

Abstract
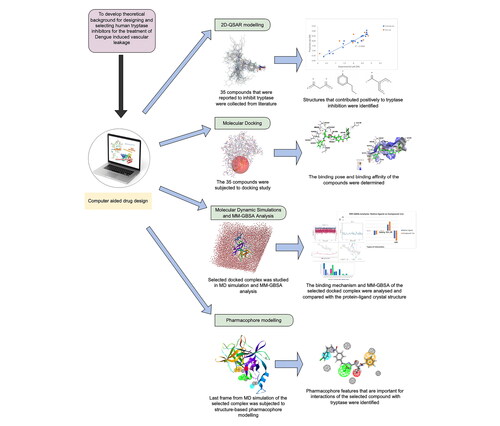
Dengue hemorrhagic fever (DHF) is severe dengue with a hallmark of vascular leakage. β-tryptase has been found to promote vascular leakage in DHF patients, which could be a potential target for DHF treatment. This study aims to develop a theoretical background for designing and selecting human β-tryptase inhibitors through computational studies. Thirty-four α-keto-[1,2,3]-oxadiazoles scaffold-based compounds were used to generate 2D-QSAR models and for molecular docking studies with β-tryptase (PDB Code 4A6L). In addition, molecular dynamics (MD) simulation and molecular mechanics generalised born surface area (MM-GBSA) analysis on the binding of the reported most active compound, compound 11e, towards β-tryptase were performed. Finally, a structure-based pharmacophore model was generated. The selected 2D-QSAR models have statistically proven good models by internal and external validation as well as the y-randomization test. The docking results of compound 11e showed lower CDOCKER energy than the 4A6L co-crystallised ligand and a similar binding pattern as the 4A6L co-crystallised ligand. From molecular dynamics simulation, 4A6L in compound 11e bound state has RMSD below 2 Å throughout the 500 ns simulation, indicating the docked complex is stable. Besides, MM-GBSA analysis suggested the 4A6L-compound 11e docked complex (−66.04 Kcal/mol) is structurally as stable as the 4A6L-native ligand co-crystallized structure (−66.84 Kcal/mol). The best pharmacophore model identified features included hydrogen bond acceptor, ionic interaction, hydrophobic interaction, and aromatic ring, which contribute to the inhibitory potency of a compound. This study supplied insight and knowledge for developing novel chemical compounds with improved inhibition of β-tryptase.
Communicated by Ramaswamy H. Sarma
Acknowledgements
The authors would like to thank the members of Cell Signalling Laboratory, Faculty of Medicine and Health Sciences, Universiti Putra Malaysia, for supporting this study. The authors would also like to thank the International Islamic University of Malaysia-IIUM through Research Management Centre Grant 2020 (RMCG20-008-0008) and Universiti Kebangsaan Malaysia (UKM) for providing Discovery Studio® 3.1 software. The authors are also thankful to Atta-ur-Rahman Institute for Natural Product Discovery (AuRIns) and the Faculty of Applied Science, Universiti Teknologi Mara, for the support in molecular dynamics simulation.
Disclosure statement
There are no competing interests to declare.