
Abstract
In the present paper we propose a novel blind docking protocol based on Autodock-Vina. The developed docking protocol can provide binding site identification and binding pose prediction at the same time, by a systematical exploration of the protein volume performed with several preliminary docking calculations. In our opinion, this protocol can be successfully applied during the first steps of the virtual screening pipeline, because it provides binding site identification and binding pose prediction at the same time without visual evaluation of the binding site. After the binding pose prediction, MM/GBSA re-scoring rescoring procedures has been applied to improve the accuracy of the protein–ligand bound state. The FRAD protocol has been tested on 116 protein–ligand complexes of the Heat Shock Protein 90 – alpha, on 176 of Human Immunodeficiency virus protease 1, and on more than 100 protein–ligand system taken from the PDBbind dataset. Overall, the FRAD approach combined to MM/GBSA re-scoring can be considered as a powerful tool to increase the accuracy and efficiency with respect to other standard docking approaches when the ligand-binding site is unknown.
Communicated by Ramaswamy H. Sarma
1. Introduction
Discovery and development of a new drug is a complex process which requires a lot of time and resources. The main goal of drug discovery is to obtain compounds that successfully interact with therapeutic targets (Batool et al., Citation2019; Hughes et al., Citation2011). Computer Aided Drug Design (CADD) approaches are widely used to increase the efficiency of the drug design (Macalino et al., Citation2015). CADD techniques represents a fundamental tool in the preliminary stages of drug discovery pipeline (Surabhi & Singh, Citation2018; Veselovsky & Ivanov, Citation2003). Computational drug design tools can be divided into ligand-based drug design (LBDD) and structure-based drug design (SBDD). The choice between these two methods dependents on the available information about the biological target (Aparoy et al., Citation2012). SBDD approach uses 3 D structure of the protein target for the screening of potential ligands (Batool et al., Citation2019). On the other side, LBDD approach is employed where 3 D structure of the protein–ligand complex is missing, and computational modelling can be applied to develop theoretical predictive models starting from the chemical description of the ligands with known biological activity (Bacilieri & Moro, Citation2006; Śledź & Caflisch, Citation2018). In this framework, molecular docking is one of the most powerful techniques of SBDD (Gupta et al., Citation2018). The aim of molecular docking is to predict the experimental binding mode of a small molecule within the binding site of the selected target receptor (Ferreira et al., Citation2015; Hernndez-Santoyo et al., Citation2013; Preto et al., Citation2018). A search algorithm and an energy scoring function are the basic tools of a docking methodology for generating and evaluating the ligand conformations (Li et al., Citation2019; Novič et al., Citation2016; Prieto-Martínez et al., Citation2018). The docking procedure has already demonstrated its ability to correctly predict the ligand binding pose starting from the preliminary knowledge of the binding region (Hetényi & Van Der Spoel, Citation2006; Seeliger & De Groot, Citation2010). In this case, a small docking box is selected around the protein binding site in order to facilitate the docking by focusing sampling of the translational, rotational, and torsional degrees of freedom of the ligand (Morris et al., Citation2008). However, there are situation in which the information about binding site is missing and it becomes necessary to explore the entire protein surface by docking algorithms (Hetényi & Van Der Spoel, Citation2006). Several methods have been developed to address this issue (Ghersi & Sanchez, Citation2009; Grosdidier et al., Citation2009; Hassan et al., Citation2017). AutoDock Vina (Vina) (Trott & Olson, Citation2010) is a widely employed technique when information about the binding site is available, ensuring a successful docking procedure. When the protein binding pocket is unidentified, Vina can execute the so-called blind docking (BD). In BD, the target is included into a single simulation box covering the entire surface of the protein (Muscat et al., Citation2020). This method has many limitations because it is difficult to exhaustively sample the whole energy landscape in a fixed number of steps to find the best ligand conformation (Hassan et al., Citation2017). Another approach to address the above-mentioned issue consists in reducing the search space, focusing only in some areas of the protein. The SiteHound algorithm (Hernandez et al., Citation2009) predicts the location of potential binding site and after that, and multiple independent docking procedures on smaller boxes centered on predicted binding sites can be performed. The results show that the docking focused on a small number of predicted binding sites reduces the computational time required to obtain the solution and generates more accurate results in terms of correct pose prediction in comparison to BD. However, if the real binding site is not included in the boxes in which the docking is carried out, the procedure could lead to an incorrect result. The main limitation of the two analyzed methods consists in a too inaccurate sampling box for the BD method and uncertain prediction of the binding pocket for the SiteHound algorithm. In this work, a fragmented docking protocol (FRAD) has been developed to improve the performance of the blind docking performed by Autodock-Vina. The main idea is to divide the protein volume in multiple overlapping boxes. A docking calculation has been performed for each of the previously-mentioned boxes, in order to cover the entire protein surface. The predicted binding poses have been collected, obtaining an ensemble of several protein–ligand binding conformations. Then, the protein–ligand bound states have been ranked considering the corresponding energy scores estimated by Autodock-Vina. The partition in several boxes allows the systematical and exhaustive exploration of the whole protein surface, which improves the discovery of ligand conformations adopted within each examined box. In addition, the complete investigation of the whole protein structure, intrinsically leads to discover of the real binding site. The FRAD molecular docking has been applied to 116 crystal structures of Heat Shock Protein 90 – alpha (Hsp90α), 176 of Human Immunodeficiency virus protease 1 (HIV-1 PR), and more than 150 protein–ligand complex coming from the PDBbind dataset (Z. Liu et al., Citation2015; R. Wang et al., Citation2005 ).
2. Material and methods
2.1. Proteins dataset
Two proteins have been considered for testing the protocol: The Heat Shock Protein 90 – alpha (Hsp90α) and the Human Immunodeficiency virus protease 1 (HIV-1 PR). The main reason for the choice of the latter proteins is because they are among the most widely used proteins in literature for docking purposes (Ahmed et al., Citation2013; Al-Sha’er & Taha, Citation2012; Chang et al., Citation2010; Mey et al., Citation2016; Mobaraki et al., Citation2019; Penkler & Tastan Bishop, Citation2019).
Heat Shock Protein is a globular chaperone protein that assists other proteins to fold correctly and allows cells to survive in extremely heat conditions. It can preserve the integrity of the cells if they are exposed to high temperatures by regulating the flux of calcium ions, maintaining the chromosomal stability and safeguarding the endoplasmic reticulum proteins homeostasis. If on the one hand, Hsp90 aid the human body to survive at elevate temperature, on the other hand, can stabilize the proteins necessary for tumor growth. For this reason, Hsp90 inhibitors are mainly investigated as anti-cancer drugs (Prodromou et al., Citation1997). Hsp90α is an isoform of Hsp and it is located in the cytoplasm of the eukaryotic cells. It consists of four structural domains: N-terminal domain, middle region, C-terminal domain and a linker region that connect the first two domains. The binding pocket is situated in the N-terminal domain, which is predominantly constituted by hydrophobic residues. The pocket is a binding site for many molecules included antibiotics like radicicol and geldanamycin, which have anti-tumor activity and ATP (Garrido et al., Citation2001; Prodromou & Pearl, Citation2003).
HIV-1 PR is a retroviral aspartyl protease, an enzyme which acts in peptide bond hydrolysis in retroviruses, which is necessary for the life cycle of the retrovirus HIV that causes AIDS. HIV-1 PR is constituted from 99 amino acid and exists as a homodimer with only one active site. The binding pocket is located between the identical subunits. Each monomer consist of a wide β-sheet region (a loop of glycine) which partly constitutes the binding site of the substrate and one of the two fundamental residues of aspartyl, Asp-25 and Asp-25′ which are at the bottom of the cavity (Brik & Wong, Citation2003; Deeks et al., Citation1997). The HIV-1 PR enzyme activity can be inhibited from HIV protease inhibitors by blocking the active site of the protease.
The docking protocol has been also tested on 100 protein–ligand complex coming from the PDBbind dataset (Z. Liu et al., Citation2015; R. Wang et al., Citation2005).
2.2. Dataset Preparation
FD and BD methods have been carried out on the same set of complexes extracted from the Protein Data Bank (PDB) (Berman et al., Citation2000). The dataset of Hsp90α is composed of 116 crystal structures, while the dataset of HIV-1 PR is composed of 176 crystal structures. As normal procedure, all the waters molecules have been removed from each PDB entry. Missing residues and atoms have been reconstructed to the main protein chain utilizing the program Modeller (Webb & Sali, Citation2016). The particular cases where the protein or ligand have a PDB record of double spatial positions of the atoms have been treated selecting only one of the positions pair. Hydrogens and Gasteiger charges have been added both the ligand and the protein employing python scripts from MGLTools (Morris et al., Citation2009; Sanner, Citation1999).
2.3. Docking procedure
2.3.1. FRAD protocol
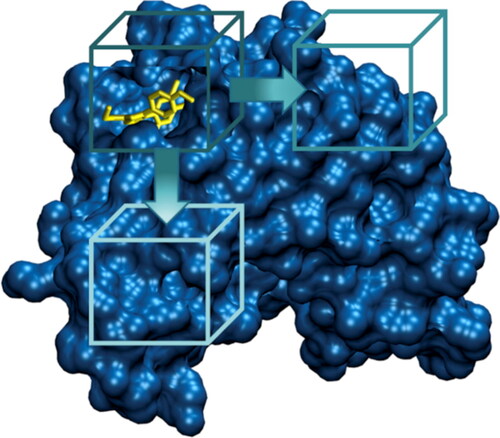
In the FRAD protocol, the protein volume has been divided in multiple overlapping boxes, as shown in . The size of each box has been chosen as twice the ligand size and the overlap between different boxes has been set to fifty percent. A docking calculation has been performed by Autodock-Vina (Trott & Olson, Citation2010) for each of the previously-mentioned boxes, in order to cover the entire protein surface. The exhaustiveness value has been set to 8. The first ten predicted binding poses have been collected, obtaining an ensemble of several protein–ligand binding conformations. Then, the protein–ligand bound states have been ranked considering the corresponding energy scores estimated by Autodock-Vina.
Figure 1. Main idea of FD: a docking procedure is performed in each box which moves on the whole protein surface. Protein is represented in blue, ligand in yellow.
2.3.2. Blind docking protocol
A single docking calculation has been performed by using Autodock-Vina (Trott & Olson, Citation2010) on the whole protein volume. The exhaustiveness value has been set to 8. Results of FRAD and BD methods have been compared by root mean squared deviation (RMSD) of ligand heavy atoms of the solution for FRAD and BD with respect to the experimental ligand structures. In literature a value of RMSD lower or equal to 0.2 nm is recommended as a limit value for a good pose reproduction (Cole et al., Citation2005; Gabb et al., Citation1997; Lape et al., Citation2010; Nissink et al., Citation2002; Zaheer-ul-Haq et al., Citation2010). The RMSD comparison has been made employing the ligand conformation to which Vina attributed as the best in the affinity value ranking.
2.4. MM-GBSA rescoring
Docking algorithms generate binding poses of compounds and rank them by means of scoring functions (Du et al., Citation2016; Kairys et al., Citation2019; Vuignier et al., Citation2010). However, docking scores and experimental binding affinities usually do not correlate, because screening large numbers of compounds in a reasonable time requires the use of approximate scoring function. Hence, docking results can be improved employing post-processing strategies (Quiroga & Villarreal, Citation2016; Rastelli & Pinzi, Citation2019). The MM-GBSA methods has been successfully applied in literature as re-scoring procedure after molecular docking (Chen et al., Citation2016; Genheden & Ryde, Citation2015; Sun, Li, Shen, et al., Citation2014; Zhang et al., Citation2017). The MM-GBSA parameters were set accordingly to previous literature (Muscat et al., Citation2020; Su et al., Citation2015). In detail, the bondi, mbondi, and mbondi2 radii sets were prepared using the antechamber program of AmberTools19 (Salomon-Ferrer et al., Citation2013). The optimized version GBneck (GBneck2) (Nguyen et al., Citation2013) was used as GB model. The default setting of MM-GBSA surface tension (α = 0.0072 kcal/mol Å2) and the non-polar free energy correction term (β = 0) were applied. The dielectric constant of the medium was set to 80. Successful rescoring is obtained if the lowest binding free energy value belongs to the pose with a RMSD less than 0.2 nm.
3. Results
In this section, results regarding the FRAD and BD docking procedures are reported to compare performances of both methods as applied to ligand pose discovery in Hsp90α and in HIV-1 PR datasets. As a first result, we’ve shown if the binding poses have been correctly predicted. Then if the rescoring method produces improvements to the results. RMSD Tables concerning the use of FRAD and BD on 116 Hsp90α and 176 HIV-1 PR proteins with co-crystallized ligands are reported in Supplementary Information (Tables S1 and S2). The FRAD protocol has been also tested on 100 protein–ligand systems taken from the PDBbind dataset (Z. Liu et al., Citation2015; R. Wang et al., Citation2005), as reported in Table S3.
3.1. FRAD vs BD in protein–ligand complexes of Hsp90α
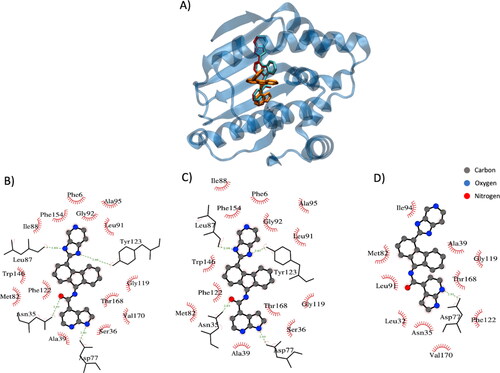
As described in the previous sections, several binding ligand poses have been generated for each protein and the pose considered for the validation is the one which Vina has attributed the maximum affinity value. The example shows in qualitatively shows the FRAD ability to produce more accurate poses with respect to the BD method. Moreover, exhibits a comparison between the protein–ligand interactions in crystallized complex (), in complex generated employing the FRAD method () and in complex generated by the BD method (). It is worth mentioning that FRAD approach can identify the exact number of h-bonds with the correct amino acids and is able to conserve a high fidelity of the hydrophobic contacts (), if compared with BD method ().
Figure 2. Example of docking result employing FRAD and BD methods (PDB: 2yki). (A) Protein is represented in blue, the ligand in the crystal structures in red, the ligand conformation generated with FRAD method in cyan (RMSD is 0.04 nm) and the ligand conformation generated with BD method in orange (RMSD is 1.55 nm). (B) Protein–ligand interactions in crystallized complex. (C) Protein–ligand interactions in the generated with FRAD method complex. (D) Protein–ligand interactions in the generated with BD method complex. Elements in figure (A), (B) and (C) are the conformation of the ligands, hydrogen bonds in green and hydrophobic residue names.
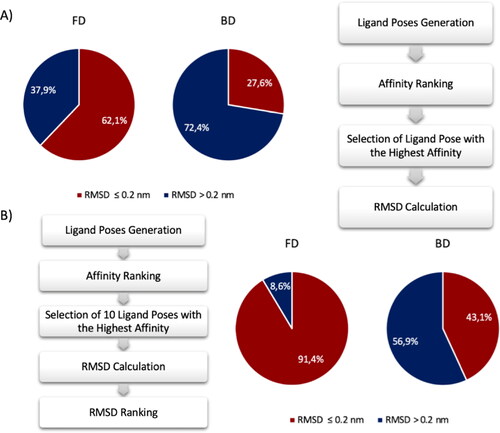
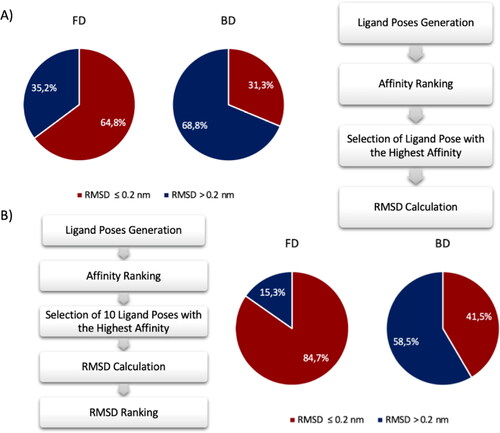
In order to have a clear picture of the FRAD and BD performance, pie chart is selected as graphical tool to highlight the accuracy of both methods. The pie chart in . Percentage of poses generated with RMSD lesser and greater than 0.2 nm compared the crystal structure pose using the FRAD and BD method. Results of FRAD methods are shown in the pie chart on the left and results of BD method are shown in the pie chart on the right. Red and blue indicate the poses with RMSD lesser and greater than 0.2 nm, respectively. (A) The comparison has been made considering the poses with the highest affinity value. An accurate ligand conformation has been discovered in 62.1% of cases using FRAD method and 27.6% using BD method. (B) The comparison has been made considering the poses with the best RMSD value between first 10 top poses. An accurate ligand conformation has been discovered in 91.4% of cases using FRAD method and 43.1% using BD method. A shows the percentages of generated ligand poses with RMSD value lesser and greater than 0.2 nm in comparison to the experimental ligand conformation. Considering the output conformation with greatest affinity, BD finds the correct pose prediction in only 26.7% of attempts, whereas FRAD finds the correct prediction in the 62.1% of attempts. The ligand conformation with the greatest affinity identified by the BD approach is not always the one closest to the real pose of the ligand. In this framework, the first ten conformations have been selected and the RMSD analysis between them and the original ligand has been computed. The hypothesis is to find the pose with the lowest RMSD value between the top ten records of Vina’s output conformations in order to evaluate if there is a pose predicted correctly with an RMSD less than 0.2 nm. shows the pie chart percentages of the obtained results considering the pose with lowest RMSD between the first ten poses. Interestingly, an improvement in the performance is notable for both FRAD and BD methods. The results highlight that the correct pose is generated among the ten top poses and the correct identification of it can be achieved through rescoring methodologies. In detail, FRAD finds in 91.4% of times the poses with RMSD value lower than 0.2 nm, while BD discovers in 43.1% of times the correct conformation.
Figure 3. Percentage of poses generated with RMSD lesser and greater than 0.2 nm compared the crystal structure pose using the FRAD and BD method. Results of FRAD methods are shown in the pie chart on the left and results of BD method are shown in the pie chart on the right. Red and blue indicate the poses with RMSD lesser and greater than 0.2 nm, respectively. (A) The comparison has been made considering the poses with the highest affinity value. An accurate ligand conformation has been discovered in 62.1% of cases using FRAD method and 27.6% using BD method. (B) The comparison has been made considering the poses with the best RMSD value between first 10 top poses. An accurate ligand conformation has been discovered in 91.4% of cases using FRAD method and 43.1% using BD method.
In Supplementary Information, shows the relationship between the improving in performance with the increase in the number of considered poses.
3.2. FRAD vs BD in protein–ligand complexes of HIV-1 PR
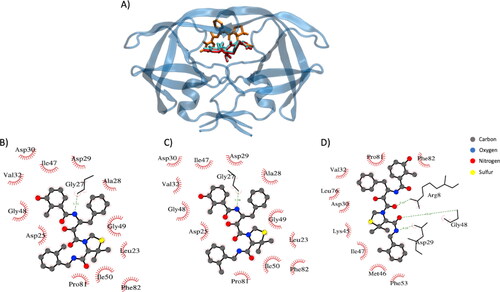
The second structure considered for the Vina FRAD and BD method is HIV-1 PR protein. In this context, the docking procedure has been performed 176 different ligand-protein complexes. As applied before for the Hsp90α protein, several binding ligand poses have been generated for each protein and the pose considered for the validation is the one which Vina has attributed the maximum affinity value with the protein. shows how an accurate pose has been predicted using FRAD method compared to the BD method. Moreover, exhibits a comparison between the protein–ligand interactions in crystallized complex (), in complex generated employing the FRAD method () and in complex generated employing BD method (). It is worth mentioning that FRAD method is able to identify the exact number of h-bonds with the correct amino acids and is able to conserve a high fidelity of the hydrophobic contacts (), if compared with BD method ().
Figure 4. Example of docking result employing FRAD and BD methods (PDB: 1msn). (A) Protein is represented in blue, the ligand in the crystal structures in red, the ligand conformation generated with FRAD method in cyan (RMSD is 0.03 nm) and the ligand conformation generated with BD method in orange (RMSD is 2.08 nm). (B) Protein–ligand interactions in crystallized complex. (C) Protein–ligand interactions in the generated with FRAD method complex. (D) Protein–ligand interactions in the generated with BD method complex. Elements in figure (A), (B) and (C) are the conformation of the ligands, hydrogen bonds in green and hydrophobic residue names.
The pie chart in shows the percentages of generated ligand poses with RMSD value lesser and greater than 0.2 nm in comparison to the crystal structure ligand conformation. Considering the output conformations with greatest affinity, BD finds the correct pose prediction in only 31.3% of attempts, while FRAD finds the correct prediction in the 64.8% of attempts. Considering also this second dataset, the ligand conformation which Vina assigns the greatest affinity is not always the one closest to the real pose of the ligand. In this framework, the first ten conformations have been selected and the RMSD analysis between them and the experimental ligand has been computed. The hypothesis is to find the pose with the lowest RMSD value between the top ten records of Vina’s output conformations in order to evaluate if there is a pose predicted correctly with an RMSD less than 0.2 nm. shows the pie chart percentages of the obtained results considering the pose with lowest RMSD between the first ten poses. As pointed out in the previous section, an improvement in the performance is notable for both FRAD and BD methods. The results highlight that the correct pose is generated among the ten top poses and the correct identification of it can be achieved through rescoring methodologies. In detail, FRAD finds in 84.7% of times the poses with RMSD value lower than 0.2 nm, while BD discovers in 41.5%. of times the correct conformation.
Figure 5. Percentage of poses generated with RMSD lesser and greater than 0.2 nm compared the crystal structure pose using the FRAD and BD method. Results of FRAD methods are shown in the pie chart on the left and results of BD method are shown in the pie chart on the right. Red and blue indicate the poses with RMSD lesser and greater than 0.2 nm, respectively. (A) The comparison has been made considering the poses with the highest affinity value. An accurate ligand conformation has been discovered in 64.8% of cases using FRAD method and 31.3% using BD method. (B) The comparison has been made considering the poses with the best RMSD value between first 10 top poses. An accurate ligand conformation has been discovered in 84.7% of cases using FRAD method and 41.5% using BD method.
In Supplementary Information, exhibits the relationship between the improving in performance with the increase in the number of poses considered. A rescoring procedure has been performed in order to evaluate if MM-GBSA is able to attribute the best affinity value to the conformation with the lowest RMSD value, namely, to further validate and improve the FRAD method.
3.3. MM-GBSA rescoring
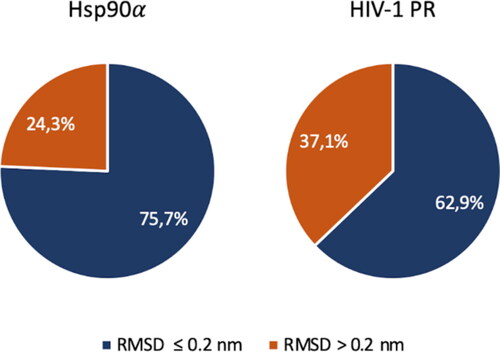
The evaluation of Vina's pose made considering the RMSD as principal parameter for the ranking is a commonly used protocol (Castro-Alvarez et al., Citation2017; Cavasotto & Abagyan, Citation2004; Hu et al., Citation2004; Prieto-Martínez et al., Citation2018), but the pose with the lowest RMSD does not always correspond to the one with greater affinity. The rescoring procedure may have a strong influence in the BD protocol, since the FRAD protocol is supposed to be performed within a variable protein region. To address this issue, a rescoring protocol on outgoing poses has been accomplished. Binding free energy has been calculated using the MM-GBSA method on the first ten protein–ligand complex whose affinity is the highest calculated by Vina. Ten protein–ligand complexes has been considered for the calculation. In the rescoring results for Hsp90α and HIV-1 PR structures are represented. In 75.7% of cases the greatest binding affinity estimated with MM-GBSA corresponds to ligand pose with a RMSD lesser than 0.2 nm compared the ligand in the crystal structure. The percentage of correct poses prediction is higher comparing the results in which the ranking is done with the affinity estimated by Vina. In HIV-1 PR the performance is about the same as that obtained by considering the docking affinities.
Figure 6. Rescoring results. The percentage of the poses generated with RMSD lesser and greater than 0.2 nm compared the crystal structure pose using FRAD method are shown for Hsp90α and HIV-1 PR structures. Results of Hsp90α are shown in the pie chart on the left and results of HIV-1 PR are shown in the pie chart on the right. Blue and orange indicate the poses with RMSD lesser and greater than 0.2 nm, respectively. An accurate ligand conformation has been discovered in 75.7% of cases in Hsp90α and 62.9% in HIV-1 PR.
4. Discussion
Protein–ligand docking is a powerful tool in drug discovery to predict binding modes and affinities of ligand (Ben-Shimon & Niv, Citation2015; Hetényi & van der Spoel, Citation2002; Hetényi & Van Der Spoel, Citation2006). The blind docking is a common strategy employed when the binding site of a target is unknown. However, BD has many limitations because it is unlikely to exhaustively sample the whole energy landscape to find the correct ligand pose (Hetényi & Van Der Spoel, Citation2011). One of the problems which may lead in a lack of accuracy in the blind docking results is due to an incorrect identification of the binding site (Brown & Jagt, Citation2004; Ghersi & Sanchez, Citation2009). Nowadays, the research community involves conspicuous effort in developing new software, like METADOCK2 (Imbernón et al., Citation2021), which increases the blind docking accuracy and efficiency in comparison with the Vina’s protocol. The speed up of this approach is due to the adoption of metaheuristic schemes. The strategies proposed usually employ specific software to find the active site in the target and then docking the ligands into the discovered binding site (Blaszczyk et al., Citation2016; Ghersi & Sanchez, Citation2009; Lin et al., Citation2016; Y. Liu et al., Citation2020). In particular, algorithms like SiteHound (Hernandez et al., Citation2009), CurPocket (Y. Liu et al., Citation2020) and COACH (Yang et al., Citation2013) have allowed an increased docking accuracy due to a more precise identification of the ligand binding site. These approaches have demonstrated how the blind docking results without information of the correct binding site are characterized by a very low success probability. However, if the true binding site is not included in the boxes in which the docking is carried out, the procedure will lead to an incorrect result. Another possibility is to perform a preliminary BD calculation to predict the binding site, and then a re-docking focused in the correct protein region (Agrawal et al., Citation2019). Despite a slight increase in accuracy, the computation cost of this procedure is higher and does not fully address the correct binding site prediction. The main goal of this work is to improve the BD procedure through a docking protocol called FRAD. In the FRAD protocol, the protein volume has been divided in multiple overlapping boxes. A docking calculation has been performed by Autodock-Vina (Trott & Olson, Citation2010) for each of the previously-mentioned boxes, in order to cover the entire protein surface. The first predicted binding poses have been collected, obtaining an ensemble of several protein–ligand binding conformations. Then, the protein–ligand bound states have been ranked considering the corresponding energy scores estimated by Autodock-Vina. A first comparison has been carried out between the RMSD values obtained considering only the ligand pose with the lowest docking energy. In this context, the performances obtained with the FRAD method are better than the BD method in both protein–ligand complexes analyzed. From another point of view, both the FRAD and BD methods increased their performance if we take into consideration the first ten poses, crowing nevertheless FRAD algorithm as the best one even in this case. The method of performing the docking procedure in several boxes that translate on the protein has proven to be a winning strategy in finding the lowest RMSD (<0.2 nm) conformation of the protein–ligand complex, namely the closest to the crystal structure. The improvement induced by this method has been confirmed by comparing the performances obtained in other works in which a BD has been carried on Hsp90α and HIV1-PR (Mey et al., Citation2016; Mobley et al., Citation2014).
A second analysis has been accomplished in order to evaluate the performance of Vina's scoring function, as done before in literature (Gaillard, Citation2018; Quiroga & Villarreal, Citation2016). Docking algorithms, like Vina, have been developed with the goal to screening large numbers of compounds in a reasonable time, for this reason, they use approximate scoring functions (Graves et al., Citation2008). It is reasonable to hypothyze that the best pose, namely the one with the minimum RMSD from the crystal structure, is not always the first in the rank. Indeed, it has become general opinion that docking results should be improved by means of more rigorous post-docking processing strategies (Palacio-Rodríguez et al., Citation2019; Quiroga & Villarreal, Citation2016; Rastelli & Pinzi, Citation2019). Several works have employed re-scoring approaches to predict binding affinities and afterwards compare these results with the docking scores (Lyne et al., Citation2006; Sgobba et al., Citation2012; Thompson et al., Citation2008). In this context, a study on Protein kinase proved that the correlation between calculated binding free energies with MM-GBSA and MM-PBSA methods and experimental values is higher than using docking scores (Sun, Li, Tian, et al., Citation2014). Instead, in a set of β-Amyloid Cleaving Enzyme 1 (BACE-1) the re-scoring docking poses using MM-GBSA did not improve the correlation with experimental affinities due to the ligand dataset (El Khoury et al., Citation2019). It is important to mention that BACE-1 ligands dataset consisted of macrocycles which have multiple flexible bonds that generate a large conformational space and for this reason require a more accurate MM-GBSA protocol(El Khoury et al., Citation2019). In recent literature, the performance of MM-PBSA and MM-GBSA rescoring (E. Wang et al., Citation2019; Z. Wang et al., Citation2019; Weng et al., Citation2019) have been evaluated also in protein–protein docking showing that MM-GBSA may be a good choice for predicting the binding affinities and identifying correct binding structures (Chen et al., Citation2016). Within this context, we’ve shown how the MM-GBSA rescoring is able to further improve the accuracy of the docking predictions. Recently, the same procedure of MM-GBSA rescoring has been applied to the three top scored docking poses showing that the results have improved compared the case in which only the best scored docking pose is considered (Rastelli et al., Citation2010; Sun, Li, Shen, et al., Citation2014). The method is a good compromise between efficiency and speed since it has been applied on minimized protein–ligand complexes. Future challenges could consist in finding more rigorous rescoring methods in order to improve the accuracy with a reasonable computational effort. Within this framework, the integration of binding pose prediction and affinity estimations based on artificial intelligence techniques could be implemented in order to improve the accuracy and sensitivity of the presented blind docking protocol.
5. Conclusions
The docking protocol developed in this study has demonstrated high efficiency in predicting protein–ligand docking when binding sites are a priori not known. It is worth to mention that the FRAD approach is a simple, fast, and easy to implement. FRAD is accurate in term of binding site identification and RMSD of the lowest energy docked pose. Nevertheless, the idea of performing a re-scoring on the FRAD docking results by methods estimating the free binding energy such as MM/GBSA proved to be a good solution for improvement. The adoption of the FRAD protocol has brought consistent increase of the ligand pose prediction accuracy with a reasonable computational effort, bringing an overall improvement in comparison to the BD procedure.
Authors’ contributions
GG and MAD conceived the research. GG, and ADG did the molecular dynamics simulations. GG, ADG, BM, DP, AD and MAD analyzed and rationalized the data. All authors wrote the paper and critically commented to the manuscript. All authors read and approved the final manuscript.
SI-03.pdf
Download PDF (381.3 KB)Disclosure statement
No potential conflict of interest was reported by the authors.
Funding
The present study has been developed as part of the VIRTUOUS project funded by the European Union’s Horizon 2020 research and innovation program under the Maria Sklodowska-Curie-RISE Grant Agreement No 872181 (https://www.virtuoush2020.com/). This work was also supported by a grant from the Swiss National Supercomputing Centre (CSCS).
Additional information
Funding
References
- Agrawal, P., Singh, H., Srivastava, H. K., Singh, S., Kishore, G., & Raghava, G. P. S. (2019). Benchmarking of different molecular docking methods for protein–peptide docking. BMC Bioinformatics, 19(S13), 426. https://doi.org/10.1186/s12859-018-2449-y
- Ahmed, L., Rasulev, B., Turabekova, M., Leszczynska, D., & Leszczynski, J. (2013). Receptor- and ligand-based study of fullerene analogues: Comprehensive computational approach including quantum-chemical, QSAR and molecular docking simulations. Organic & Biomolecular Chemistry, 11(35), 5798–5808. https://doi.org/10.1039/c3ob40878g
- Al-Sha'er, M. A., & Taha, M. O. (2012). Application of docking-based comparative intermolecular contacts analysis to validate Hsp90α docking studies and subsequent in silico screening for inhibitors. Journal of Molecular Modeling, 18(11), 4843–4863. https://doi.org/10.1007/s00894-012-1479-z
- Aparoy, P., Kumar Reddy, K., & Reddanna, P. (2012). Structure and ligand based drug design strategies in the development of Novel 5- LOX inhibitors. Current Medicinal Chemistry, 19(22), 3763–3778. https://doi.org/10.2174/092986712801661112
- Bacilieri, M., & Moro, S. (2006). Ligand-based drug design methodologies in drug discovery process: An overview. Current Drug Discovery Technologies, 3(3), 155–165. https://doi.org/10.2174/157016306780136781
- Batool, M., Ahmad, B., & Choi, S. (2019). A structure-based drug discovery paradigm. International Journal of Molecular Sciences, 20(11), 2783. https://doi.org/10.3390/ijms20112783
- Ben-Shimon, A., & Niv, M. Y. (2015). AnchorDock: Blind and flexible anchor-driven peptide docking. Structure (London, England : 1993), 23(5), 929–940. https://doi.org/10.1016/j.str.2015.03.010
- Berman, H. M., Westbrook, J., Feng, Z., Gilliland, G., Bhat, T. N., Weissig, H., Shindyalov, I. N., & Bourne, P. E. (2000). The protein data bank. Nucleic Acids Research, 28(1), 235–242. https://doi.org/10.1093/nar/28.1.235
- Blaszczyk, M., Kurcinski, M., Kouza, M., Wieteska, L., Debinski, A., Kolinski, A., & Kmiecik, S. (2016). Modeling of protein–peptide interactions using the CABS-dock web server for binding site search and flexible docking. Methods (San Diego, Calif.), 93, 72–83. https://doi.org/10.1016/j.ymeth.2015.07.004
- Brik, A., & Wong, C. H. (2003). HIV-1 protease: Mechanism and drug discovery. Organic & Biomolecular Chemistry, 1(1), 5–14. https://doi.org/10.1039/b208248a
- Brown, W. M., & Jagt, D. L. V. (2004). Creating artificial binding pocket boundaries to improve the efficiency of flexible ligand docking. Journal of Chemical Information and Computer Sciences, 44(4), 1412–1422. https://doi.org/10.1021/ci049853r
- Castro-Alvarez, A., Costa, A. M., & Vilarrasa, J. (2017). The Performance of several docking programs at reproducing protein–macrolide-like crystal structures. Molecules, 22(1), 136. https://doi.org/10.3390/molecules22010136
- Cavasotto, C. N., & Abagyan, R. A. (2004). Protein flexibility in ligand docking and virtual screening to protein kinases. Journal of Molecular Biology, 337(1), 209–225. https://doi.org/10.1016/j.jmb.2004.01.003
- Chang, M. W., Ayeni, C., Breuer, S., & Torbett, B. E. (2010). Virtual screening for HIV protease inhibitors: A comparison of AutoDock 4 and Vina. PLoS One, 5(8), e11955. https://doi.org/10.1371/journal.pone.0011955
- Chen, F., Liu, H., Sun, H., Pan, P., Li, Y., Li, D., & Hou, T. (2016). Assessing the performance of the MM/PBSA and MM/GBSA methods. 6. Capability to predict protein–protein binding free energies and re-rank binding poses generated by protein–protein docking. Physical Chemistry Chemical Physics: PCCP, 18(32), 22129–22139. https://doi.org/10.1039/c6cp03670h
- Cole, J. C., Murray, C. W., Nissink, J. W. M., Taylor, R. D., & Taylor, R. (2005). Comparing protein–ligand docking programs is difficult. Proteins, 60(3), 325–332. https://doi.org/10.1002/prot.20497
- Deeks, S. G., Smith, M., Holodniy, M., & Kahn, J. O. (1997). HIV-1 protease inhibitors. A review for clinicians. JAMA, 277(2), 145–153. https://doi.org/10.1001/jama.277.2.145
- Du, X., Li, Y., Xia, Y. L., Ai, S. M., Liang, J., Sang, P., Ji, X. L., & Liu, S. Q. (2016). Insights into protein–ligand interactions: Mechanisms, models, and methods. International Journal of Molecular Sciences, 17(2), 144. https://doi.org/10.3390/ijms17020144
- El Khoury, L., Santos-Martins, D., Sasmal, S., Eberhardt, J., Bianco, G., Ambrosio, F. A., Solis-Vasquez, L., Koch, A., Forli, S., & Mobley, D. L. (2019). Comparison of affinity ranking using AutoDock-GPU and MM-GBSA scores for BACE-1 inhibitors in the D3R Grand Challenge 4. Journal of Computer-Aided Molecular Design, 33(12), 1011–1020. https://doi.org/10.1007/s10822-019-00240-w
- Ferreira, L., dos Santos, R., Oliva, G., & Andricopulo, A. (2015). Molecular docking and structure-based drug design strategies. Molecules (Basel, Switzerland), 20(7), 13384–13421. https://doi.org/10.3390/molecules200713384
- Gabb, H. A., Jackson, R. M., & Sternberg, M. J. E. (1997). Modelling protein docking using shape complementarity, electrostatics and biochemical information. Journal of Molecular Biology, 272(1), 106–120. https://doi.org/10.1006/jmbi.1997.1203
- Gaillard, T. (2018). Evaluation of AutoDock and AutoDock Vina on the CASF-2013 benchmark. Journal of Chemical Information and Modeling, 58(8), 1697–1706. https://doi.org/10.1021/acs.jcim.8b00312
- Garrido, C., Gurbuxani, S., Ravagnan, L., & Kroemer, G. (2001). Heat shock proteins: Endogenous modulators of apoptotic cell death. Biochemical and Biophysical Research Communications, 286(3), 433–442. https://doi.org/10.1006/bbrc.2001.5427
- Genheden, S., & Ryde, U. (2015). The MM/PBSA and MM/GBSA methods to estimate ligand-binding affinities. Expert Opinion on Drug Discovery, 10(5), 449–461. https://doi.org/10.1517/17460441.2015.1032936
- Ghersi, D., & Sanchez, R. (2009). Improving accuracy and efficiency of blind protein–ligand docking by focusing on predicted binding sites. Proteins, 74(2), 417–424. https://doi.org/10.1002/prot.22154
- Graves, A. P., Shivakumar, D. M., Boyce, S. E., Jacobson, M. P., Case, D. A., & Shoichet, B. K. (2008). Rescoring docking hit lists for model cavity sites: Predictions and experimental testing. Journal of Molecular Biology, 377(3), 914–934. https://doi.org/10.1016/j.jmb.2008.01.049
- Grosdidier, A., Zoete, V., & Michielin, O. (2009). Blind docking of 260 protein–ligand complexes with EADock 2.0. Journal of Computational Chemistry, 30(13), 2021–2030. https://doi.org/10.1002/jcc.21202
- Gupta, M., Sharma, R., & Kumar, A. (2018). Docking techniques in pharmacology: How much promising? Computational Biology and Chemistry, 76, 210–217. https://doi.org/10.1016/j.compbiolchem.2018.06.005
- Hassan, N. M., Alhossary, A. A., Mu, Y., & Kwoh, C. K. (2017). Protein–ligand blind docking using QuickVina-W with inter-process spatio-temporal integration. Scientific Reports, 7(1), 15451. https://doi.org/10.1038/s41598-017-15571-7
- Hernandez, M., Ghersi, D., & Sanchez, R. (2009). SITEHOUND-web: A server for ligand binding site identification in protein structures. Nucleic Acids Research, 37(Web Server issue), W413–W416. https://doi.org/10.1093/nar/gkp281
- Hernndez-Santoyo, A., Yair, A., Altuzar, V., Vivanco-Cid, H., & Mendoza-Barrer, C. (2013). Protein–protein and protein–ligand docking. In Protein engineering – Technology and application. InTech. https://doi.org/10.5772/56376
- Hetényi, C., & van der Spoel, D. (2002). Efficient docking of peptides to proteins without prior knowledge of the binding site. Protein Science, 11(7), 1729–1737. https://doi.org/10.1110/ps.0202302
- Hetényi, C., & Van Der Spoel, D. (2006). Blind docking of drug-sized compounds to proteins with up to a thousand residues. FEBS Letters, 580(5), 1447–1450. https://doi.org/10.1016/j.febslet.2006.01.074
- Hetényi, C., & Van Der Spoel, D. (2011). Toward prediction of functional protein pockets using blind docking and pocket search algorithms. Protein Science, 20(5), 880–893. https://doi.org/10.1002/pro.618
- Hu, X., Balaz, S., & Shelver, W. H. (2004). A practical approach to docking of zinc metalloproteinase inhibitors. Journal of Molecular Graphics & Modelling, 22(4), 293–307. https://doi.org/10.1016/j.jmgm.2003.11.002
- Hughes, J., Rees, S., Kalindjian, S., & Philpott, K. (2011). Principles of early drug discovery. British Journal of Pharmacology, 162(6), 1239–1249. https://doi.org/10.1111/j.1476-5381.2010.01127.x
- Imbernón, B., Serrano, A., Bueno-Crespo, A., Abellán, J. L., Pérez-Sánchez, H., & Cecilia, J. M. (2021). METADOCK 2: A high-throughput parallel metaheuristic scheme for molecular docking. Bioinformatics (Oxford, England), 37(11), 1515–1520. https://doi.org/10.1093/bioinformatics/btz958
- Kairys, V., Baranauskiene, L., Kazlauskiene, M., Matulis, D., & Kazlauskas, E. (2019). Binding affinity in drug design: Experimental and computational techniques. Expert Opinion on Drug Discovery, 14(8), 755–768. https://doi.org/10.1080/17460441.2019.1623202
- Lape, M., Elam, C., & Paula, S. (2010). Comparison of current docking tools for the simulation of inhibitor binding by the transmembrane domain of the sarco/endoplasmic reticulum calcium ATPase. Biophysical Chemistry, 150(1–3), 88–97. https://doi.org/10.1016/j.bpc.2010.01.011
- Li, J., Fu, A., & Zhang, L. (2019). An overview of scoring functions used for protein–ligand interactions in molecular docking. Interdisciplinary Sciences, Computational Life Sciences, 11(2), 320–328. https://doi.org/10.1007/s12539-019-00327-w
- Lin, Y. F., Cheng, C. W., Shih, C. S., Hwang, J. K., Yu, C. S., & Lu, C. H. (2016). MIB: Metal ion-binding site prediction and docking server. Journal of Chemical Information and Modeling, 56(12), 2287–2291. https://doi.org/10.1021/acs.jcim.6b00407
- Liu, Y., Grimm, M., Dai, W., Tao, Hou, M., Chun, Xiao, Z. X., & Cao, Y. (2020). CB-Dock: A web server for cavity detection-guided protein–ligand blind docking. Acta Pharmacologica Sinica, 41(1), 138–144. https://doi.org/10.1038/s41401-019-0228-6
- Liu, Z., Li, Y., Han, L., Li, J., Liu, J., Zhao, Z., Nie, W., Liu, Y., & Wang, R. (2015). PDB-wide collection of binding data: Current status of the PDBbind database. Bioinformatics (Oxford, England), 31(3), 405–412. https://doi.org/10.1093/bioinformatics/btu626
- Lyne, P. D., Lamb, M. L., & Saeh, J. C. (2006). Accurate prediction of the relative potencies of members of a series of kinase inhibitors using molecular docking and MM-GBSA scoring. Journal of Medicinal Chemistry, 49(16), 4805–4808. https://doi.org/10.1021/jm060522a
- Macalino, S. J. Y., Gosu, V., Hong, S., & Choi, S. (2015). Role of computer-aided drug design in modern drug discovery. Archives of Pharmacal Research, 38(9), 1686–1701. https://doi.org/10.1007/s12272-015-0640-5
- Mey, A. S. J. S., Juárez-Jiménez, J., Hennessy, A., & Michel, J. (2016). Blinded predictions of binding modes and energies of HSP90-α ligands for the 2015 D3R grand challenge. Bioorganic & Medicinal Chemistry, 24(20), 4890–4899. https://doi.org/10.1016/j.bmc.2016.07.044
- Mobaraki, N., Hemmateenejad, B., Weikl, T. R., & Sakhteman, A. (2019). On the relationship between docking scores and protein conformational changes in HIV-1 protease. Journal of Molecular Graphics & Modelling, 91, 186–193. https://doi.org/10.1016/j.jmgm.2019.06.011
- Mobley, D. L., Liu, S., Lim, N. M., Wymer, K. L., Perryman, A. L., Forli, S., Deng, N., Su, J., Branson, K., & Olson, A. J. (2014). Blind prediction of HIV integrase binding from the SAMPL4 challenge. Journal of Computer-Aided Molecular Design, 28(4), 327–345. https://doi.org/10.1007/s10822-014-9723-5
- Morris, G. M., Huey, R., Lindstrom, W., Sanner, M. F., Belew, R. K., Goodsell, D. S., & Olson, A. J. (2009). AutoDock4 and AutoDockTools4: Automated docking with selective receptor flexibility. Journal of Computational Chemistry, 30(16), 2785–2791. https://doi.org/10.1002/jcc.21256
- Morris, G. M., Huey, R., & Olson, A. J. (2008). UNIT using AutoDock for ligand-receptor docking. Current Protocols in Bioinformatics, 24(1), 8–14. https://doi.org/10.1002/0471250953.bi0814s24
- Muscat, S., Pallante, L., Stojceski, F., Danani, A., Grasso, G., & Deriu, M. A. (2020). The impact of natural compounds on S-shaped Aβ42 fibril: From molecular docking to biophysical characterization. International Journal of Molecular Sciences, 21(6), 2017. https://doi.org/10.3390/ijms21062017
- Nguyen, H., Roe, D. R., & Simmerling, C. (2013). Improved generalized born solvent model parameters for protein simulations. Journal of Chemical Theory and Computation, 9(4), 2020–2034. https://doi.org/10.1021/ct3010485
- Nissink, J. W. M., Murray, C., Hartshorn, M., Verdonk, M. L., Cole, J. C., & Taylor, R. (2002). A new test set for validating predictions of protein–ligand interaction. Proteins, 49(4), 457–471. https://doi.org/10.1002/prot.10232
- Novič, M., Tibaut, T., Anderluh, M., Borišek, J., & Tomašič, T. (2016). The comparison of docking search algorithms and scoring functions: An overview and case studies. In S. Dastmalchi, M. Hamzeh-Mivehroud, & B. Sokouti (Eds.), Methods and algorithms for molecular docking-based drug design and discovery (pp. 99–127). IGI Global. https://doi.org/10.4018/978-1-5225-0115-2.ch004
- Palacio-Rodríguez, K., Lans, I., Cavasotto, C. N., & Cossio, P. (2019). Exponential consensus ranking improves the outcome in docking and receptor ensemble docking. Scientific Reports, 9(1), 5142. https://doi.org/10.1038/s41598-019-41594-3
- Penkler, D. L., & Tastan Bishop, Ö. (2019). Modulation of Human Hsp90α Conformational Dynamics by Allosteric Ligand Interaction at the C-Terminal Domain. Scientific Reports, 9(1), 1600. https://doi.org/10.1038/s41598-018-35835-0
- Preto, J., Gentile, F., Winter, P., Churchill, C., Omar, S. I., & Tuszynski, J. A. (2018). Molecular dynamics and related computational methods with applications to drug discovery. In L. Bonilla, E. Kaxiras, R. Melnik (Eds.), Coupled Mathematical Models for Physical and Biological Nanoscale Systems and Their Applications. BIRS-16w5069 2016. Springer Proceedings in Mathematics and Statistics, vol. 232 (pp. 267–285). Springer. https://doi.org/10.1007/978-3-319-76599-0_14
- Prieto-Martínez, F. D., Arciniega, M., & Medina-Franco, J. L. (2018). Acoplamiento molecular: Avances recientes y retos. TIP Revista Especializada en Ciencias Químico-Biológicas, 21. https://doi.org/10.22201/fesz.23958723e.2018.0.143
- Prodromou, C., & Pearl, L. (2003). Structure and functional relationships of Hsp90. Current Cancer Drug Targets, 3(5), 301–323. https://doi.org/10.2174/1568009033481877
- Prodromou, C., Roe, S. M., Piper, P. W., & Pearl, L. H. (1997). A molecular clamp in the crystal structure of the N-terminal domain of the yeast Hsp90 chaperone. Nature Structural Biology, 4(6), 477–482. https://doi.org/10.1038/nsb0697-477
- Quiroga, R., & Villarreal, M. A. (2016). Vinardo: A scoring function based on Autodock Vina improves scoring, docking, and virtual screening. PLoS One, 11(5), e0155183. https://doi.org/10.1371/journal.pone.0155183
- Rastelli, G., Del Rio, A., Degliesposti, G., & Sgobba, M. (2010). Fast and accurate predictions of binding free energies using MM-PBSA and MM-GBSA. Journal of Computational Chemistry, 31(4), 797–810. https://doi.org/10.1002/jcc.21372
- Rastelli, G., & Pinzi, L. (2019). Refinement and rescoring of virtual screening results. Frontiers in Chemistry, 7, 498. https://doi.org/10.3389/fchem.2019.00498
- Salomon-Ferrer, R., Case, D. A., & Walker, R. C. (2013). An overview of the Amber biomolecular simulation package. Wiley Interdisciplinary Reviews: Computational Molecular Science, 3(2), 198–210. https://doi.org/10.1002/wcms.1121
- Sanner, M. F. (1999). Python: A programming language for software integration and development. Journal of Molecular Graphics & Modelling, 17(1), 57–61.
- Seeliger, D., & De Groot, B. L. (2010). Ligand docking and binding site analysis with PyMOL and Autodock/Vina. Journal of Computer-Aided Molecular Design, 24(5), 417–422. https://doi.org/10.1007/s10822-010-9352-6
- Sgobba, M., Caporuscio, F., Anighoro, A., Portioli, C., & Rastelli, G. (2012). Application of a post-docking procedure based on MM-PBSA and MM-GBSA on single and multiple protein conformations. European Journal of Medicinal Chemistry, 58, 431–440. https://doi.org/10.1016/j.ejmech.2012.10.024
- Śledź, P., & Caflisch, A. (2018). Protein structure-based drug design: From docking to molecular dynamics. Current Opinion in Structural Biology, 48, 93–102. https://doi.org/10.1016/j.sbi.2017.10.010
- Su, P.-C., Tsai, C.-C., Mehboob, S., Hevener, K. E., & Johnson, M. E. (2015). Comparison of radii sets, entropy, QM methods, and sampling on MM-PBSA, MM-GBSA, and QM/MM-GBSA ligand binding energies of F. tularensis enoyl-ACP reductase (FabI). Journal of Computational Chemistry, 36(25), 1859–1873. https://doi.org/10.1002/jcc.24011
- Sun, H., Li, Y., Shen, M., Tian, S., Xu, L., Pan, P., Guan, Y., & Hou, T. (2014). Assessing the performance of MM/PBSA and MM/GBSA methods. 5. Improved docking performance using high solute dielectric constant MM/GBSA and MM/PBSA rescoring. Physical Chemistry Chemical Physics, 16(40), 22035–22045. https://doi.org/10.1039/c4cp03179b
- Sun, H., Li, Y., Tian, S., Xu, L., & Hou, T. (2014). Assessing the performance of MM/PBSA and MM/GBSA methods. 4. Accuracies of MM/PBSA and MM/GBSA methodologies evaluated by various simulation protocols using PDBbind data set. Physical Chemistry Chemical Physics, 16(31), 16719–16729. https://doi.org/10.1039/c4cp01388c
- Surabhi, S., & Singh, B. (2018). Computer aided drug design: An overview. Journal of Drug Delivery and Therapeutics, 8(5), 504–509. https://doi.org/10.22270/jddt.v8i5.1894
- Thompson, D. C., Humblet, C., & Joseph-McCarthy, D. (2008). Investigation of MM-PBSA rescoring of docking poses. Journal of Chemical Information and Modeling, 48(5), 1081–1091. https://doi.org/10.1021/ci700470c
- Trott, O., & Olson, A. J. (2010). AutoDock Vina: Improving the speed and accuracy of docking with a new scoring function, efficient optimization, and multithreading. Journal of Computational Chemistry, 31(2), 455–461. https://doi.org/10.1002/jcc.21334
- Veselovsky, A. V., & Ivanov, A. S. (2003). Strategy of computer-aided drug design. Current Drug Targets. Infectious Disorders, 3(1), 33–40. https://doi.org/10.2174/1568005033342145
- Vuignier, K., Schappler, J., Veuthey, J. L., Carrupt, P. A., & Martel, S. (2010). Drug-protein binding: A critical review of analytical tools. Analytical and Bioanalytical Chemistry, 398(1), 53–66. https://doi.org/10.1007/s00216-010-3737-1
- Wang, R., Fang, X., Lu, Y., Yang, C.-Y., & Wang, S. (2005). The PDBbind database: Methodologies and updates. Journal of Medicinal Chemistry, 48(12), 4111–4119. https://doi.org/10.1021/jm048957q
- Wang, E., Sun, H., Wang, J., Wang, Z., Liu, H., Zhang, J. Z. H., & Hou, T. (2019). End-point binding free energy calculation with MM/PBSA and MM/GBSA: Strategies and applications in drug design. Chemical Reviews, 119(16), 9478–9508. https://doi.org/10.1021/acs.chemrev.9b00055
- Wang, Z., Wang, X., Li, Y., Lei, T., Wang, E., Li, D., Kang, Y., Zhu, F., & Hou, T. (2019). farPPI: A webserver for accurate prediction of protein–ligand binding structures for small-molecule PPI inhibitors by MM/PB(GB)SA methods. Bioinformatics (Oxford, England), 35(10), 1777–1779. https://doi.org/10.1093/bioinformatics/bty879
- Webb, B., & Sali, A. (2016). Comparative protein structure modeling using MODELLER. Current Protocols in Bioinformatics, 54(1), 5.6.1–5.6.37. https://doi.org/10.1002/cpbi.3
- Weng, G., Wang, E., Wang, Z., Liu, H., Zhu, F., Li, D., & Hou, T. (2019). HawkDock: A web server to predict and analyze the protein–protein complex based on computational docking and MM/GBSA. Nucleic Acids Research, 47(W1), W322–W330. https://doi.org/10.1093/nar/gkz397
- Yang, J., Roy, A., & Zhang, Y. (2013). Protein–ligand binding site recognition using complementary binding-specific substructure comparison and sequence profile alignment. Bioinformatics, 29(20), 2588–2595. https://doi.org/10.1093/bioinformatics/btt447
- Zaheer-Ul-Haq, Halim, S. A., Uddin, R., & Madura, J. D. (2010). Benchmarking docking and scoring protocol for the identification of potential acetylcholinesterase inhibitors. Journal of Molecular Graphics and Modelling, 28(8), 870–882. https://doi.org/10.1016/j.jmgm.2010.03.007
- Zhang, X., Perez-Sanchez, H., C., & Lightstone, F. (2017). A comprehensive docking and MM/GBSA rescoring study of ligand recognition upon binding antithrombin. Current Topics in Medicinal Chemistry, 17(14), 1631–1639. https://doi.org/10.2174/1568026616666161117112604