess anti-diabetic properties so it is worth studying whether the compounds’ glycemic control is a result of the inhibition of the SGLT-2 protein (Fitrianda et al., 2017; Supkamonseni et al., 2014). Moreover, among all of Centella asiatica’s phytochemicals, ASA, AST, MAD, and MAA are of primary interest since these are considered to be the plant’s most biologically active compounds (Hashim et al., 2011). Hence, even with poor oral bioavailability, AST, MAD and RUT were still included in the subsequent analysis. Other methods of drug administration could still be explored.
The online server, Molsoft (Molsoft, Citation2018) was also used to quantify the drug-likeness of a molecule. It calculates the drug-likeness score of the compounds using an algorithm based on the subject’s pharmacokinetic properties. These scores are then compared to the drug-likeness scores of known drugs (positives) and non-drugs (negatives) from their own set of databases. A drug-likeness score around 0 to 2 describes a drug-like molecule while a score between −6 to −1 corresponds to a non-drug compound. The drug-likeness scores of the majority of the ligands except for MYR are within the prescribed range (), indicating drug-like characteristics that are similar to most of the commercially approved drugs.
Molecular docking
The binding conformation and orientation of the phytochemicals were virtually the same in all three experiments for each ligand. From the 21 ligands, five exceeded the estimated binding energy of at least one of the references (). AST (–9.833 kcal/mol), MAD (–10.236 kcal/mol), and, RUT (–10.25 kcal/mol) had docking scores higher than all the reference drugs (–8.873 kcal/mol to −9.947 kcal/mol) suggesting that these three compounds have stronger binding affinities towards SGLT-2, while methyl brahmate (MEB) (–9.513 kcal/mol) and castilliferol (CAS) (–9.897 kcal/mol) only scored higher than one reference compound, DAP (–8.873 kcal/mol).
Table 2. Average docking and MM-PBSA scores of the 21 phytochemicals from the three trials.
Eleven compounds had stronger MM/PBSA binding free energies than at least one of the references. The top five compounds with the highest docking scores still obtained stronger MM/PBSA binding energies than the references (–11.595 kcal/mol to −12.168 kcal/mol). RUT also still obtained the highest energy (–15.266 kcal/mol) from all phytochemicals tested. We can observe from the results that the MM/PBSA energies do not necessarily follow the docking scores. For example, BET got the second highest MM/PBSA energy (–14.700 kcal/mol) but was not part of the top compounds with the best docking scores. This is also apparent on some compounds including centellasapogenol (CEN), isothankunic acid (ISO), madasiatic acid (MAA), myricetin (MYR), and quercetin (QUE), which all had favorable MM/PBSA energies. The difference in the scores could be due to the inclusion of polar and non-polar solvation energies which resulted to a more negative binding energy in the MM/PBSA method (Thompson et al., Citation2008). Only the ligands with a stronger binding affinity than at least one of the references, evaluated either by the docking scoring function or MM/PBSA, were included in the succeeding experiments. These are AST, BET, CAS, CEN, ISO, MAA, MAD, MEB, MYR, QUE, and RUT.
Ligand – receptor interactions
The majority of the residues in the binding site of SGLT-2 that interacted with the three references, also interacted with the eleven candidate ligands (). Most of interactions are hydrophobic and polar in nature, with only a few electrostatic interactions were present (). Hydrophobic interactions are the most common type of interactions in protein-ligand complexes, and usually determine the overall stability of a drug-receptor interactions (de Freitas & Schapira, Citation2017). Gln 428 and the external gate residue Tyr 263, which both hold critical roles in binding activity, were observed to form polar and hydrophobic interactions with the majority ligands, respectively ( and ). In contrast, there were no visible interaction with the gate residue Met 73 in any of the compounds, but interactions with the other extracellular gate residues, such as Tyr 87 and Phe 424, were present in some (). MAD was the only ligand to miss a contact with any of the external gate residues. Electrostatic interaction with Glu 68 was also observed to be consistent with all the ligands and the references. Aside from its electrostatic contribution, Glu 68 also produced hydrogen bonding (H-bonding) interactions with all the eleven candidate ligands (). H-bonding have been shown in studies to have significant importance in conferring stability to protein-ligand interactions (Li et al., 2018; Wang et al., Citation2011). Glu 68 produced three H-bonds with ISO, two H-bonds with CAN, DAP, MYR, QUE, and RUT, and one H-bond with the rest of the ligands except for AST and MEB – the only two compounds that Glu 68 failed to pair with (). H-bond contacts were also present on other residues including Ala 63, Asn 260, Asn 267, Asp 189, Gln 428, Glu 88, Ser 66, Ser 368, Thr 431, Tyr 138, while CEN, MYR, and QUE formed H-bond connections with Gln 428 – one of the important residues.
Table 3. Common interacting residues among the ligands with the highest docking scores and the references within 4.0 Å.
Table 4. H-bond interactions of the top candidate ligands.
Molecular dynamics simulations
To evaluate the stability of the resulting complexes from molecular docking, a time dependent MD simulation was conducted for 100 ns using GROMACS 2019.3. Two types of RMSD were used to quantify how much the complex has changed over the simulation time. Complex-RMSD () describes the movement of both the ligand and the protein backbone while ligand-RMSD () shows the fluctuation of the ligand alone inside the binding pocket – both of which are used to describe the stability. tabulates the average complex-RMSD and ligand-RMSD on the last 10 ns of the simulation.
Figure 4. Complex-RMSD of the top five ligands and the three references docked against the binding site of SGLT-2. References: (A) CAN, (B) DAP, (C) EMP. Candidate ligands: (D) AST, (E) BET, (F) CAS, (G) CEN, (H) ISO, (I) MAA, (J) MAD, (K) MEB, (L) MYR, (M) QUE, (N) RUT.
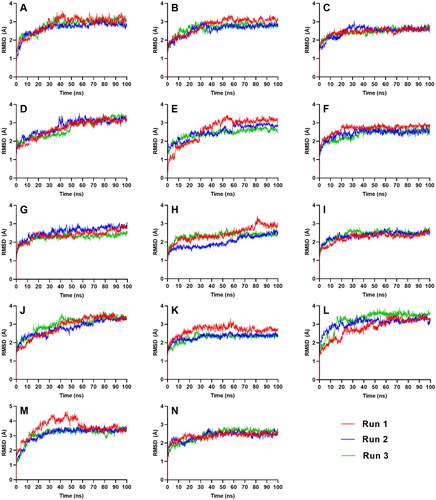
Figure 5. Ligand-RMSD of the top five ligands and the three references docked against the binding site of SGLT-2. References: (A) CAN, (B) DAP, (C) EMP. Candidate ligands: (D) AST, (E) BET, (F) CAS, (G) CEN, (H) ISO, (I) MAA, (J) MAD, (K) MEB, (L) MYR, (M) QUE, (N) RUT.
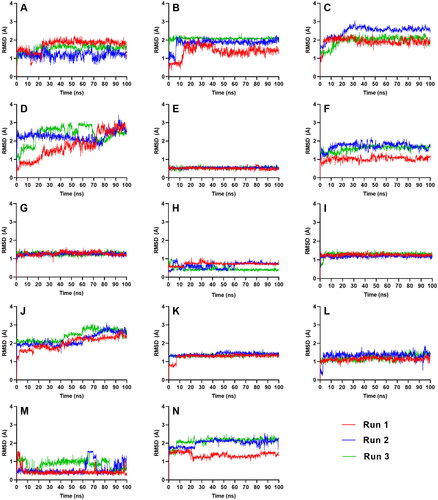
Table 5. Complex-RMSD and Ligand-RMSD of the top eleven promising compounds.
All complexes except for AST and MAD equilibrated within the simulation time of 100 ns, achieving an RMSD range between 1 to 3 Å. CAS, CEN, MAA, MEB, and RUT stabilized the quickest at around 40 ns (), while BET, MYR, and QUE equilibrated at around 60 ns. ISO took a longer time before it reached equilibrium at 80 ns in two of the three runs.
If we look at the average number of H-bond pairs from all three runs (), CEN, MYR, MEB and QUE, had consistent number of H-bonds throughout the simulation. MEB even had a gradual increase of H-bonding towards the end which further conferred the rigidity of the complex. BET, CAS, and MAA may have fewer H-bonding, but these molecules are smaller, so the number of its H-bond contacts were just enough to hold the ligands in place. ISO formed the least number of H-bond pairs that may have affected the complex’s stabilization. Meanwhile, RUT’s H-bonds connections sharply decreased at the 40 ns mark but remained consistent afterwards.
Figure 6. Average number of intermolecular hydrogen bond interactions from the three runs throughout the entire simulation. References: (A) CAN, (B) DAP, (C) EMP. Candidate ligands: (D) AST, (E) BET, (F) CAS, (G) CEN, (H) ISO, (I) MAA, (J) MAD, (K) MEB, (L) MYR, (M) QUE, (N) RUT.
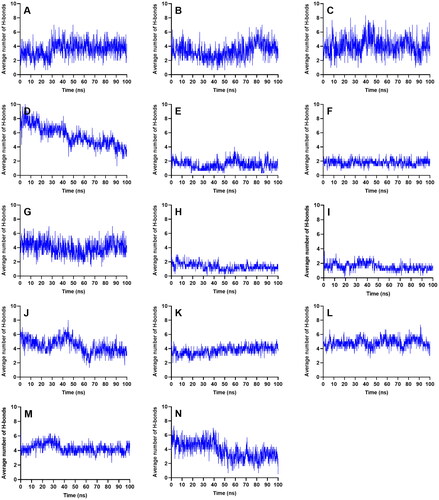
AST and MAD had increasing complex-RMSD () towards the latter part of the simulation, and large and erratic fluctuations were also observed in their ligand-RMSD (). The number of H-bonds contacts of AST continued to slope downwards (), and while MAD registered a steady number of H-bonds pairs, it was relatively low considering its size so it may have needed more H-bond connections to fully secure itself inside the binding pocket of SGLT-2. The large structures of AST and MAD may have caused the ligands to form interactions outside the binding region.
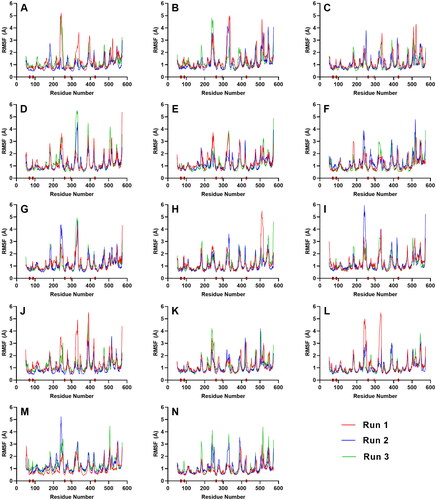
RMSF plots are then used to describe the flexibility of each amino acid residue during molecular dynamics simulations (Benson & Daggett, Citation2012). It was observed that the RMSF values of the residues constituting the active site of SGLT-2 were only slightly changed (). The low values at the troughs describe the residues’ reduced movements which prevented the binding site to undergo drastic structural changes and made it less susceptible to rearrangement. Since the shape of the site was not altered during the simulation, this allowed the ligands to stay and fully latch themselves inside the cavity. The brown bars on the x-axis represent the position of the critical residues including Gln 428, and the gate residues, Met 73, Tyr 263, Phe 424, and Tyr 87. As seen in , all these residues had low RMSF values. It follows that the ligands that sufficiently formed contacts with the aforementioned active site residues also had constrained movements.
Figure 7. RMSF plot of the top five ligands and the three references docked against the binding site of SGLT-2. References: (A) CAN, (B) DAP, (C) EMP. Candidate ligands: (D) AST, (E) BET, (F) CAS, (G) CEN, (H) ISO, (I) MAA, (J) MAD, (K) MEB, (L) MYR, (M) QUE, (N) RUT. The brown bars in the x axis represent the location of critical residues including Gln 428, and the gate residues, Met 73, Tyr 263, Phe 424, and Tyr 87.
MM/PBSA analysis
The binding affinities (Equation (3)) of the eleven complexes were estimated from the average MM/PBSA binding free energies using the last 10 ns (last 100 frames) of the trajectories.
Five ligands (AST, BET, CEN, MEB, RUT) obtained more negative MM/PBSA energies than at least one of the references. MEB (–31.07 kcal/mol) and BET (–29.60 kcal/mol) registered the strongest binding energies and surpassed the energies of all the references (EMP: –18.113 kcal/mol, DAP: –21.16 kcal/mol, CAN: –28.763 kcal/mol) (). CEN got the third highest binding free energy (–24.64 kcal/mol) but it was not able to match the energy of CAN (–28.763 kcal/mol). AST (–19.10 kcal/mol) and RUT (–18.59 kcal/mol) only managed to score higher than the reference EMP (–18.11 kcal/mol), while CAS, ISO, MAA, MAD, MYR, and QUE failed to surpass the binding energies of any of the reference compounds.
Table 6. Average binding free energies of the top ligands.
The magnitude of the energetic components of MEB closely resembles the values obtained by the two references, CAN and DAP. The strong van der Waals interactions of MEB, BET, and CEN compensated for their weak electrostatic forces that led to a considerably high binding affinity. ISO and MAA had similar electrostatic, polar solvation, and SASA energies as MEB, BET, and CEN, but their van der Waals energies were not as strong. It was also observed that molecules with higher polar surface area ( (PSA), Supplementary Materials) such as AST, MYR, and QUE, and RUT tend to have higher polar solvation energies but more negative electrostatic energies. Polar solvation energy, along with the non-polar solvation energy, constitutes the energy required to transfer a solute from a vacuum into the solvent. It describes the work done for moving a partial charge of a solute in a reference medium to another point in a solvent medium (Izairi & Kamberaj, Citation2017; Kumari et al., 2014). It is expected that molecules with large dipole moments or high polar surface area (PSA) would require more work in the desolvation process. This could explain the high polar solvation energies of AST, MAD, MYR, QUE, and RUT. BET, CEN, and MEB are not as polar as all the other ligands ( (PSA), Supplementary Materials) and only required relatively low polar solvation energies in the coupling process (). RUT obtained one of the most negative electrostatic energies, which can be correlated to its high dipole moment – the highest among all ligands. When all the energetic terms are totaled, RUT was still able to surpass the binding energy of one of the references, EMP, hence RUT can also be considered as a potential candidate molecule.
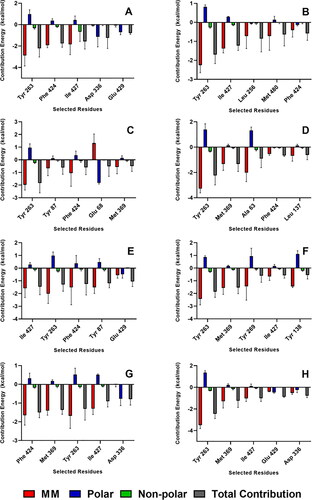
Decomposition of the binding free energy on a per residue basis of the ligands with the strongest MM/PBSA energies (AST, BET, CEB, MEB, RUT) is shown in , while the decomposition of top contributing residues into energy contributions from molecular mechanics energy, polar solvation energy, and non-polar solvation energy is presented in . The residues Tyr 263, Phe 424, Met 369, and Ile 427 were among the highest contributors to the observed negative binding free energies (). It is also notable to add that Tyr 263 and Phe 424 are part of the gate residues, which are important in the inhibition mechanism of SGLT-2. Tyr 263 consistently imparted the strongest molecular mechanics energies which largely compensated the unfavorable positive solvation energy (). Residues Tyr 263, Phe 424, Met 369, and Ile 427 also contributed positive polar solvation energies which favors complex dissociation. Nevertheless, the total energy contribution from the three energetic terms still favored complex formation driven by the strong intermolecular interactions between the ligand and the protein. On the other hand, Asp 336 and Gln 429 provided the most negative polar solvation energies in the RUT-receptor complex. Asp 336 and Gln 429 also imparted favorable polar solvation energies in the MEB-receptor and BET-receptor complexes, respectively.
Figure 8. Decomposition of the binding free energy on a per residue basis of the complexes with the strongest MM/PBSA energies. References: (A) CAN, (B) DAP, (C) EMP. Final candidate ligands: (D) AST, (E) BET, (F) CEN, (G) MEB, (H) RUT.
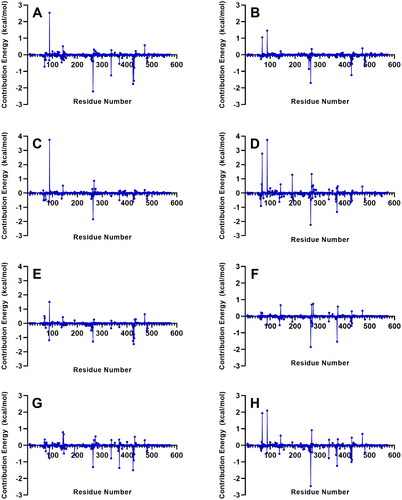
Figure 9. Residues with the highest energy contribution from the complexes with the strongest MM/PBSA energies. Also shown is the decomposition of the binding free energy on a per-residue basis into contributions from the molecular mechanics (van der Waals + electrostatic) energy, polar solvation energy, and non-polar solvation energy. References: (A) CAN, (B) DAP, (C) EMP. Final candidate ligands: (D) AST, (E) BET, (F) CEN, (G) MEB, (H) RUT.
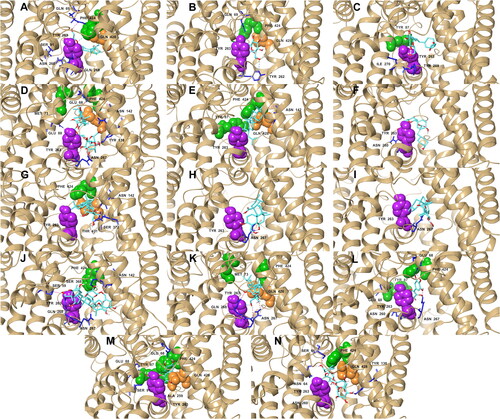
Geometric clustering within 2.0 Å RMSD was done on the last 10 ns of the simulations to generate representative structures of each complex (). These representative structures were used to determine ligand-residue contacts. BET, CEN, ISO, MEB, QUE, retained the hydrophobic and polar interactions with the important residues that were also present in their initial docking pose (). BET and QUE kept their interactions with Phe 424, Tyr 87, Tyr 263, and Gln 428; CEN and MEB with Phe 424, Tyr 263, and Gln 428; and ISO with Tyr 263. AST and RUT were unsuccessful to sustain an interaction with Tyr 87; nonetheless, AST formed new interactions with two external gate residues, Met 73 and Phe 424. MEB also established a hydrophobic interaction with Met 73, the external gate residue that did not form any connections with any of the ligands before the MD simulations. MYR lost its interaction with Gln 428 but still interacted with three gate residues. CAS, MAA and MAD also lost their initial contacts with all the important residues except Tyr 263, but MAD made a new connection with Phe 424. Interactions with important residues Tyr 263, Phe 424, and Gln 428 were also noticeably present in the ligands with the strongest MM/PBSA energies (AST, BET, CEN, MEB, and RUT). These observations point towards the high potential of AST, BET, CEN, MEB, and RUT as inhibitors of SGLT-2.
Figure 10. Representative structures of the most populated cluster of conformations that are within 2.0 Å RMS of each other in the last 10 ns of the trajectory: hydrogen bonds (pink); aromatic hydrogen bonds (blue); pi-pi stacking (black). References: (A) CAN, (B) DAP, (C) EMP. Candidate ligands: (D) AST, (E) BET, (F) CAS, (G) CEN, (H) ISO, (I) MAA, (J) MAD, (K) MEB, (L) MYR, (M) QUE, (N) RUT. Extracellular (Phe 424, Tyr 87, Met 73) and intracellular (Tyr 263) gate residues are represented with green and purple spheres, respectively. Gln 428, the residue with the most critical role in the absorption mechanism, is represented with orange spheres.
Conclusion
Molecular docking experiments were performed on 21 phytochemicals from Centella asiatica, a known folk-medicinal plant, and three reference drugs, onto the SGLT-2 receptor. This revealed 11 compounds (AST, BET, CAS, CEN, ISO, MAA, MAD, MEB, MYR, QUE, and RUT) with better docking scores or MM/PBSA energies than at least one of the references. 100-ns MD simulations were done on these 11 complexes to determine their stability, binding energies, and interacting residues. RMSD and RMSF trajectory analyses elucidated the stable behavior of the ligands in the binding region. MM/PBSA free energy calculations using the last 10 ns of the simulations showed that AST, BET, CEN, MEB, and RUT had stronger binding free energies than at least one of the references. Compounds with higher polar surface area tend to have weaker affinity towards the protein because of the larger polar solvation energy requirement during coupling. Detailed interaction analysis revealed the importance of the residues Tyr 263, Phe 424, Met 369, and Ile 427 for their molecular mechanics energy contributions, and Asp 336 and Glu 429, for their favorable polar solvation energies.
All these demonstrate the potential of AST, BET, CEN, MEB, and RUT as SGLT-2 inhibitors. The five candidates matched the binding affinities of known SGLT-2 inhibitor drugs, and formed favorable interactions with known critical residues of the receptor. Among the five, however, BET, CEN, and MEB have better absorption, distribution, metabolism, excretion properties and better equilibration profiles than AST and RUT. Their trajectories inside SGLT-2 are also comparable to those of the reference molecules. Thus, further in vitro and in vivo studies using BET, CEN and MEB as inhibitors against the SGLT-2 protein are recommended.
| Abbreviations | ||
| ADMET | = | absorption, distribution, metabolism, excretion, toxicity |
| ASA | = | asiatic acid |
| AST | = | asiaticoside |
| BET | = | betulinic acid |
| BAY | = | bayogenin |
| CAN | = | canagliflozin |
| CEN | = | centellasapogenol |
| CAS | = | castilliferol |
| DAP | = | dapagliflozin |
| EMP | = | empagliflozin |
| GROMACS | = | Groningen Machine for Chemical imulations |
| ISO | = | isothankunic acid |
| MAA | = | madasiatic acid |
| MAD | = | madecassoside |
| MD | = | molecular dynamics |
| MEA | = | methyl asiatate |
| MEB | = | methyl brahmate |
| MM/PBSA | = | molecular mechanics Poisson–Boltzmann surface area |
| MYR | = | myricetin |
| Ro5 | = | Lipinski’s Rule of Five |
| QUE | = | quercetin |
| RUT | = | rutin |
| SGLT-2 | = | Sodium-glucose co-transporter 2 |
Supplementary_Material.pdf
Download PDF (4 MB)Acknowledgments
The authors want to thank the Computing and Archiving Research Environment of the Department of Science and Technology – Advanced Science and Technology Institute (COARE-DOST-ASTI), the Engineering Research and Development for Technology of the Department of Science and Technology (ERDT-DOST), and the University of the Philippines Diliman – Office of the Vice-Chancellor for Research and Development (UP-OVCRD) for their support to this research.
Disclosure statement
The authors have no conflicts of interest to report.
References
- Athanasiadis, E., Cournia, Z., & Spyrou, G. (2012). ChemBioServer: A web-based pipeline for filtering, clustering and visualization of chemical compounds used in drug discovery. Bioinformatics (Oxford, England), 28(22), 3002–3003. https://doi.org/10.1093/bioinformatics/bts551
- Benson, N. C., & Daggett, V. (2012). A comparison of multiscale methods for the analysis of molecular dynamics simulations. The Journal of Physical Chemistry B, 116(29), 8722–8731. https://doi.org/10.1021/jp302103t
- Bersales, L. G. (2018). Deaths in the Philippines, 2016. Philippine Statistics Authority [WWW Document]. Retrieved October 17, 2020 from https://psa.gov.ph/content/deaths-philippines-2016
- Centers for Disease Control and Prevention. (2020). National diabetes statistics report, 2020. Centers for Disease Control and Prevention, US Department of Health and Human Services.
- Chauhan, P. K., Pandey, I. P., & Dhatwalia, V. K. (2010). Evaluation of the anti-diabetic effect of ethanolic and methanolic extracts of Centella asiatica leaves extract on alloxan induced diabetic rats. Advances in Biological Research, 4, 27–30.
- Choi, C.-I. (2016). Sodium-glucose cotransporter 2 (SGLT2) inhibitors from natural products: Discovery of next-generation antihyperglycemic agents. Molecules, 21(9), 1136. https://doi.org/10.3390/molecules21091136
- Dash, B. K., Faruquee, H. M., Biswas, S. K., Alam, M. K., Sisir, S. M., & Prodhan, U. K. (2011). Antibacterial and antifungal activities of several extracts of Centella asiatica L. against some human pathogenic microbes. Life Sciences and Medicine Research, 2011, 1–5.
- de Freitas, R. F., & Schapira, M. (2017). A systematic analysis of atomic protein–ligand interactions in the PDB. MedChemComm, 8(10), 1970–1981. https://doi.org/10.1039/C7MD00381A
- Deng, D., Xu, C., Sun, P., Wu, J., Yan, C., Hu, M., & Yan, N. (2014). Crystal structure of the human glucose transporter GLUT1. Nature, 510(7503), 121–125.
- Fitrianda, E., Sukandar, E. Y., Elfahmi, E., & Adnyana, I. K. (2017). Antidiabetic activity of extract, fractions, and asiaticoside compound isolated from Centella asiatica Linn. Leaves in alloxan-induced diabetic mice. Asian Journal of Pharmaceutical and Clinical Research, 10(10), 268–272. https://doi.org/10.22159/ajpcr.2017.v10i10.20419
- Genheden, S., & Ryde, U. (2015). The MM/PBSA and MM/GBSA methods to estimate ligand-binding affinities. Expert Opinion on Drug Discovery, 10(5), 449–461. https://doi.org/10.1517/17460441.2015.1032936
- George, M., & Joseph, L. (2009). Anti-allergic, anti-pruritic, and anti-inflammatory activities of Centella asiatica extracts. African Journal of Traditional, Complementary and Alternative Medicines, 6(4), 554–559. https://doi.org/10.4314/ajtcam.v6i4.57206.
- Gohil, K. J., Patel, J. A., & Gajjar, A. K. (2010). Pharmacological review on Centella asiatica: A potential herbal cure-all. Indian Journal of Pharmaceutical Sciences, 72(5), 546–556. https://doi.org/10.4103/0250-474X.78519
- Hanwell, M. D., Curtis, D. E., Lonie, D. C., Vandermeersch, T., Zurek, E., & Hutchison, G. R. (2012). Avogadro: An advanced semantic chemical editor, visualization, and analysis platform. Journal of Cheminformatics, 4(1), 17. https://doi.org/10.1186/1758-2946-4-17
- Hashim, P., Sidek, H., Helan, M., Sabery, A., Palanisamy, U. D., & Ilham, M. (2011). Triterpene composition and bioactivities of Centella asiatica. Molecules (Basel, Switzerland), 16(2), 1310–1322. https://doi.org/10.3390/molecules16021310
- Hsia, D. S., Grove, O., & Cefalu, W. T. (2017). An update on SGLT2 inhibitors for the treatment of diabetes mellitus. Current Opinion in Endocrinology, Diabetes, and Obesity, 24, 73.
- Hung, H.-Y., Qian, K., Morris-Natschke, S. L., Hsu, C.-S., & Lee, K.-H. (2012). Recent discovery of plant-derived anti-diabetic natural products. Natural Product Reports, 29(5), 580–606. https://doi.org/10.1039/c2np00074a
- Izairi, R., & Kamberaj, H. (2017). Comparison study of polar and nonpolar contributions to solvation free energy. Journal of Chemical Information and Modeling, 57(10), 2539–2553. https://doi.org/10.1021/acs.jcim.7b00368
- Jugran, A. K., Rawat, S., Devkota, H. P., Bhatt, I. D., & Rawal, R. S. (2020). Diabetes and plant-derived natural products: From ethnopharmacological approaches to their potential for modern drug discovery and development. PhytotherapyResearch,5(1), 223–245. https://doi.org/10.1002/ptr.6821
- Kabir, A. U., Samad, M. B., D’Costa, N. M., Akhter, F., Ahmed, A., & Hannan, J. M. A. (2014). Anti-hyperglycemic activity of Centella asiatica is partly mediated by carbohydrase inhibition and glucose-fiber binding. BMC Complementary and Alternative Medicine, 14(1), 1–14. https://doi.org/10.1186/1472-6882-14-31
- Kalra, S. (2014). Sodium glucose co-transporter-2 (SGLT2) inhibitors: A review of their basic and clinical pharmacology. Diabetes Therapy: Research, Treatment and Education of Diabetes and Related Disorders, 5(2), 355–366. https://doi.org/10.1007/s13300-014-0089-4
- Kim, S., Thiessen, P. A., Bolton, E. E., Chen, J., Fu, G., Gindulyte, A., Han, L., He, J., He, S., Shoemaker, B. A., Wang, J., Yu, B., Zhang, J., & Bryant, S. H. (2016). PubChem substance and compound databases. Nucleic Acids Research, 44(D1), D1202–D1213. https://doi.org/10.1093/nar/gkv951
- Kumari, R., Kumar, R., Consortium, O. S. D. D., & Lynn, A. (2014). g_mmpbsa-a GROMACS tool for high-throughput MM-PBSA calculations . Journal of Chemical Information and Modeling, 54(7), 1951–1962. https://doi.org/10.1021/ci500020m
- Lemkul, J. (2018). From proteins to perturbed hamiltonians: A suite of tutorials for the GROMACS-2018 molecular simulation package [Article v1. 0]. Living Journal of Computational Molecular Science, 1, 5068.
- Li, J., Liu, Z., Zhao, Y., Zhu, X., Yu, R., Dong, S., & Wu, H. (2018). Novel natural angiotensin converting enzyme (ACE)-inhibitory peptides derived from sea cucumber-modified hydrolysates by adding exogenous proline and a study of their structure–activity relationship. Marine Drugs, 16(8), 271. https://doi.org/10.3390/md16080271
- Lipinski, C. A. (2004). Lead-and drug-like compounds: The rule-of-five revolution. Drug Discovery Today: Technologies, 1(4), 337–341. https://doi.org/10.1016/j.ddtec.2004.11.007
- Morris, G. M., Huey, R., Lindstrom, W., Sanner, M. F., Belew, R. K., Goodsell, D. S., & Olson, A. J. (2009). AutoDock4 and AutoDockTools4: Automated docking with selective receptor flexibility. Journal of Computational Chemistry, 30(16), 2785–2791. https://doi.org/10.1002/jcc.21256
- Molsoft, L. L. C. (2018). Drug-Likeness and molecular property prediction. http://molsoft.com/mprop/ (Accessed on May 16, 2021)
- Petit, J., Meurice, N., Kaiser, C., & Maggiora, G. (2012). Softening the rule of Five-where to draw the line? Bioorganic & Medicinal Chemistry, 20(18), 5343–5351. https://doi.org/10.1016/j.bmc.2011.11.064
- Pittella, F., Dutra, R. C., Junior, D. D., Lopes, M. T., & Barbosa, N. R. (2009). Antioxidant and cytotoxic activities of Centella asiatica (L) Urb. International Journal of Molecular Sciences, 10(9), 3713–3721. https://doi.org/10.3390/ijms10093713
- Rizvi, S., Shakil, S., Biswas, D., Shakil, S., Shaikh, S., Bagga, P., & Kamal, M. A. (2013). Invokana (Canagliflozin) as a dual inhibitor of AChE and SGLT2: Advancement in Alzheimer’s disease-diabetes type 2 linkage via an enzoinformatics study. CNS Neurol Disord Drug Targets, 18, 18.
- Roglic, G. (2016). WHO global report on diabetes: A summary. International Journal of Noncommunicable Diseases, 1(1), 3. https://doi.org/10.4103/2468-8827.184853
- Rose, P. W., Prlić, A., Altunkaya, A., Bi, C., Bradley, A. R., Christie, C. H., Costanzo, L. D., Duarte, J. M., Dutta, S., & Feng, Z. (2016). The RCSB protein data bank: Integrative view of protein, gene and 3D structural information. Nucleic Acids Research, 45(D1), D271–D281. https://doi.org/10.1093/nar/gkw1000.
- Schrödinger. (2020a). Schrödinger release 2020-3: Maestro. Schrödinger, LLC.
- Schrödinger. (2020b). Schrödinger release 2020-3: QikProp. Schrödinger, LLC.
- Sha, S., Polidori, D., Farrell, K., Ghosh, A., Natarajan, J., Vaccaro, N., Pinheiro, J., Rothenberg, P., & Plum-Mörschel, L. (2015). Pharmacodynamic differences between canagliflozin and dapagliflozin: Results of a randomized, double-blind, crossover study. Diabetes, Obesity and Metabolism, 17(2), 188–197. https://doi.org/10.1111/dom.12418
- Shaikh, F., & Siu, S. W. (2016). Identification of novel natural compound inhibitors for human complement component 5a receptor by homology modeling and virtual screening. Medicinal Chemistry Research: An International Journal for Rapid Communications on Design and Mechanisms of Action of Biologically Active Agents, 25, 1564–1573. https://doi.org/10.1007/s00044-016-1591-1
- Singh, M., & Kumar, A. (2018). Risks associated with SGLT2 inhibitors: An overview. Current Drug Safety, 13(2), 84–91. https://doi.org/10.2174/1574886313666180226103408
- Supkamonseni, N., Thinkratok, A., Meksuriyen, D., & Srisawat, R. (2014). Hypolipidemic and hypoglycemic effects of Centella asiatica (L.) extract in vitro and in vivo. Indian Journal of Experimental Biology, 52, 965–971.
- Tamura, Y., Miyagawa, H., Yoshida, T., & Chuman, H. (2015). Binding interaction of SGLT with sugar and thiosugar by the molecular dynamics simulation. Biochimica et Biophysica Acta (BBA) - Biomembranes, 1848(11), 2799–2804. https://doi.org/10.1016/j.bbamem.2015.08.001
- Tentolouris, A., Vlachakis, P., Tzeravini, E., Eleftheriadou, I., & Tentolouris, N. (2019). SGLT2 inhibitors: A review of their antidiabetic and cardioprotective effects. International Journal of Environmental Research and Public Health, 16(16), 2965. https://doi.org/10.3390/ijerph16162965
- Thompson, D. C., Humblet, C., & Joseph-McCarthy, D. (2008). Investigation of MM-PBSA rescoring of docking poses. Journal of Chemical Information and Modeling, 48(5), 1081–1091. https://doi.org/10.1021/ci700470c
- Vanommeslaeghe, K., Hatcher, E., Acharya, C., Kundu, S., Zhong, S., Shim, J., Darian, E., Guvench, O., Lopes, P., Vorobyov, I., & Mackerell, A. D. (2010). CHARMM general force field: A force field for drug-like molecules compatible with the CHARMM all-atom additive biological force fields. Journal of Computational Chemistry, 31(4), 671–690. https://doi.org/10.1002/jcc.21367
- Veeresham, C. (2012). Natural products derived from plants as a source of drugs. Journal of Advanced Pharmaceutical Technology & Research, 3(4), 200–201. https://doi.org/10.4103/2231-4040.104709
- Wang, X., Wu, S., Xu, D., Xie, D., & Guo, H. (2011). Inhibitor and substrate binding by angiotensin-converting enzyme: Quantum mechanical/molecular mechanical molecular dynamics studies. Journal of Chemical Information and Modeling, 51(5), 1074–1082. https://doi.org/10.1021/ci200083f
- Watanabe, A., Choe, S., Chaptal, V., Rosenberg, J. M., Wright, E. M., Grabe, M., & Abramson, J. (2010). The mechanism of sodium and substrate release from the binding pocket of vSGLT. Nature, 468(7326), 988–991. https://doi.org/10.1038/nature09580
- Wright, E. M., Loo, D. D., & Hirayama, B. A. (2011). Biology of human sodium glucose transporters. Physiological Reviews, 91(2), 733–794. https://doi.org/10.1152/physrev.00055.2009
- Zhang, M.-Q., & Wilkinson, B. (2007). Drug discovery beyond the 'rule-of-five' . Current Opinion in Biotechnology, 18(6), 478–488. https://doi.org/10.1016/j.copbio.2007.10.005