
Abstract
In this study, the impact of four P-gp mutations (G185V, G830V, F978A and ΔF335) on drug-binding and efflux-related signal-transmission mechanism was comprehensively evaluated in the presence of ligands within the drug-binding pocket (DBP), experimentally related with changes in their drug efflux profiles. The severe repacking of the transmembrane helices (TMH), induced by mutations and exacerbated by the presence of ligands, indicates that P-gp is sensitive to perturbations in the transmembrane region. Alterations on drug-binding were also observed as a consequence of the TMH repacking, but were not always correlated with alterations on ligands binding mode and/or binding affinity. Finally, and although all P-gp variants holo systems showed considerable changes in the intracellular coupling helices/nucleotide-binding domain (ICH-NBD) interactions, they seem to be primarily induced by the mutation itself rather than by the presence of ligands within the DBP. The data further suggest that the changes in drug efflux experimentally reported are mostly related with changes on drug specificity rather than effects on signal-transmission mechanism. We also hypothesize that an increase in the drug-binding affinity may also be related with the decreased drug efflux, while minor changes in binding affinities are possibly related with the increased drug efflux observed in transfected cells.
Communicated by Ramaswamy H. Sarma
1. Introduction
Over-expression of membrane efflux pumps is intimately related to multidrug-resistance (MDR) phenomenom in cancer cells (Kapoor et al., Citation2021). Among them, P-glycoprotein (P-gp, ABCB1) is the most studied so far, and it is capable to extrude a wide range of neutral and charged hydrophobic compounds (Kapoor et al., Citation2021), through an ATP-dependent mechanism (Verhalen et al., Citation2017).
P-glycoprotein architecture comprises two transmembrane domains (TMDs), each one formed by six transmembrane α-helices (TMHs) packed in a pseudo 2-fold symmetry, and two cytoplasmic nucleotide-binding domains (NBDs), with both P-gp halves connected through a small peptide sequence (the ‘linker’; residues 627–688) (Aller et al., Citation2009). The transmembrane and cytoplasmic domains of the P-gp efflux pump are physically linked to the respective NBD by coils bridging TMH6/NBD1 and TMH12/NBD2, and by short intracellular coupling helices (ICHs), located between TMHs 2/3 (ICH1-NBD1), 4/5 (ICH2-NBD2), 8/9 (ICH3-NBD2) and 10/11 (ICH4-NBD1), the latter involved in the TMD-NBD communication through non-covalent interactions (Clouser & Atkins, Citation2022; Bonito et al., Citation2020). Additionally, some studies indicated that ICHs may also play important roles in P-gp folding and maturation (Loo et al., Citation2013; Loo & Clarke, Citation2013). Oppositely, the drug-binding pocket (DBP) is located within the TMHs of both N- and C-terminal P-gp halves, and contains, at least, three distinct drug-binding sites (DBS): the modulator site (M-site) located at the top of DBP, and the substrate-binding sites (SBSs) H and R, named after Hoechst33342 and Rhodamine-123, respectively, located in the opposite side next to the inner leaflet of the lipid bilayer (Ferreira et al., Citation2013).
Although decades of research focus on the molecular basis of drug promiscuity and efflux, the mechanisms of drug specificity and efflux-related signal-transmission are still unclear, thus hampering the development of novel, more potent and selective P-gp modulators able to overcome MDR. Herein, one interesting approach is the study of P-gp variants experimentally related to altered drug-resistance phenotypes and/or changes in the basal and drug-stimulated ATPase activity. Our previous work (Bonito et al., Citation2020), reported the impact of four hP-gp variants involved in MDR—G185V (Choi et al., Citation1988; Safa et al., Citation1990; Rao, Citation1995; Ramachandra et al., Citation1996; Loo & Clarke, Citation1995; Omote et al., Citation2004), G830V (Omote et al., Citation2004; Loo & Clarke, Citation1994), F978A (Mittra et al., Citation2017; Loo & Clarke, Citation1993) and ΔF335 (Harker et al., Citation1983; Harker & Sikic, Citation1985; Chen et al., Citation1997; Chen et al., Citation2000)—on the P-gp structure in the absence of ligands within the DBP (apo systems). In this paper, in order to understand if the P-gp conformational changes induce by these P-gp mutations affect drug-binding and efflux-related signal-transmission mechanism, the structural impact of these P-gp mutations was assessed in the presence of molecules, experimentally described as having alterations in their efflux profiles in these variants, bound to the reported DBSs (holo systems).
Similar to our previous work, as the glycine mutations G185V (TMH3) and G830V (TMH9) are located at the SBSs H and R, respectively, both variants will be named SBS-variants. On the other hand, the F978A (TMH12) and ΔF335 (TMH6) mutations, both lying at the M-site, will be referred as M-site variants. Following, the ligands were chosen considering: (i) the location of the mutation at the TMDs, (ii) the experimental and/or computational evidences for its putative DBS (Ferreira et al., Citation2013; Shapiro & Ling, Citation1997; Tamai & Safa, Citation1991; Dey et al., Citation1997; Loo et al., Citation2005; Loo & Clarke, Citation2005; Loo et al., Citation2003a), and (iii) opposite changes in the drug efflux profiles by the same P-gp mutation (Bonito et al., Citation2020). Thus, colchicine (COL) and vinblastine (VIN), both predicted to bind at the H-site, were the substrates chosen for the G185V (H-site). Likewise, doxorubicin (DOX) and actinomycin D (ACT), both predicted to interact with the R-site, were the substrates selected for the G830V (R-site). On the other hand, since the mutations at the M-site (F978A and ΔF335) seem to strongly affect the SBSs properties and drug-binding (Bonito et al., Citation2020), the structural effects of these mutations were analyzed in the presence of ligands in all three sites. Therefore, the P-gp modulator cyclosporine A (CYC, M-site), and the substrates ACT (R-site) and VIN (H-site) were the molecules selected for the F978A mutation. However, to the best of our knowledge, there is no information about a P-gp substrate that binds at the H-site and has altered drug-resistance profile in transfected cells harboring the ΔF335 mutation. That said, DOX (R-site) and two P-gp modulators, CYC and valspodar (VLS), both predicted to bind at the M-site, were the ligands chosen for this P-gp variant.
2. Material and methods
2.1. Initial structures
From our previous study, five MD apo systems were used as template to build the MD holo systems, the refined and validated hP-gp wild-type (WT) and four hP-gp variants models (G185V, G830V, F978A and ΔF335). In each one the transporter is embedded in a lipid bilayer with 469 molecules of 1-palmitoyl-2-oleoylphosphatidylcholine (POPC), for which the lipid parameterization by Poger et al. was used (Poger & Mark, Citation2010; Poger et al., Citation2010) as previously described (Bonito et al., Citation2020).
From our docking studies previously published (Bonito et al., Citation2020), the top-ranked docking poses for the chosen ligands were selected and used to assemble the WT and variants MD holo systems. The molecules were parameterized in the PRODRG (Schüttelkopf & van Aalten, Citation2004) online server and manually curated, according to the GROMOS96 force field (Bonvin et al., Citation2000; Chandrasekhar et al., Citation2005; Daura et al., Citation1998; Scott et al., Citation1999), using AM1-BCC partial charges calculated with antechamber 1.27 (Wang et al., Citation2006; Lemkul et al., Citation2010). The protein–ligand–membrane systems were then solvated with 63.810 ± 31 single-point charge (SPC) water molecules and neutralized with 11 chlorine ions, for a total of 16 MD holo systems. These MD systems were the starting point for several short MD runs.
2.2. Free-energy calculations and analysis
After a short energy minimization run to minimize clashes between the ligand and the protein, a total of five replicates of 20 ns MD runs were performed for each system (N = 16), in a total of 1.6 µs of simulation time (0.1 µs each system). For each replicate initial velocities were assigned from a Maxwell-Boltzmann distribution at 303 K. To enhance sampling, for the last two replicates distinct snapshots were retrieved from the first MD run (at 18 and 19 ns, respectively). Only the last 10 ns of simulation time were considered as production runs and used for further analysis (RMSD plots for all systems are available as Supporting Information, Figures S1–S2). Relative free-energies of binding (ΔGbind) were calculated using the g_mmpbsa tool (Kumari et al., 2014), with an implicit membrane correction for polar solvation energies (Ferreira et al., Citation2015). As in a previous paper, gmx bundle, gmx hbond (van der Spoel et al., Citation2006) and g_contacts (Blau & Grubmuller, Citation2013) tools were used to further evaluate changes in the transmembrane (TM) helical bundle, hydrogen bond (HB) networks and protein interactions, respectively.
Figure 1. Superimposition of the colchicine-bound systems at the H-site for WT (blue, licorice) and G185V variant (dark yellow, ball-and-stick).
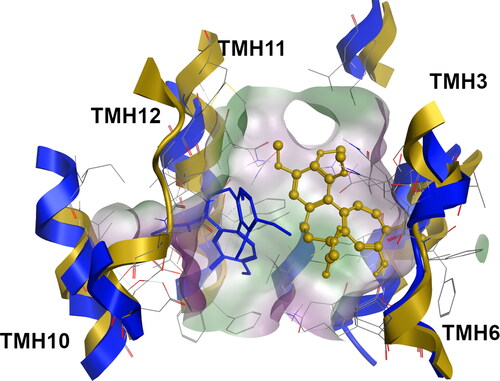
Figure 2. Superimposition of the doxorubicin-bound systems at the R-site for the WT (blue, licorice), G830V (dark yellow, ball-and-stick) and ΔF335 (gray, ball-and-stick) variants.
2.3. MD simulation parameters
All MD simulations were done with GROMACS v2016.x package (Abraham et al., Citation2015). All NVT equilibration runs were performed at 303 K using the Velocity-rescale (V-rescale) (Bussi et al., Citation2007) thermostat. The Nosé-Hoover (Nosé & Klein, Citation1983; Hoover, Citation1985) thermostat and the Parrinello-Rahman (Parrinello & Rahman, Citation1981) barostat for temperature (303 K) and pressure (1 bar), respectively, were applied in all NpT runs. Due to the presence of membrane, pressure equilibration was achieved through a semi-isotropic pressure coupling, with the systems’ compressibility set to 4.5 × 10−5 bar−1. All bond lengths were constrained using the LINCS (Hess et al., Citation1997; Hess, Citation2008) or SETTLE (Miyamoto & Kollman, Citation1992) (for water molecules) algorithms. The Particle Mesh Ewald (PME) with cubic interpolation (Essmann et al., Citation1995; Darden et al., Citation1993) was employed with a cut-off radius of 12 Å for both electrostatic and van der Waals interactions, and an FFT grid spacing of 0.16 for long range electrostatics. Group-based and Verlet (Pronk et al., Citation2013) cut-off schemes were applied for the calculation of non-bonded interactions on CPU or GPU, respectively.
3. Results and discussion
The analysis that will be presented and discussed in the following subsections relates to the behavior of the human P-gp WT and variants studied containing different allocrites inside the drug-binding sites.
3.1. Structural analysis of transmembrane domains of the hP-gp variants
To give additional insights on drug specificity and efflux-related signal-transmission mechanism, the structural impact of four selected P-gp mutations (G185V, G830V, F978A and ΔF335), experimentally related to changes in drug efflux and substrate specificity, were analyzed in the presence of molecules bound to each site inside the DBP (holo systems). The molecules chosen are described in literature as having altered efflux profiles in these P-gp variants—G185V (COL, VIN), G830V (ACT, DOX), F978A (ACT, VIN, CYC) and ΔF335 (CYC, VLS, DOX; ).
Table 1. Structural impact of mutations in the TMH repacking for the holo hP-gp variantsTable Footnotea.
Mutations (Bonito et al., Citation2020) and/or the presence of ligands within the DBP may directly affect the TMDs architecture (Kapoor et al., Citation2013). Thus, the rearrangement of the transmembrane helices (TMHs) was assessed using the gmx bundle for the P-gp variants holo systems and compared to the WT (holo system). All the significant changes in the bundle parameters are summarized in the Supporting Information (Tables S1–S24 and Figures S3–S50). To organize and simplify the results described below, the studied P-gp systems will be encoded and referred as ‘variant-DBS1_ligand-DBS2’, where DBS1 is the DBS where the mutation is located and DBS2 the DBS where the molecule is reported to bind.
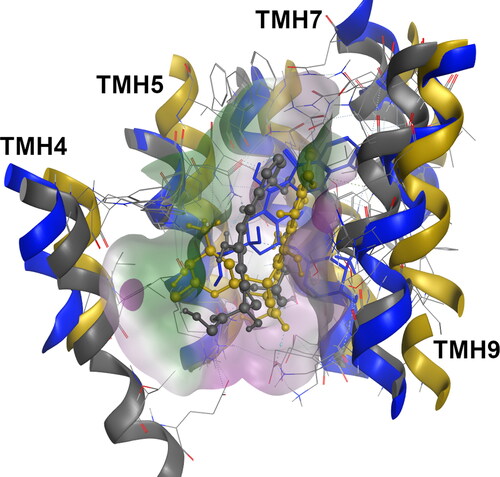
Figure 3. Superimposition of the actinomycin-bound systems at the R-site for the WT (blue, licorice) and G830V variant (dark yellow, ball and stick).
Overall, the bundle parameters calculated for all P-gp variants were found to have distinct shifts from the WT, when in the presence of the respective ligands. Most remarkably, the changes were larger when the helices that form the DBP portals (4/6 and 10/12) and the ‘crossing helices’ (4/5 and 10/11), directly connecting the TMD1 to NBD2 and TMD2 to NBD1, respectively (Kim & Chen, Citation2018), were involved. However, distinguishable structural effects in the TMH rearrangement were still observed between P-gp variants. The SBS-variants G185V and G830V, in the presence of the respective ligands, revealed deeper changes in the TMH repacking affecting both P-gp halves, and thus not limited to its surrounding environment. Firstly, COL in the G185V-H_COL-H system induced a stronger impact in the TMDs architecture than VIN in the G185V-H_VIN-H system. Similarly, the F978A variant showed a striking TMH repacking, more severe in the F978A-M_CYC-M and F978A-M_ACT-R systems. On the other hand, the total repacking observed in the ΔF335 variant seemed to be similar in all holo ΔF335 systems.
The structural impact of a specific ligand in two different P-gp variants was determined (). Herein, a stronger effect in the TMH repacking was observed in the G185V-H_VIN-H and G830V-R_DOX-R systems than those observed for the M-site variants in the F978A-M_VIN-H and ΔF335-M_DOX-R systems, respectively. On the other hand, ACT induced a similar impact in the TMH rearrangement in both G830V-R_ACT-R and F978A-M_ACT-R systems. Moreover, CYC seemed to have larger shifts in the bundle parameters in the F978A-M_CYC-M system than for the ΔF335-M_CYC-M system. Finally, when compared to the P-gp variants apo systems (Bonito et al., Citation2020), more severe changes in the TMDs architecture were observed for all holo P-gp variants. Nonetheless, the presence of ligands in the M-site variants seemed to have a larger effect in the TMH repacking than those reported for the respective apo systems.
Altogether, the analysis of the helical bundle indicates that the studied P-gp mutations, when in the presence of ligands within the DBP, (i) severely affect the native spatial position of the helices that form the DBP portals, which may alter the access of drugs to the internal cavity or their release to the membrane (Kapoor et al., Citation2021), and (ii) induces changes in the helical bundle concerning the ‘crossing helices’, crucial for the NBD dimerization process upon ATP-binding (Kim & Chen, Citation2018). Additionally, the presence of molecules seems to induce a more severe impact in the TMHs rearrangement than those observed in the respective apo systems (Bonito et al., Citation2020), indicating a TMDs adjustment and response to the presence of ligands not circumscribed to the TM helix (or site) where the mutation is located.
3.2. Characterization of protein–ligand interactions
Since all the above mutations changed the TMDs architecture in variable degrees, their impact on the ligand-binding affinity was further evaluated. Relative free-energies of binding (ΔGbind) were estimated using the g_mmpbsa (only available in GROMACS v5.x) tool and compared to the WT holo systems (Supporting Information, Figures S51–S54).
Although a severe TMH repacking was observed in all P-gp variants, the binding affinity was affected differently (). While no significant changes were found in the G185V-H_COL-H system, a substantial increase in binding affinity was observed in both G185V-H_VIN-H and F978A-M_VIN-H systems. Oppositely, no considerable effects were observed in the calculated ΔGbind values for the G830V-R_DOX-R and ΔF335-M_DOX-R systems. No significant changes in binding affinities were also found for the G830V-R_ACT-R system (although a significant change on the molecules’ binding mode was observed, as discussed below). On the other hand, although observing a slight increase in the ACT-binding affinity in the F978A-M_ACT-R system, no clear conclusions could be made about the possible changes in the molecule’s affinity. Finally, an increase on the ligand-binding affinities was observed in the F978A-M_CYC_M, ΔF335-M_CYC-M and ΔF335-M_VLS-M systems.
Table 2. Structural impact of mutations in the ligand-binding affinity for the P-gp variants holo systems.
To understand how the severe TMH rearrangement observed in all P-gp variants affected ligand-binding affinity differently, specific protein-ligand interactions were additionally assessed using the g_contacts (from GROMACS v4.6.7) and gmx hbond tools, and again compared to the WT protein holo systems. Mean contact frequencies ≥ 0.5 and variations above 10% were considered as significant and are depicted in SI (Supporting Information, Tables S25–S27).
According to our previous studies, the TMH repacking induced by these P-gp mutations led to severe changes in both structure and residues distribution at the DBSs, thus having a strong impact on size and polarity (Bonito et al., Citation2020). Herein, and concerning the G185V-H_COL-H system, colchicine, initially located at the bottom of the H-site, shifted from its original binding position in WT, losing interactions with TMHs 10/12 while establishing new ones with TMHs 6/11 ().
However, and according to the ratio between the number of significant protein-ligand contacts and the average of the total number of contacts (hereby named contact efficiency ratio—CE, Supporting Information, Table S28), similar values were observed in both WT-H_COL-H and G185V-H_COL-H systems, thus indicating that although COL established new contact points with residues from other TMHs, no significant impact in binding affinity was observed for this ligand. In the same way, a similar CE ratio was observed between the WT-R_DOX-R and the variant holo systems G830V-R_DOX-R and ΔF335-M_DOX-R.
Regarding doxorubicin, a common binding mode between the WT and both G830V and ΔF335 variants was observed, in which the tetracyclic core is found in close contact with hydrophobic surface patches—by interacting with residues from TMHs 7 and 9—in the respective pockets (). These results indicate that the binding mode for DOX is not substantially affected by these mutations, and no significant alterations in the DOX binding affinity were observed for these variants. Oppositely, a great increase in the vinblastine contact efficiency ratio was observed in both G185V-H_VIN-H and F978A-M_VIN-H systems. The visual inspection showed that VIN was deeply buried in the H-site in both P-gp variants, which may account for stronger interactions and, concomitantly, to the higher binding affinity observed for this molecule.
Quite interestingly, a complete distinct binding mode were observed in the ACT in the G830V-R_ACT-R system. The visual inspection revealed severe alterations in the ACT-binding mode, with a large region of the molecule protruding from the R-site towards the middle of the DBP and, thus exposed to the surrounding environment (). Since the contact efficiency (CE) ratio was higher in the G830V-R_ACT-R system than in WT-R_ACT-R but without significant change in binding energy, the loss of contacts between a large region of the molecule and the protein may explain the minor impact in the ACT ΔGbind values observed for this variant, that is, the molecule changes the interaction pattern loosing interactions by making fewer but stronger ones. Oppositely, a visual inspection of the F978A variant showed that ACT remained deeply buried in the R-site as observed in WT, thus showing a similar contact efficiency ratio and, as expected, no changes in the ACT binding affinity.
Lastly, an increase in the CE ratio was observed for both F978A-M_CYC-M and ΔF335-M_CYC-M systems. Again, visual inspection showed that the structural changes at the M-site in the F978A variant prompted CYC to change the conformation of its macrocycle, shifting one of the isoleucine side-chains into the more hydrophobic environment at the center of the macrocyclic core () and therefore improving the CYC binding affinity.
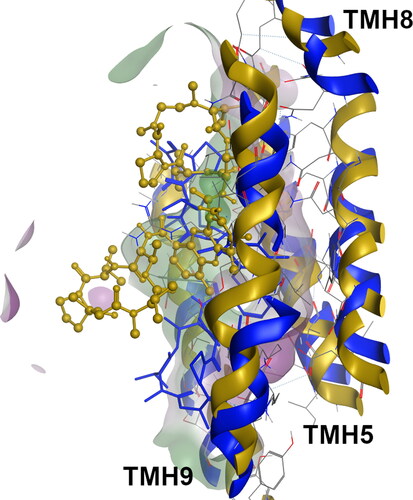
Figure 4. Superimposition of the cyclosporine-bound systems at the M-site for the WT (blue, licorice) and F978A variant (dark yellow, ball-and-stick).
Regarding the ΔF335-M_CYC-M system, no considerable changes in the CYC binding mode was observed in respect to WT, with all hydrophobic groups being protected from the solvent. However, the increase in the CYC binding affinity observed for these variants allow to infer that small changes of conformation in a large ligand may play important roles in binding affinity. Likewise, valspodar in the ΔF335-M_VLS-M system was also found to be deeply buried inside the M-site, which favors the reinforcement of the VLS-ΔF335 interactions, and consequently, its binding affinity. These results could be explained by the lower impact in the M-site volume and polarity reported for the F978A and ΔF335 variants apo systems, when compared to the SBSs where the structural impact of these mutations was higher (Bonito et al., Citation2020).
Altogether, the above data indicate that these P-gp mutations seem to have an impact on the binding energy, ligand-binding modes and/or on the protein-ligand interactions. Moreover, although the mutations have an undeniably distinct but strong structural impact in the TMH repacking, similar effects on the binding energy observed for the same molecule across two different mutations (VIN-H in G185V and F978A; ACT-R in G830V and F978A; DOX-R in G830V and ΔF335; CYC-M in F978A and ΔF335), suggest that the molecular properties of these ligands may play important contributions (i) to their binding affinity rather than the severity of the TMH repacking induced by these mutations, or (ii) in the way P-gp repacks its transmembrane helices when in the presence of these ligands. These remarks seem to be independent of the DBS where the molecule binds.
3.3. Interactions between coupling helices and nucleotide-binding domains
All the herein considered P-gp variants are experimentally described as having altered drug efflux profiles and/or changes in the basal and/or drug-stimulated ATPase activity (Choi et al., Citation1988; Safa et al., Citation1990; Rao, Citation1995; Ramachandra et al., Citation1996; Loo & Clarke, Citation1995; Omote et al., Citation2004; Loo & Clarke, Citation1994; Mittra et al., Citation2017; Loo & Clarke, Citation1993; Harker et al., Citation1983; Harker & Sikic, Citation1985; Chen et al., Citation1997; Chen et al., Citation2000) (for additional details, please refer to (Bonito et al., Citation2020)). Therefore, as the ICH-NBD residue interactions are increasingly considered to be key players in signal-transmission and efflux-related conformational changes (Loo & Clarke, Citation2013; Wise, Citation2012; Pajeva et al., Citation2013; Loo et al., Citation2003b; Loo & Clarke, Citation2016a), a thorough evaluation of the ICH-NBD contacts was performed and compared to the WT holo systems. The total number of contacts was estimated by the gmx hbond module (Supporting Information, Figures S55–S62).
Overall, all P-gp holo systems revealed changes in the mean total number of contacts at the ICH-NBD signal-transmission interfaces. However, while in most cases a reinforcement in the total number of contacts was observed at the ICHs-NBD1 interfaces, a negative impact in the total number of contacts was observed in the ICHs interacting with NBD2 ().
Table 3. Structural impact of mutations in the total number of contacts at the ICHs-NBD interfaces for the P-gp variants holo systems.
As all P-gp mutations in the presence of ligands seemed to affect the total number of contacts at the ICH-NBD interfaces, we aimed for the identification of which residue pairs were mostly involved through the evaluation of their mean contact frequencies, using the g_contacts tool. Only mean contact frequencies ≥ 0.5 and variations above 10% were considered meaningful (Supporting Information, Tables S28–S32). For clarity purposes, the analysis of each ICH-NBD interface will be described separately:
3.3.1. ICH-NBD1 interfaces
Starting from the N-terminal P-gp halve, the first ICH-NBD interface is the ICH1-NBD1. Herein, all P-gp variants holo systems showed a significant increase in the mean contact frequencies between I160 and the NBD1 residue L443, described as important for ATP-binding (Loo & Clarke, Citation2016a; Kwan & Gros, Citation1998; Ferreira et al., Citation2017). On the contrary, a negative impact in the residue interactions was observed in most of the P-gp variants holo systems between D164-K405, also involved in the TMD-NBD communication pathway (Kapoor et al., Citation2013). When concerning the hydrogen bond network, a negative effect in the HB lifetime was also observed in both G830V-R_ACT-R and F978A-M_ACT-R systems as well as in all ΔF335 variant holo systems. A decrease in the HB lifetime was also inferred from the G185V-H_VIN-H, but when in the presence of colchicine a significant reinforcement of the HB network in the G185V-H_COL-H system was observed.
On the other hand, all the P-gp variants showed clear differences in the residue interactions at the ICH4-NBD1, the second signal-transmission interface involving NBD1. Regarding the SBS-variants, similar effects in the residue interactions were observed in the G185V holo systems, mainly reinforcing the contact frequencies between Q912-R467, with R467 being involved in the propagation of the conformational changes (Ward et al., Citation2013). Oppositely, a strong negative impact in the mean contact frequencies was observed for the residue pair T911-R467. A negative variation in the contact frequencies was also verified to occur for the residue pairs S909-Q441 and Q912-R464, with S909 being involved in the activation and ATPase stimulation when in the presence of drugs and/or lipids (Loo & Clarke, Citation2016a, Citationb). In the G830V-R_ACT-R system, the presence of ACT again induced a strong negative effect in the contacts involving the R905-Q438, T911-R467 and Q912-R464 residue pairs. Quite interestingly, mutational studies had already suggested that R905 is a pivotal residue in the activation and ATPase stimulation when in the presence of drugs and/or lipids (Loo & Clarke, Citation2016a, Citationb). Surprisingly, an opposite trend was found for both M-site variants. Both the F978A and ΔF335 holo systems showed a significant reinforcement in the mean contact frequencies between the R905-Y401/Q438 and V908/T911-R467 residue pairs, with Y401 (NBD1) again being important for ATP binding (Kim & Chen, Citation2018). Regarding hydrogen bonds, the G830V-R_ACT-R system was the only one that showed a dramatic increase in the HB lifetime while a negative impact in the calculated HB parameters was observed in all other P-gp variants holo systems.
3.3.2. ICH-NBD2 interfaces
At the C-terminal P-gp halve, the first signal-transmission interface is the ICH2-NBD2. Herein, all P-gp variants holo systems showed a decrease in the mean contact frequencies between the residues A266 (ICH2) and F1086 (NBD2), a critical residue pair for coupling of ATP-binding to conformational changes in the TMDs (Loo et al., Citation2013; Loo & Clarke, Citation2013). Nonetheless, substantial differences in residue pair interactions were observed between the P-gp variants. For both G185V-H_COL-H and G185V-H_VIN-H systems, the presence of these ligands affected the R262-F1086 (positive variation) and I265-R1110 (negative variation) residue pairs, being R262 (ICH2) an important residue in the propagation of the conformational changes (Ward et al., 2013). However, a noteworthy reinforcement in the mean contact frequencies between T263-Q1118 and T263-D1200 residue pairs was observed only for both G185V holo systems. Accordingly, Q1118 is an important residue for ATP-binding/hydrolysis and Mg2+ binding since mutations in this residue, located within the Q-loop, blocked drug-stimulated ATPase activity (Altenberg et al., Citation1994; Prajapati & Sangamwar, Citation2014). In contrast, the presence of ACT and DOX in the G830V-R_ACT-R and G830V-R_DOX-R systems had an overall negative impact in the mean contact frequencies involving the R262-Q1081 and T263-S1117 residue pairs. Regarding the M-site variants, a negative variation in the mean contact frequencies was observed in the F978A variant holo systems, namely between R1110 (NBD2) and residues I265/F267 (ICH2), described as involved in P-gp maturation and activity (Loo et al., Citation2013; Loo & Clarke, Citation2013; Loo & Clarke, Citation2015). Similarly, the presence of DOX in the ΔF335-M_DOX-R system negatively affected most of the identified ICH-NBD contact points.
Changes in the ICH-NBD hydrogen-bond network were additionally observed for the G185V and M-site variants holo systems. While the G185V-H_COL-H system showed a decreased in the HB lifetime, VIN on the other hand reinforced the calculated HB parameters in the G185V-H_VIN-H system. Opposite effects were observed between CYC and VLS in their respective P-gp variants: while the HB lifetime was negatively affected by the presence of CYC in both F978A-M_CYC-M and ΔF335-M_CYC-M systems, the presence of VLS in the ΔF335-M_VLS-M system instead reinforced the HB network.
Finally, most of the P-gp holo systems also showed negative effects in the residue interactions at the ICH3-NBD2, the second signal-transmission interface interacting with NBD2. Most particularly, a significant decrease in the mean contact frequencies was observed between the residue pairs V801/S802 and the NBD2 residue Y1087, which is thought to play an important role in P-gp activity and folding (Loo et al., Citation2003b). Moreover, the Y1087 residue was also predicted to be the key residue in the ICH3-NBD2 communication (Pajeva et al., Citation2013). Similarly, and concerning the HB network, a general decrease in the HB lifetime was observed in the holo systems of the G185V and M-site variants.
Altogether the results indicate that the studied P-gp mutations holo systems (i) have long-range effects in the residue interactions at the ICH-NBD signal-transmission interfaces, (ii) induce asymmetries in the ICH-NBD residue interactions and (iii) affect the mean contact frequencies of important residue pairs experimentally related to drug-stimulated ATPase activity, P-gp maturation and/or folding.
Mechanistic insights
P-glycoprotein over-expression is one of the most relevant multidrug-resistance mechanisms in cancer cells (Juliano & Ling, Citation1976). To develop compounds with high selectivity and efficacy towards P-gp, a deeper knowledge underlying the mechanisms of drug specificity and efflux-related signal-transmission is needed. In our previous work, a refined hP-gp WT model and four P-gp variants (G185V, G830V, F978A and ΔF335), experimentally related to changes in substrate specificity, basal and drug-stimulated ATPase activity were built in the apo state inward-facing conformation, and their structural impact in the TMDs and ICH-NBD interactions was thoroughly evaluated (Bonito et al., Citation2020). In the present study, these P-gp models were used to assess the structural effects of these mutations on drug-binding and efflux-related signal-transmission mechanism in the presence of ligands within the DBP, that are also experimentally described as having altered efflux patterns.
Taken together, our data indicate that these P-gp mutations in the presence of ligands within the DBP induce structural effects in the TMDs by mainly affecting (i) the DBP portals 4/6 and 10/12 with the possibility for compromising the access and release of substrates, and (ii) the ‘crossing helices’ 4/5 and 10/11 able of compromising the NBD dimerization upon ATP-binding. Although these findings are in agreement with those reported in our P-gp apo systems (Bonito et al., Citation2020), the impact on the TMH repacking seems to be stronger in the P-gp holo systems, suggesting that P-gp remains sensitive to the presence of ligands. Additionally, our results are also in agreement with the experimental studies performed by Loo and Clarke (Loo et al., Citation2003b) and with the recent biochemical studies by Clouser and co-workers (Clouser & Atkins, Citation2022; Clouser et al., Citation2021). Accordingly, substrates bind to P-gp through a ‘substrate-induced fit mechanism’, where the size and shape of the substrate induce rearrangements in the TMHs (Loo et al., Citation2003b). Furthermore, the rearrangement of the TMH residues’ side-chains due to repacking affect the protein–ligand interactions and/or binding affinity (local effects). On the other hand, the presence of mutations and/or ligands induces variations in the total number of contacts at the ICH-NBD interfaces, possibly affecting the TMD-NBD communication, and consequently altering drug efflux (long-range effects). Lastly, the impact in the mean contact frequencies of specific ICH-NBD residue pairs, experimentally described as involved in the signal-transmission mechanism, folding and ATP-binding (Loo & Clarke, Citation2015), offers a possible explanation for the alterations in drug efflux reported for these P-gp variants.
Interestingly, most of the P-gp variants holo systems showed asymmetrical changes in the total number of contacts at the ICH-NBD interfaces, namely a reinforcement in the number of contacts at the ICHs-NBD1, while a decrease in the contacts was observed in the ICHs connecting NBD2. Two experimental studies recently demonstrated that (i) the presence of cholesterol in the membrane induces asymmetries between NBDs, involving the ICH-NBD interfaces (Clouser et al., Citation2021), and (ii) there are common regions affected in a similar manner, but diverging in the post-hydrolysis state, especially in ICHs 3 and 4. Concerning the former, if taken into account that cholesterol is also a P-gp substrate (Clouser et al., Citation2021), the latter corroborates our hypothesis that the presence of the selected ligands within the DBP exacerbate the asymmetries in the contacts at the ICH-NBD interfaces. The fact by which similar changes in the residue’s interactions at the ICH-NBD interfaces were also reported in these P-gp variants apo systems (Bonito et al., Citation2020), especially concerning the M-site variants, additionally suggests that (i) such alterations can be induced solely by the mutation, independently of the presence of molecules within the DBP, and (ii) such alterations can be further exacerbated by the presence of molecules bound at the SBSs.
Nevertheless, important structural differences were observed between the P-gp variants holo systems. Regarding the G185V (TMH3) mutation, experimental data showed that it conferred increased resistance to COL in transfected cells, with an increase in the COL-stimulated ATPase activity, and a 3.6-fold decrease in COL binding. Oppositely, a decreased resistance to VIN was reported for this P-gp variant, with a 3.8- to 5.5-fold increase in the VIN binding (Loo & Clarke, Citation1994). Although no clear conclusions could be made about the possible changes in the COL binding affinity in the G185V-H_COL-H system, an indubitably increase in the VIN binding affinity was observed in the G185V-H_VIN-H system, in agreement with the experimental data.
Nonetheless, the presence of COL induced a reinforcement of the residue’s interactions and HB network at the ICH1-NBD1, something that was not observed when in the presence of VIN. As ICH1 establishes interactions with the A-loop residues of NBD1 and the adenine group of ATP (Becker et al., Citation2009), we hypothesized that the reinforcement of the ICH1-NBD1 interactions may be related to the increase in COL-stimulated ATPase activity reported in the experimental studies and, therefore, with the increased resistance to COL. Furthermore, a recent study emphasized the role of the conserved ICH1-NBD1 interface as critical for the cross-talk between the TMD-NBD domains in ABCG2, another relevant ABC transporter in the MDR in cancer cells (Ferreira et al., Citation2017). On the other hand, the decrease in the VIN efflux experimentally observed in this P-gp variant, in our models, is solely due to the sharp increase in the VIN binding affinity. A previous experimental study performed by Loo and Clarke (Loo & Clarke, Citation1993) focused on another single point mutation, F335A, which correlated the increase in the VIN binding affinity within the DBS with a decrease in the VIN efflux. According to the authors, the release of VIN during the transport cycle was postulated to be impaired due to its increased affinity at the DBS, and the herein published model corroborates those assumptions.
Located at the opposite P-gp halve, the G830V mutation (TMH9) in the presence of DOX (G830V-R_DOX-R) did not seem to significantly affect DOX binding affinity, as observed in the G185V-H_COL-H system. Although no more conclusions could be retrieved from our data, it clearly suggests that even minor effects in the ligands binding affinity may be related with the increased drug efflux. In contrast, and despite the overall increase in the contact efficiency ratio—which was expected to contribute for increasing the ACT-binding affinity—, the severe changes in ACT binding mode observed in the G830V-R_ACT-R system seemed to act as a ‘counter-weight’, having as consequence a non-significant change in the ΔGbind values. Therefore, we suggested that in this specific case, the decrease in the ACT efflux does not seem to be related with the increase binding affinity as observed with VIN, but to the ACT unfavorable binding mode at the R-site, which may reduce the ability for the G830V variant in transport ACT. This hypothesis is supported by at least one experimental study, where only a slight decrease resistance to ACT (0.29-fold) in respect to WT was observed in this P-gp variant (Loo & Clarke, Citation1994).
Overall, it becomes clear that the binding of substrates at the H and R sites has similar effects on drug efflux in both glycine variants. Additionally, these findings also corroborate our previous studies, where we suggested that G185 and G830 residues, located at opposite halves have equivalent roles in P-gp function, likely more involved in drug binding (Bonito et al., Citation2020). Finally, and although no significant changes in the DOX-binding affinity in the ΔF335-M_DOX-R system, both F978A (TMH12) and ΔF335 (TMH6) mutations tend to induce an increase on ligand-binding affinities, which seems to be independent of the DBS where the molecules interact. Therefore, we hypothesize that for certain ligands, changes in drug binding affinity within the DBS are a possible mechanism underlying the decreased drug efflux.
Authors’ Contributions
C.A.B., R.J.F. and D.J.V.A.S. conceived the experiment(s), C.A.B. conducted the experiment(s), C.A.B., R.J.F., and D.J.V.A.S. analyzed the results and wrote the paper. C.A.B., R.J.F., D.J.V.A.S., M.J.U.F., J.-P.G. and M.N.D.S.C. reviewed the manuscript. All authors agreed with the final version of the manuscript.
Supplemental Material
Download PDF (9.3 MB)Disclosure statement
Ricardo J. Ferreira is affiliated with Red Glead Discovery AB (Lund, Sweden). All other authors declare no competing interests.
Additional information
Funding
References
- Kapoor, K., Pant, S., & Tajkhorshid, E. (2021). Active participation of membrane lipids in inhibition of multidrug transporter P-glycoprotein. Chemical Science, 12(18), 6293–6306. https://doi.org/10.1039/D0SC06288J
- Verhalen, B., Dastvan, R., Thangapandian, S., Peskova, Y., Koteiche, H. A., Nakamoto, R. K., Tajkhorshid, E., & Mchaourab, H. S. (2017). Energy transduction and alternating access of the mammalian ABC transporter P-glycoprotein. Nature, 543(7647), 738–741. https://doi.org/10.1038/nature21414
- Aller, S. G., Yu, J., Ward, A., Weng, Y., Chittaboina, S., Zhuo, R., Harrell, P. M., Trinh, Y. T., Zhang, Q., Urbatsch, I. L., & Chang, G. (2009). Structure of P-glycoprotein reveals a molecular basis for poly-specific drug binding. Science (New York, N.Y.), 323(5922), 1718–1722. https://doi.org/10.1126/science.1168750
- Clouser, A. F., & Atkins, W. M. (2022). Long range communication between the drug-binding sites and nucleotide binding domains of the efflux transporter ABCB1. Biochemistry, 61(8), 730–740. https://doi.org/10.1021/acs.biochem.2c00056
- Bonito, C. A., Ferreira, R. J., Ferreira, M.-J U., Gillet, J.-P., Cordeiro, M. N. D. S., & Dos Santos, D. J. V. A. (2020). Theoretical insights on helix repacking as the origin of P-glycoprotein promiscuity. Scientific Reports, 10(1), 9823. https://doi.org/10/s41598-020-66587-5
- Abraham, M. J., Murtola, T., Schulz, R., Páll, S., Smith, J. C., Hess, B., & Lindahl, E. (2015). GROMACS: High performance molecular simulations through multi-level parallelism from laptops to supercomputers. SoftwareX, 1–2, 19–25. https://doi.org/10.1016/j.softx.2015.06.001
- Altenberg, G. A., Vanoye, C. G., Horton, J. K., & Reuss, L. (1994). Unidirectional fluxes of rhodamine 123 in multidrug-resistant cells: Evidence against direct drug extrusion from the plasma membrane. Proceedings of the National Academy of Sciences of the United States of America, 91(11), 4654–4657. https://doi.org/10.1073/pnas.91.11.4654
- Blau, C., & Grubmuller, H. (2013). g_contacts: Fast contact search in bio-molecular ensemble data. Computer Physics Communications, 184(12), 2856–2859. https://doi.org/10.1016/j.cpc.2013.07.018
- Bonvin, A. M., Mark, A. E., & van Gunsteren, W. F. (2000). The GROMOS96 benchmarks for molecular simulation. Computer Physics Communications, 128(3), 550–557. https://doi.org/10.1016/S0010-4655(99)00540-8
- Bussi, G., Donadio, D., & Parrinello, M. (2007). Canonical sampling through velocity rescaling. Journal of Chemical Physics, 126(1), 014101–014107. https://doi.org/10.1063/1.2408420
- Chandrasekhar, I., Bakowies, D., Glättli, A., Hünenberger, P., Pereira, C., & van Gunsteren, W. F. (2005). Molecular dynamics simulation of lipid bilayers with GROMOS96: Application of surface tension. Molecular Simulation , 31(8), 543–548. https://doi.org/10.1080/08927020500134243
- Chen, G., Durán, G. E., Steger, K. A., Lacayo, N. J., Jaffrézou, J. P., Dumontet, C., & Sikic, B. I. (1997). Multidrug-resistant human sarcoma cells with a mutant P-glycoprotein, altered phenotype, and resistance to cyclosporins. The Journal of Biological Chemistry, 272(9), 5974–5982. https://doi.org/10.1074/jbc.272.9.5974
- Chen, K., Lacayo, N. J., Durán, G. E., Cohen, D., & Sikic, B. I. (2000). Loss of cyclosporin and azidopine binding are associated with altered ATPase activity by a mutant P-glycoprotein with deleted phe(335). Molecular Pharmacology, 57(4), 769–777. https://doi.org/10.1124/mol.57.4.769
- Choi, K. H., Chen, C. J., Kriegler, M., & Roninson, I. B. (1988). An altered pattern of cross-resistance in multidrug-resistant human cells results from spontaneous mutations in the mdr1 (P-glycoprotein) gene. Cell, 53(4), 519–529. https://doi.org/10.1016/0092-8674(88)90568-5
- Darden, T., York, D., & Pedersen, L. (1993). Particle mesh Ewald: An N⋅log(N) method for Ewald sums in large systems. Journal of Chemical Physics. , 98(12), 10089–10092. https://doi.org/10.1063/1.464397
- Daura, X., Mark, A. E., & Van Gunsteren, W. F. (1998). Parametrization of aliphatic CHn united atoms of GROMOS96 force field. Journal of Computational Chemistry, 19(5), 535–547. https://doi.org/10.1002/(SICI)1096-987X(19980415)19:5 < 535::AID-JCC6 > 3.0.CO;2-N
- Dey, S., Ramachandra, M., Pastan, I., Gottesman, M. M., & Ambudkar, S. V. (1997). Evidence for two nonidentical drug-interaction sites in the human P-glycoprotein. Proceedings of the National Academy of Sciences of the United States of America, 94(20), 10594–10599. https://doi.org/10.1073/pnas.94.20.10594
- Essmann, U., Perera, L., Berkowitz, M. L., Darden, T., Lee, H., & Pedersen, L. G. (1995). A smooth particle mesh Ewald method. Journal of Chemical Physics, 103(19), 8577–8593. https://doi.org/10.1063/1.470117
- Ferreira, R. J., Bonito, C. A., Ferreira, M. J. U., & dos Santos, D. J. V. A. (2017). About P-glycoprotein: a new drugable domain is emerging from structural data. WIREs Computational Molecular Science, 7(5), e1316. https://doi.org/10.1002/wcms.1316
- Ferreira, R. J., Ferreira, M.-J. U., & dos Santos, D. J. V. A. (2015). Do adsorbed drugs onto P-glycoprotein influence its efflux capability? Physical Chemistry Chemical Physics : PCCP, 17(34), 22023–22034. https://doi.org/10.1039/C5CP03216D
- Ferreira, R. J., Ferreira, M.-J. U., dos., & Santos, D. J. V. A. (2013). Molecular docking characterizes substrate-binding sites and efflux modulation mechanisms within P-glycoprotein. Journal of Chemical Information and Modeling, 53(7), 1747–1760. https://doi.org/10.1021/ci400195v
- Harker, W. G., MacKintosh, F. R., & Sikic, B. I. (1983). Development and characterization of a human sarcoma cell line, MES-SA, sensitive to multiple drugs. Cancer Research, 43(10), 4943–4950.
- Harker, W. G., & Sikic, B. I. (1985). Multidrug (pleiotropic) resistance in doxorubicin-selected variants of the human sarcoma cell line MES-SA. Cancer Research, 45(9), 4091–4096.
- Hess, B. (2008). P-LINCS: A parallel linear constraint solver for molecular simulation. Journal of Chemical Theory and Computation, 4(1), 116–122. https://doi.org/10.1021/ct700200b
- Hess, B., Bekker, H., Berendsen, H. J., & Fraaije, J. G. E. M. (1997). LINCS: A linear constraint solver for molecular simulations. Journal of Computational Chemistry, 18(12), 1463–1472. https://doi.org/10.1002/(SICI)1096-987X(199709)18:12 < 1463::AID-JCC4 > 3.0.CO;2-H
- Hoover, W. (1985). Canonical dynamics: Equilibrium phase-space distributions. Physical Review. A, General Physics, 31(3), 1695–1697. https://doi.org/10.1103/PhysRevA.31.1695
- Kapoor, K., Bhatnagar, J., Chufan, E. E., & Ambudkar, S. V. (2013). Mutations in intracellular loops 1 and 3 lead to misfolding of human P-glycoprotein (ABCB1) that can be rescued by cyclosporine A, which reduces its association with chaperone Hsp70. The Journal of Biological Chemistry, 288(45), 32622–32636. https://doi.org/10.1074/jbc.M113.498980
- Kim, Y., & Chen, J. (2018). Molecular structure of human P-glycoprotein in the ATP-bound, outward-facing conformation. Science (New York, N.Y.), 359(6378), 915–919. https://doi.org/10.1126/science.aar7389
- Kumari, R., Kumar, R., & Lynn, A. (2014). g_mmpbsa —A GROMACS tool for high-throughput MM-PBSA calculations. Journal of Chemical Information and Modeling, 54(7), 1951–1962. https://doi.org/10.1021/ci500020m
- Kwan, T., & Gros, P. (1998). Mutational analysis of the P-glycoprotein first intracellular loop and flanking transmembrane domains. Biochemistry, 37(10), 3337–3350. https://doi.org/10.1021/bi972680x
- Lemkul, J. A., Allen, W. J., & Bevan, D. R. (2010). Practical considerations for building GROMOS-compatible small-molecule topologies. Journal of Chemical Information and Modeling, 50(12), 2221–2235. https://doi.org/10.1021/ci100335w
- Loo, T. W., Bartlett, M. C., & Clarke, D. M. (2013). Human P-glycoprotein contains a greasy ball-and-socket joint at the second transmission interface. The Journal of Biological Chemistry, 288(28), 20326–20333. https://doi.org/10.1074/jbc.M113.484550
- Loo, T. W., Bartlett, M. C., & Clarke, D. M. (2005). ATP hydrolysis promotes interactions between the extracellular ends of transmembrane segments 1 and 11 of human multidrug resistance P-glycoprotein. Biochemistry, 44(30), 10250–10258. https://doi.org/10.1021/bi050705j
- Loo, T. W., Bartlett, M. C., & Clarke, D. M. (2003a). Simultaneous binding of two different drugs in the binding pocket of the human multidrug resistance P-glycoprotein. The Journal of Biological Chemistry, 278(41), 39706–39710. https://doi.org/10.1074/jbc.M308559200
- Loo, T. W., Bartlett, M. C., & Clarke, D. M. (2003b). Substrate-induced conformational changes in the transmembrane segments of human P-glycoprotein. Direct evidence for the substrate-induced fit mechanism for drug binding. The Journal of Biological Chemistry, 278(16), 13603–13606. https://doi.org/10.1074/jbc.C300073200
- Loo, T. W., & Clarke, D. M. (1993). Functional consequences of phenylalanine mutations in the predicted transmembrane domain of P-glycoprotein. The Journal of Biological Chemistry, 268(27), 19965–19972.
- Loo, T. W., & Clarke, D. M. (1994). Functional consequences of glycine mutations in the predicted cytoplasmic loops of P-glycoprotein. The Journal of Biological Chemistry, 269(10), 7243–7248.
- Loo, T. W., & Clarke, D. M. (1995). Rapid purification of human P-glycoprotein mutants expressed transiently in HEK 293 cells by nickel-chelate chromatography and characterization of their drug-stimulated ATPase activities. The Journal of Biological Chemistry, 270(37), 21449–21452. https://doi.org/10.1074/jbc.270.37.21449
- Loo, T. W., & Clarke, D. M. (2005). Recent progress in understanding the mechanism of P-glycoprotein-mediated drug efflux. The Journal of Membrane Biology, 206(3), 173–185. https://doi.org/10.1007/s00232-005-0792-1
- Loo, T. W., & Clarke, D. M. (2013). A salt bridge in intracellular loop 2 is essential for folding of human P-glycoprotein. Biochemistry, 52(19), 3194–3196. https://doi.org/10.1021/bi400425k
- Loo, T. W., & Clarke, D. M. (2015). The transmission interfaces contribute asymmetrically to the assembly and activity of human P-glycoprotein. The Journal of Biological Chemistry, 290(27), 16954–16963. https://doi.org/10.1074/jbc.M115.652602
- Loo, T. W., & Clarke, D. M. (2016a). Drugs modulate interactions between the first nucleotide-binding domain and the fourth cytoplasmic loop of human P-glycoprotein. Biochemistry, 55(20), 2817–2820. https://doi.org/10.1021/acs.biochem.6b00233
- Loo, T. W., & Clarke, D. M. (2016b). P-glycoprotein ATPase activity requires lipids to activate a switch at the first transmission interface. Biochemical and Biophysical Research Communications, 472(2), 379–383. https://doi.org/10.1016/j.bbrc.2016.02.124
- Mittra, R., Pavy, M., Subramanian, N., George, A. M., O'Mara, M. L., Kerr, I. D., & Callaghan, R. (2017). Location of contact residues in pharmacologically distinct drug binding sites on P-glycoprotein. Biochemical Pharmacology, 123, 19–28. https://doi.org/10.1016/j.bcp.2016.10.002
- Miyamoto, S., & Kollman, P. A. (1992). Settle: An analytical version of the SHAKE and RATTLE algorithm for rigid water models. Journal of Computational Chemistry, 13(8), 952–962. https://doi.org/10.1002/jcc.540130805
- Nosé, S., & Klein, M. L. (1983). Constant pressure molecular dynamics for molecular systems. Molecular Physics, 50(5), 1055–1076. https://doi.org/10.1080/00268978300102851
- Omote, H., Figler, R. A., Polar, M. K., & Al-Shawi, M. K. (2004). Improved energy coupling of human P-glycoprotein by the glycine 185 to valine mutation. Biochemistry, 43(13), 3917–3928. https://doi.org/10.1021/bi035365l
- Pajeva, I. K., Hanl, M., & Wiese, M. (2013). Protein contacts and ligand binding in the inward-facing model of human P-glycoprotein. ChemMedChem, 8(5), 748–762. https://doi.org/10.1002/cmdc.201200491
- Parrinello, M., & Rahman, A. (1981). Polymorphic transitions in single crystals: A new molecular dynamics method. Journal of Applied Physics, 52(12), 7182–7190. https://doi.org/10.1063/1.328693
- Poger, D., & Mark, A. E. (2010). On the validation of molecular dynamics simulations of saturated and cis -monounsaturated phosphatidylcholine lipid bilayers: A comparison with experiment. Journal of Chemical Theory and Computation, 6(1), 325–336. https://doi.org/10.1021/ct900487a
- Poger, D., Van, Gunsteren, W. F., & Mark, A. E. (2010). A new force field for simulating phosphatidylcholine bilayers. Journal of Computational Chemistry, 31(6), 1117–1125. https://doi.org/10.1002/jcc.21396
- Prajapati, R., & Sangamwar, A. T. (2014). Translocation mechanism of P-glycoprotein and conformational changes occurring at drug-binding site: Insights from multi-targeted molecular dynamics. Biochimica et Biophysica Acta, 1838(11), 2882–2898. https://doi.org/10.1016/j.bbamem.2014.07.018
- Pronk, S., Páll, S., Schulz, R., Larsson, P., Bjelkmar, P., Apostolov, R., Shirts, M. R., Smith, J. C., Kasson, P. M., van der Spoel, D., Hess, B., & Lindahl, E. (2013). GROMACS 4.5: A high-throughput and highly parallel open source molecular simulation toolkit. Bioinformatics (Oxford, England), 29(7), 845–854. https://doi.org/10.1093/bioinformatics/btt055
- Ramachandra, M., Ambudkar, S. V., Gottesman, M. M., Pastan, I., & Hrycyna, C. A. (1996). Functional characterization of a glycine 185-to-valine substitution in human P-glycoprotein by using a vaccinia-based transient expression system. Molecular Biology of the Cell, 7(10), 1485–1498. https://doi.org/10.1091/mbc.7.10.1485
- Rao, U. S. (1995). Mutation of glycine 185 to valine alters the ATPase function of the human P-glycoprotein expressed in Sf9 cells. The Journal of Biological Chemistry, 270(12), 6686–6690.
- Safa, A. R., Stern, R. K., Choi, K., Agresti, M., Tamai, I., Mehta, N. D., & Roninson, I. B. (1990). Molecular basis of preferential resistance to colchicine in multidrug-resistant human cells conferred by Gly-185––Val-185 substitution in. Proceedings of the National Academy of Sciences of the United States of America, 87(18), 7225–7229. https://doi.org/10.1073/pnas.87.18.7225
- Schüttelkopf, A. W., & van Aalten, D. M. F. (2004). PRODRG: A tool for high-throughput crystallography of protein-ligand complexes. Acta Crystallographica. Section D, Biological Crystallography, 60(Pt 8), 1355–1363. https://doi.org/10.1107/S0907444904011679
- Scott, W. R. P., Hünenberger, P. H., Tironi, I. G., Mark, A. E., Billeter, S. R., Fennen, J., Torda, A. E., Huber, T., Krüger, P., & van Gunsteren, W. F. (1999). The GROMOS biomolecular simulation program package. The Journal of Physical Chemistry A, 103(19), 3596–3607. https://doi.org/10.1021/jp984217f
- Shapiro, A. B., & Ling, V. (1997). Positively cooperative sites for drug transport by P-glycoprotein with distinct drug specificities. European Journal of Biochemistry, 250(1), 130–137. https://doi.org/10.1111/j.1432-1033.1997.00130.x
- Tamai, I., & Safa, A. R. (1991). Azidopine noncompetitively interacts with vinblastine and cyclosporin A binding to P-glycoprotein in multidrug resistant cells. Journal of Biological Chemistry, 266(25), 16796–16800. https://doi.org/10.1016/S0021-9258(18)55371-0
- van der Spoel, D., van Maaren, P. J., Larsson, P., & Tîmneanu, N. (2006). Thermodynamics of hydrogen bonding in hydrophilic and hydrophobic media. The Journal of Physical Chemistry. B, 110(9), 4393–4398. https://doi.org/10.1021/jp0572535
- Wang, J., Wang, W., Kollman, P. A., & Case, D. A. (2006). Automatic atom type and bond type perception in molecular mechanical calculations. Journal of Molecular Graphics & Modelling, 25(2), 247–260. https://doi.org/10.1016/j.jmgm.2005.12.005
- Ward, A. B., Szewczyk, P., Grimard, V., Lee, C.-W., Martinez, L., Doshi, R., Caya, A., Villaluz, M., Pardon, E., Cregger, C., Swartz, D. J., Falson, P. G., Urbatsch, I. L., Govaerts, C., Steyaert, J., & Chang, G. (2013). Structures of P-glycoprotein reveal its conformational flexibility and an epitope on the nucleotide-binding domain. Proceedings of the National Academy of Sciences of the United States of America, 110(33), 13386–13391. https://doi.org/10.1073/pnas.1309275110
- Wise, J. G. (2012). Catalytic transitions in the human MDR1 P-glycoprotein drug binding sites. Biochemistry, 51(25), 5125–5141. https://doi.org/10.1021/bi300299z
- Juliano, R. L., & Ling, V. (1976). A surface glycoprotein modulating drug permeability in Chinese hamster ovary cell mutants. Biochimica Et Biophysica Acta, 455(1), 152–162. https://doi.org/10.1016/0005-2736(76)90160-7
- Clouser, A. F., Alam, Y. H., & Atkins, W. M. (2021). Cholesterol asymmetrically modulates the conformational ensemble of the nucleotide-binding domains of P-glycoprotein in lipid nanodiscs. Biochemistry, 60(1), 85–94. https://doi.org/10.1021/acs.biochem.0c00824
- Becker, J.-P., Depret, G., Van Bambeke, F., Tulkens, P. M., & Prévost, M. (2009). Molecular models of human P-glycoprotein in two different catalytic states. BMC Structural Biology, 9(1), 18. https://doi.org/10.1186/1472-6807-9-3
- Ferreira, R. J., Bonito, C. A., Cordeiro, M. N. D. S., Ferreira, M.-J U., & Dos Santos, D. J. V. A. (2017). Structure-function relationships in ABCG2: Insights from molecular dynamics simulations and molecular docking studies. Scientific Reports, 7(1), 15534. https://doi.org/10.1038/s41598-017-15452-z