

Abstract
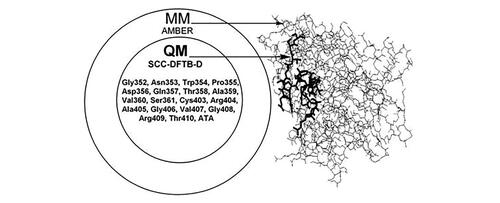
Some members of Yersinia (Y), a genus of bacteria in the family Yersiniaceae, are pathogenic in humans, causing a range of health problems, from gastrointestinal syndromes to the plague. The Y protein tyrosine phosphatase (PTP) YopH is a crucial virulence determinant, considering the vital roles of PTPs in the intracellular signal transduction pathways and cell cycle control. The structural understanding of YopH as a cellular target in pathogenic conditions caused by Y infection is a prerequisite for designing potent and selective YopH inhibitors. Thus, by using molecular docking simulations, the open and closed conformations of the so-called ‘WPD loop’ (352-Gly-Asn-Trp-Pro-Asp-Gln-Thr-Ala-Val-Ser-361), located nearby the active site (403-Cys-Arg-Ala-Gly-Val-Gly-Arg-Thr-410) in YopH structure, are shown to be relevant for recognition by carboxylic acid derivatives, and the closed conformation is a more preferable receptor in terms of the quantitative correlation with experimental data. In both cases, aurintricarboxylic acid (ATA) has the greatest affinity to YopH. Consequently, a quantum mechanics/molecular mechanics (QM/MM) molecular model is derived to see into the extent of the ATA-induced open-closed conformational change. Active site residues and the WPD loop, as well as ATA are treated using SCC-DFTB-D (QM level), while the rest of the complex is treated using AMBER force field (MM level). The active/inactive functional behavior of YopH is explored by observing the interaction mode of ATA with the wild-type (wt)/Cys403Ser receptor and evaluating the competitive inhibition parameters. Implications of the present study for experimental research are discussed.
Communicated by Ramaswamy H. Sarma
Acknowledgement
Prof. David A. Case of Rutgers University is acknowledged for granting the author an academic license for using the molecular dynamics software package Amber 11 in combination with AmberTools 1.5.
Disclosure statement
No potential conflict of interest was reported by the author.