

Abstract
The negative impact of infectious diseases like COVID-19 on public health and the global economy is evident. This pandemic represents a significant challenge for the scientific community to develop new practical analytical methods for accurately diagnosing emerging cases. Due to their selectivity and sensitivity, new methodologies based on antigen/antibody interactions to detect COVID-19 biomarkers are necessary. In this context, the theoretical, computational modeling reduces experimental efforts and saves resources for rational biosensor design. This study proposes using molecular dynamics to predict the interactions between the Receptor Binding Domain (RBD) of the SARS-CoV-2 spike protein simplified model and a set of highly characterized antibodies. The binding free energy of the antigen/antibody complexes was calculated for the simplified models and compared against the complete SARS-CoV-2 ectodomain to validate the methodology. The structural data derived from our molecular dynamics and end-point free energy calculations showed a positive correlation between both approximations, with a 0.82 Pearson correlation coefficient; t = 3.661, df = 3, p-value = 0.03522, with a 95% confident interval. Furthermore, we identified the interfacial residues that could generate covalent bonds with a specific chemical surface without perturbing the binding dynamics to develop highly sensitive and specific diagnostic devices.
Communicated by Ramaswamy H. Sarma.
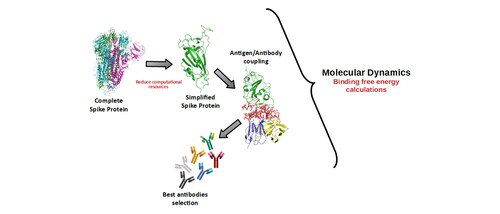
Graphical Abstract
Data and software availability
A listing of available data, construction and execution scripts and input structures files are available on the figshare repository: 10.6084/m9.figshare.15026352
All the third-party software used in this work is stated in the ‘Methods and computational tools’ section of the Manuscript. Here is a list of used software: Schrödinger Maestro v. 2019-4; Amber18 CUDA version software (Fee Waiver License for Abraham Vidal-Limon); R Statistical package v. 4.0.1, and CHARMM-GUI server was used for recover of SARS-Cov-2 spike protein full model (https://charmm-gui.org/).
Author contributions
Guillermo A. Huerta Miranda: Formal writing, Reviewing and Editing, Visualization, Investigation.: Wendy I. García-García: Reviewing and Editing, Investigation Writing – Original draft preparation.: Abraham Vidal-Limon: Conceptualization, Methodology, Writing – Original draft preparation.: Margarita Miranda-Hernández: Supervision, Project administration.
Disclosure statement
No potential conflict of interest was reported by the authors.