

Abstract
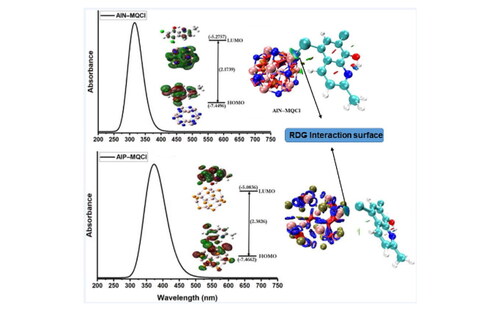
This combined Al12E12 (E = N, P) surface adsorption and docking study describes the new possibility of prospective potential probing(photophysical/optical) and therapy(medicinal/biochemical) with these adsorbent conjugates. DFT investigations were undertaken herein to help generate geometrical models and better understand the possible favorable adsorption energetics. We attempt to explain their adsorption behaviors and docking involving SARS–CoV–2 viruses (PDB)to assess their possible pharmaceutical potential against the pandemic virus (COVID–19). The adsorption behavior of 8–hydroxy–2–methylquinoline (MQ) and its halogenated derivatives, 5,7–diiodo–8–hydroxy–2–methylquinoline (MQI), 5,7–dichloro–8–hydroxy–2–methylquinoline (MQCl), and 5,7–dibromo–8–hydroxy–2–methylquinoline (MQBr), with aluminum–nitrogen (AlN), and aluminum–phosphorous (AlP) fullerene–like nanocages is reported. A decrease in the hardness of the nanoclusters when adsorbed with drug molecules resulted in an incrementally improved chemical softness (see e.g., Hard–Soft Acid Base theory) indicating that reactivity of the drug molecule in the resulting complex increases upon cluster chemical adsorption. The energy gap is found to be maximized for AlN–MQ and minimized for AlP–MQI; the reduced density gradient (RDG) iso–surfaces and AIM studies also corroborated this. Therefore, these two were found, respectively, to be the least and most electrically conductive of the species under study. We selected a simple medicinal building block (chelator)in addition to selecting the cluster based on previous literature reports. Important parameters such as gap energies and global indices were determined. We assessed NLO properties. The SARS–CoV–2 virus PDB docking data for 6VW1, 6VYO, 6WKQ, 7AD1, 7AOL, 7B3C, were enlisted as ligand targets for studies of docking (PatchDock Server) using the requisite PDB geometries (For the structure of 6VW1, kindly see reference, Citation2020; For the structure of 6VYO kindly see reference, Citation2020; For the structure of 6WKQ kindly see reference, Citation2020; For the structure of 7AD1 kindly see reference, Citation2021; For the structure of 7AOL kindly see reference, Citation2021; For the structure of 7B3C kindly see reference, Citation2021). Such findings indicate that the AlN–drug conjugation have inhibitory effect against these selected receptors.
Communicated by Ramaswamy H. Sarma
Graphical Abstract
Acknowledgements
D.G.C.is thankful to KAIST for funding and resources to help the continued operation of the Molecular Logic Gate Laboratory. D.G.C. acknowledges the KAIX program (KAIST), the recently opened KC 30 project, and Professors Hee–Seung Lee and Young–Min Rhee (Dept. of Chem., KAIST) for recent financial assistance during the fiscal years of 2020 and 2021. ––). P.S. acknowledges the KGSP of Korea. The Molecular Logic Laboratory is grateful for funding form the Korean National Research Foundation (2021R1F1A1046576). D.G.C. acknowledges financial support and resources from KAIST and the International Joint Usage Project with ICR, Kyoto University (2019–115 and 2020–124) to make our current efforts possible. Z.U. acknowledges National Research Foundation of Korea (NRF) grant funding made available from the Korea government (MSIP) (No. 2016R1A2B3011742). This work was also in part supported by the Priority Research Centers Program through the National Research Foundation of Korea (NRF) funded by the Ministry of Education (2020R1A6A1A03041954).
Disclosure statement
No potential conflict of interest was reported by the authors.