
Abstract
IQSEC2 gene mutations are associated with epilepsy, autism, and intellectual disability. The primary function IQSEC2, mediated via its Sec 7 domain, is to act as a guanine nucleotide exchange factor for ARF6. We sought to develop a molecular model, which may explain the aberrant Sec 7 activity on ARF6 of different human IQSEC2 mutations. We integrated experimental data of IQSEC2 mutants with protein structure prediction by the RaptorX server combined with molecular modeling and molecular dynamics simulations. Normally, apocalmodulin (apoCM) binds to IQSEC2 resulting in its N-terminal fragment inhibiting access of its Sec 7 domain to ARF6. An increase in Ca2+ concentration destabilizes the interaction of IQSEC2 with apoCM and removes steric hindrance of Sec 7 binding with ARF6. Mutations at amino acid residue 350 of IQSEC2 result in loss of steric hindrance of Sec 7 binding with ARF6 leading to constitutive activation of ARF6 by Sec 7. On the other hand, a mutation at amino acid residue 359 of IQSEC2 results in constitutive hindrance of Sec 7 binding to ARF6 leading to the loss of the ability of IQSEC2 to activate ARF6. These studies provide a model for dysregulation of IQSEC2 Sec 7 activity by mutant IQSEC2 proteins.
Communicated by Ramaswamy H. Sarma
1. Introduction
The IQSEC2 gene encodes a 1,488 amino acid protein (NP_001104595.1) localized to the post synaptic density of excitatory glutamatergic neurons and inhibitory GABAergic interneurons (Sah et al., Citation2020; Murphy et al., Citation2006). Numerous mutations identified in the human X-linked IQSEC2 gene (NM_001111125.2/ENST00000396435.8) are associated with severe intellectual disability, autism, and epilepsy, which is typically drug resistant (Shoubridge et al., 2020; Zerem et al., Citation2016; Shoubridge et al., Citation2019; Mignot et al., Citation2019; Zipper et al., Citation2017; Zhang et al., Citation2015; Choi et al., Citation2020). These mutations may be generally classified as nonsense or missense mutations, the latter of which are often localized to either the Sec7 or IQ domains of IQSEC2 (). The IQ domain contains a consensus sequence for binding apocalmodulin (apoCM) (Bahler & Rhoads, Citation2002) while the Sec7 domain is known to function as a guanine nucleotide exchange factor (GEF) for the small GTPase ARF6, whose global function of regulating membrane trafficking greatly influences glutamate receptor expression at the cell surface and subsequent synapse formation (Sakagami et al., Citation2008).
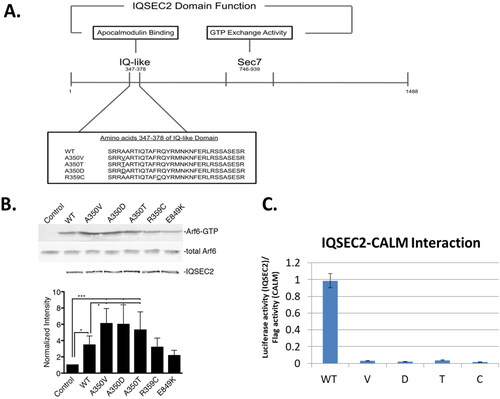
Figure 1. IQSEC2 domains and their activity. A. Schematic of the IQSEC2 gene indicating location of the IQ and Sec 7 domains. The amino acid sequence of the IQ domain mutants studied in this paper are shown. B. Basal Sec 7 activity of wild type IQSEC2 and IQ domain mutants. A representative Western blot (top) shows the amount of cellular ARF6-GTP (quantified in the bottom graph) after transfection of HEK293T cells with wild type and mutant IQSEC2 constructs as described in methods. C. Interaction strength of wild type and mutant IQSEC2 proteins with apocalmodulin, as measured in the Lumier assay (see methods and materials). Shown are the mean and SD of a minimum of 4 biological replicate wells for each pair of IQSEC2 apocalmodulin interactions. There was a greater than 50-fold difference in the interaction of wild type and all the IQ domain mutants (p < 0.000001).
Previous experimental work has led to the hypothesis that Ca2+ influx, mediated by the ligand gating of NMDA receptors, is the mechanism by which IQSEC2 GEF activity is regulated (Myers et al., Citation2012; Brown et al., Citation2016). Myers and colleagues (Myers et al., Citation2012) proposed that apoCM binds to the IQ domain of IQSEC2 under basal conditions (steady state concentration of 50–100 nM Ca2+) (Brown et al., Citation2016). During Ca2+ influx through the NMDA receptor, the Ca2+ concentration of the cell may rise transiently up to 10 μM, at which point all of the apoCM is converted to CaCM. Myers et al. provided experimental evidence that Ca2+ influx causes apoCM to detach from IQSEC2. Dissociation of apoCM from IQSEC2 is therefore thought to be necessary for the subsequent activation of ARF6.
Prior studies showed that the IQSEC2 A350V mutation does not bind apoCM (Rogers et al., Citation2019). The fact that this mutant also shows a constitutively elevated level of ARF6-GTP (Shoubridge et al., Citation2019) seems to fit the above model. However, the IQ domain R359C mutation, which also does not bind apoCM (Rogers et al., Citation2019), was not found to have constitutively elevated ARF6-GTP (Shoubridge et al., Citation2019). Rather, this mutant is unable to activate ARF6. We surmised that the reason for this discrepancy may lie in the mechanism of GDP/GTP nucleotide exchange on ARF6. Based on previous work studying other GEFs, it is thought that the binding of IQSEC2 to ARF6 increases the dissociation rate of GDP from the trimeric ARF6-GDP-IQSEC2 complex (Vetter & Wittinghofer, Citation2001; Cherfils et al., Citation1998; Renault et al., Citation2003; Jackson & Casanova, Citation2000; Goldberg, Citation1998; Béraud-Dufour et al., Citation1998; Gray et al., Citation2020; Klebe et al., Citation1995). The IQSEC2 protein inserts certain amino acids from the Sec7 domain into the nucleotide binding domain (NBD) of ARF6. This causes the affinity of ARF6 towards GDP to be reduced. The resulting binary nucleotide-free ARF6-IQSEC2 complex is stable in the absence of nucleotide. The GDP/GTP nucleotide exchange includes a series of reactions and is a reversible process (Klebe et al., Citation1995). Binding of GTP to the NBD on ARF6 is favored because of its ten-fold higher concentration in the cell. ARF6-GTP is thought to be acted on by a GTPase activating protein (GAP) and hydrolysis ensues. Through molecular modelling of multiple mutant IQSEC2 proteins we will show in this paper that an N-terminal fragment of IQSEC2 appears to be critical for determining whether the Sec7 domain has access to ARF6. This model will be used to explain the discrepant ARF6 activity of IQ domain mutants of IQSEC2.
2. Materials and methods
2.1. Experimental assessment of the interaction of IQSEC2 and its mutants with apoCM
We have previously described an assay to detect the interaction of apoCM with the IQ domain of IQSEC2 using FLAG-tagged apoCM and Renilla luciferase-tagged IQSEC2 in HEK293T cells (Rogers et al., Citation2019). The apoCM-IQSEC2 complex present in cell extracts was captured on a 96 well plate using an anti-FLAG antibody. Luciferase activity was used as an indication of the strength of the interaction. Specific mutations were introduced into the luciferase-wild type (WT) IQSEC2 vector (GenScript) ().
2.2. Generation of mutants
For the A350V (Zipper et al., Citation2017), A350T (Choi et al., Citation2020) and A350D [8] (Zhang et al., Citation2015) mutations, we changed the codon for IQSEC2 residue 350 from GCT (alanine) to GTT (valine), ACT (threonine), or GAT (aspartic acid) respectively. For the R359C (Shoubridge et al., Citation2019) mutation we changed the codon for IQSEC2 residue 359 from CGC (arginine) to TGC (cysteine).
2.3. Experimental assessment of the Sec 7 activity of human IQSEC2 carrying missense mutations in the IQ domain
We measured IQSEC2 Sec7 activity in HEK293T cells transiently transfected with IQSEC2 carrying IQ domain mutations (A350V, A350T, A350D, R359C, or E849K IQSEC2) cloned into pCAGGS (Rogers et al., Citation2019). The relative amount of ARF6 bound to GTP as compared to total ARF6 was assessed using a GGA-3 pulldown assay and western blot as previously described (Shoubridge et al., Citation2019). Bands were visualized using a LiCor Odyssey imaging system and quantified with Image Studio Lite. Each band was normalized to the untreated sham-transfected control. The data was statistically analyzed using one-way ANOVA followed by Tukey’s Multiple Comparison Test for post-hoc analysis.
2.4. Molecular modeling of apoCM-IQ domain and CaCM-IQ domain complexes
Molecular modeling of the interactions of apoCM and CaCM with the IQ domain of IQSEC2 (residues 343–370 of IQSEC2) were based on the crystal structures of apoCM and CaCM with the IQ domains present in myosin and the CaV calcium channel respectively. IQ domains are alpha helical structures. The crystal structure (2ix7.pdb) of the complex between apoCM and myosin V was used as a structural template for the generation of the complex between apoCM and the IQ domain of IQSEC2 (residues 343–370). For this purpose, the 28 amino acid residues of the Myosin V IQ domain alpha-helix starting from G791 were exchanged by in silico mutations via YASARA Structure software with the IQ domain alpha helical region of IQSEC2 corresponding to the sequence GSFLSRRAARTIQTAFRQYRMNKNFERL. The initial 3D structure of the complex of CaCM-with the IQ domain of IQSEC2 was generated by a similar process using the crystal structure of CaCM complexed with the alpha helical IQ domain of the CaV calcium ion channel (3bxl.pdb) as a structural template. The 17 amino acid residues 1819–1835 of the IQ domain of CaV were exchanged by in silico mutations with corresponding residues Gly343-Arg359 of the IQ fragment of IQSEC2. The remaining residues of the IQSEC2 IQ domain (Gln360-Leu370) were added to this structure maintaining the alpha-helical secondary structure of the IQ domain via YASARA Structure software (Krieger & Vriend, Citation2014; Krieger & Vriend, Citation2015).
2.5. Molecular modeling of the IQSEC2 Sec7 domain - ARF6 complex
The crystal structure of complex of the Sec7 fragment of IQSEC2 with ARF1 (6FAE.pdb) was used as a prototype for the generation of the IQSEC2 Sec 7-ARF6 complex by molecular modeling. The Sec7 (amino acid residues 749–1094 of IQSEC2) fragment was separated from ARF1 and the undefined loops of Sec 7 corresponding to IQSEC2 amino acid residues 1011–1016 and 1050–1054 were added to it using YASARA Structure software (Krieger & Vriend, Citation2014; Krieger & Vriend, Citation2015). The ARF6 3D structure was obtained from the 2A5D.1.A.pdb file. The superimposition of ARF6 (14–171 residues) on ARF1 in the previously described Sec7-ARF1 complex demonstrated that ARF1 and ARF6 have very close 3D structures, RMSD = 0.96 Å (Figure S1). Furthermore, as will be discussed in section 3.4 the three amino acids that dominate the stability of the ARF6-Sec7 complex are identical in ARF1 and ARF6.
2.6. Molecular dynamics relaxation to equilibrium states of the apoCM-IQ domain complex, the CaCM-IQ domain complex, the Sec7 domain and the Sec7 domain-ARF6 complex
We performed conventional molecular dynamics (cMD) simulations using AMBER20 software (Case et al., Citation2020) to assess the equilibrium stability of complexes of the IQ domain fragment of IQSEC2 with apoCM and CaCM, which had previously been generated by molecular modeling (presented above in section 2.4). The simulations were conducted using the ff14SB forcefield (Maier et al., Citation2015) in a periodic simulation cell with explicit water molecules in a tip3p model with counter ions. After preparing the input topology and structural files, the following steps were done before performing the production molecular dynamic simulations: (1) minimizing only the water, restraining the protein (20000 cycles); (2) short simulation to let water move (NPT, 310K), restraining the protein; (3) total minimization of water and protein (20000 cycles); (4) molecular dynamics of 1.4 ns to heat the system, restraining the protein (NVT, from 0 to 310K); (5) relaxing the system, restraining the protein heavy atoms (NPT, 310K, 1 ns); and (6) relaxing the system (NPT, 310K, 5 ns). In the equilibration stage, a temperature of 310 K and stable density were reached. For the actual simulations assessing complex interactions on the final stage (7) we ran production molecular dynamic simulations at 310K with a Langevin thermostat and NPT ensemble using a pressure of 1 atm and a SHAKE constraint of 2 fs. The production MD run time was varied for each complex depending on its RMSD convergence.
In order to determine the equilibrium stability of the Arf6-Sec7 complex, which has high conformational flexibility, we have used an accelerated molecular dynamic (aMD) (Hamelberg et al., Citation2004; Gedeon et al., Citation2015) simulation protocol allowing enhanced conformational sampling. The latter utilizes a bias potential function that raises the valleys’ energy levels and thus accelerates the transition between different potential energy minima. Besides the above mentioned algorithmic specificity, the aMD simulation protocol contains the same seven steps as cMD as described above controlled by the same thermodynamic parameters and force field. However, unlike cMD, the simulation periodic cell for aMD, in addition to explicit water molecules, was filled with Na + and Cl- ions in physiological concentrations. We have applied the aMD protocol for the simulation of the free Sec7 domain in order to keep the same computational approach as for the Sec7 domain-ARF6 complex.
For all complexes, the production molecular dynamics simulation was a set of sequential 100 ns steps. Every molecular dynamics step was restarted from the previous one, applying a random seeds generator. The estimated values of the standard deviation (STDEV) of RMSD fluctuations from the average value on the final equilibrated fragment of the production MD trajectory were smaller than 0.4 Å for all simulated structures (Figures S2–S4).
2.7. Conformational cluster analysis
The conformational cluster analysis of MD snapshots collected from the equilibrated fragment of production MD trajectory was conducted by CPPTRAJ software (Roe & Cheatham, Citation2013, Shao et al., Citation2007) implemented in the AMBER molecular modeling package. We used the DBSCAN (density based) clustering algorithm. The clustering process was controlled by the values for the minimum number of points to form a cluster (minpoints = 25) and the distance cutoff for forming clusters (epsilon). The latter parameter was fitted during repeated running until only one cluster centroid fraction dominated the snapshots population. Usually, the epsilon varied between a 1.5 A to 2.5 A interval. The protein 3D structure of the dominating cluster centroid was used for further structural analysis and graphical presentations. Utilizing CPPTRAJ (Roe & Cheatham, Citation2013, Shao et al., Citation2007) software implemented in AMBER20 (Case et al., Citation2020), we analyzed the RMSD convergence of all production molecular dynamic trajectories (Figures S2–S4). The conformational cluster analysis was performed as post processing on the equilibrated final fragment of the MD production trajectories to identify 3D structures of all MD simulated here, which include proteins corresponding to cluster centroids: apoCM-IQ and CaCM-IQ; Sec7 and the ARF6-Sec7 complex (Figures S5 and S6, respectively).
2.8. Calculation of binding energies of apoCM-IQwildtype and CaCM-IQwildtype complexes
Using AMBER20 software, the MMPBSA.py method (Miller et al., Citation2012) was applied for the calculation of the binding free energies of the wild type IQ domain to apoCM and CaCM in the Poisson-Boltzmann solvation model (Honig & Nicholls, Citation1995). The values of the free binding energies of apoCM and CaCM with the wild type IQ domain were calculated using the previously generated molecular dynamic trajectories. The molecular dynamic trajectory fragment of 2 ns containing 200 frames centered around the corresponding conformational cluster centroid was used in apoCM-IQ and CaCM-IQ complexes for the calculation of the average IQ free binding energies with apoCM or CaCM. The entropy calculation performed by normal mode analysis was ignored as it was computationally too expensive and is a potential source of uncertainty and a limitation of the results.
2.9. Generation of molecular models for wild type IQSEC2 (residues 1–1094) and A350V, A350D, A350T, R359C IQSEC2 mutants
We used the deep convolutional residual neural networks (ResNet) method for predicting protein 3D structure implemented in the RaptorX (Xu et al., Citation2021) server for the generation of a model of wild type IQSEC2 residues 1–760 (the server allows for no more than 1000 amino acids) and its IQ domain mutants. A fragment representing residues 749–1094 residues, containing the Sec7 domain of wild type IQSEC2, was generated and relaxed by molecular dynamics as described in sections 2.5 and 2.6. In order to construct a model for residues 1–1094 for wild type IQSEC2 and A350V, A350D, A350T and R359C IQSEC2 mutants we combined the previously generated 1–760 and 749–1094 components into one molecule. The YASARA Structure (Krieger & Vriend, Citation2014; Krieger & Vriend, Citation2015) software was used to superimpose these two fragments on residues 753–760 of a common alpha-helical fragment. Geometries of the generated wild type IQSEC2 and mutant IQSEC2 proteins (residues 1–1094) were optimized using the ff14SB force field (Maier et al., Citation2015) by YASARA Structure software in periodic simulation cells filled with explicit water molecules and Na + and Cl- ions in physiological concentration. We assessed the quality of the IQSEC2 models using the ProSA server (Wiederstein & Sippl, Citation2007) and Ramachandran plots generated by Stride server (Frishman & Argos, Citation1995), Figures S7–S11. These methods are primarily based on statistical fitting of structural parameters with experimentally well resolved 3D structures of globular proteins collected in the Protein Data Bank, so their application to verification of models of intrinsically disordered proteins such as IQSEC2 may be not optimal.
2.10. Construction of apoCM-IQ complexes with residues 1–1094 of wild type IQSEC2 and its IQ domain mutants
The complex of apoCM with the wild type IQ domain fragment was incorporated into residues 1–1094 of IQSEC2 by the superimposition of amino acid residues 348–360 on both the IQ domain of the IQSEC2 and the IQ domain of the molecular by dynamics simulated apoCM-IQ complex (section 2.4). To remove the inter-residual clashes in the formed complex the Φ torsional angle between glycine residues 342 and 343, serving as a hinge, was changed from 72° in free wild type IQSEC2 to 177.5° when apoCM was bound to wild type IQSEC2 (Figure S12). The geometry of the apoCM-IQSEC2 wild type complex was optimized by YASARA Structure under the AMBER ff14SB force field (Maier et al., Citation2015) in a periodic cell filled with explicit water molecules and physiological concentration of Na + and Cl- ions. Superimposition of the apoCM-IQ wild type complex (Section 2.4) on the IQ domain of IQSEC2 mutants (Section 2.9 and ) led to unremovable inter-residual clashes, Figures S14–S17. To resolve this problem, we docked apoCM to the IQ domain of the IQSEC2 mutants using the protein-protein docking method implemented in the HADDOCK web server (Honorato et al., Citation2021; van Zundert et al., Citation2016). The docking was conducted using the default parameters of the server (the default parameters of the HADDOCK 2.4 server are provided as a SI file). The input information requested by the HADDOCK server contains 3D structures of receptor (IQSEC2 mutant) and ligand (apoCM) as well as the sequence numbers of the amino acid residues forming binding interactions on the receptor-ligand interface surface. The latter were identified by YASARA Structure for the apoCM-IQSEC2 wild type complex previously generated and optimized by YASARA Structure, as described above. The geometries of all apoCM-IQ mutants complexes were optimized by YASARA Structure under the AMBER ff14SB force field (Maier et al., Citation2015) in a periodic cell filled with explicit water molecules and physiological concentration of Na + and Cl- ions. Finally, the apoCM-IQ binding free energies ΔG were calculated for all complexes by the FoldX web server (Schymkowitz et al., Citation2005). In order to calculate the free energy of binding of a complex AB, FoldX computes the Gibbs energies of the complex (ΔGAB) and of the two molecules A and B alone.The interaction energy is then given by: ΔGbinding = ΔGAB - (ΔGA + ΔGB). FoldX interaction energies are scaled with the solvent accessibility of the atoms involved in the interaction. In section 3.3 we will present the structures of wild type IQSEC2 and its mutants with apoCM bound to the IQ domain and their binding energies.
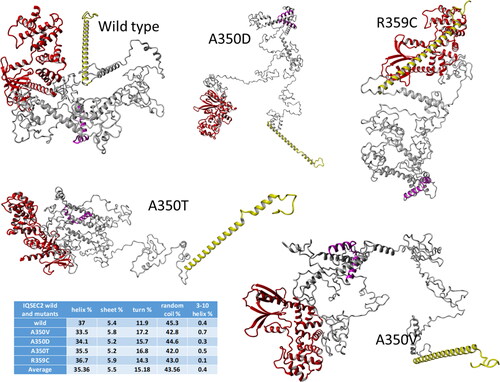
Figure 2. Molecular modeling of IQSEC2 domains. The domain structure generated by molecular modeling of fragments (1–1094 residues) of wild type IQSEC2 and its mutants demonstrates that these are Intrinsically Disordered Proteins (IDP). Colors: Sec 7 domain – red, N-terminal fragment – yellow, IQ domain – magenta, the rest of the residues are grey. The table summarizes the fraction of various secondary structures in the different proteins.
3. Results and discussion
3.1. ApoCM binding activity and ARF6-GTP levels in cells expressing mutations of IQSEC2
As mentioned above, studies in this laboratory have previously demonstrated that the A350V IQ domain mutation results in a constitutive rise in IQSEC2 Sec7 activity (Rogers et al., Citation2019), whereas the R359C IQ domain mutation results in a loss of IQSEC2 Sec 7 activity (Shoubridge et al., Citation2019). This analysis has now been expanded to include two additional human disease-causing IQ domain mutations at aa residue 350 (A350D and A350T) (Zhang et al., Citation2015; Choi et al., Citation2020). Sec7 activity of wild type and all IQ mutants was assessed by determining the relative amount of ARF6-GTP found in HEK293T cells following transient transfection with IQSEC2, as described in the Materials and Methods section. We found that all three human mutations at residue 350 resulted in a significant increase in ARF6-GTP, compared to the wild type, indicating that the Sec7 domain of these A350 mutants was constitutively activated. On the other hand, the GEF catalytic activity of the Sec7 domain for the R359C mutation was not increased relative to that of the wild type IQSEC2 (). E849K is a mutation in the Sec7 region of IQSEC2 which destroys the Sec7 activity of IQSEC2 and therefore serves as a negative control in this experiment (Shoubridge et al., Citation2010). None of the IQ domain mutant proteins was able to interact with apocalmodulin ().
3.2. Prediction of the 3D structures of IQSEC2 wild type and its mutants by molecular modeling
There is no experimentally resolved 3D structure of IQSEC2. The only available detailed information can be obtained from the X-ray crystal structure of a fragment of IQSEC2 corresponding to the Sec7-ARF1 complex (6FAE.pdb) which were used to model the seperated Sec 7 and ARF6 regions and the Sec7-ARF6 complex (sections 2.5 and 2.6). In addition, the IQ domain (residues 347–378) was expected to have an alpha helical secondary structure according to UniProtKB (Q5JU85). The remaining parts of IQSEC2 appear to be disordered as illustrated in Figure S13 representing the functional disorder profile generated for human IQSEC2 by the D2P2 platform [35] (Oates et al., Citation2013) (https://d2p2.pro). This profile shows that a very significant portion of IQSEC2, e.g. most of the residues in the N-terminal (1–740) and C-terminal fragments (1076–1488), i.e. 77.5% of the protein are expected to be disordered. Therefore, based on these high levels of disorder content, and in line with the accepted categorization for disorder in proteins as highly ordered (PPDR < 10%), moderately disordered (10%≤PPDR < 30%) and highly disordered (PPDR ≥ 30%) (Rajagopalan et al., Citation2011), human IQSEC2 can be classified as a highly disordered protein. Homology modeling servers failed to generate 3D models of the full-length IQSEC2, consistent with the experimental data and the disorder prediction results. Thus, we used the deep convolutional residual neural networks (ResNet) method for predicting protein 3D structure implemented in the RaptorX server (Xu et al., Citation2021) for the generation of the 1–760 amino acid residue (the server allows for no more than 1000 amino acids) model of wild type IQSEC2 and its IQ domain mutants. The Sec7 domain of IQSEC2 corresponding to IQSEC2 amino acid residues 749–1094, was generated and relaxed by molecular dynamics as described in sections 2.5 and 2.6. Finally, in order to construct models for amino acid residues 1–1094 of wild type IQSEC2 as well as for A350V, A350D, A350T and R359C IQSEC2 mutants we have combined on amino acid residues 753–760 of a common alpha helical fragment the previously generated 1–760 and 749–1094 components into one molecule. The YASARA Structure (Krieger & Vriend, Citation2014; Krieger & Vriend, Citation2015) software was used to superimpose them on amino acid residues 753–760 of a common alpha helical fragment as described insection 2.9. The structures of the generated wild type and mutant IQSEC2 were analyzed by YASARA Structure software and classified as Intrinsically Disordered Proteins (IDP). The highly disordered C-terminal end region of IQSEC2 containing amino acid residues 1095–1488 (Figure S13) is not included in the molecular models analyzed here. Geometries of the generated wild type and mutant IQSEC2 proteins (1–1094 residues) were fully optimized using a ff14SB force field (Maier et al., Citation2015) by YASARA Structure software in periodic simulation cells filled with explicit water molecules and Na + and Cl- ions in physiological concentration (section 2.9) and are presented in .
3.3. A model for the regulation of IQSEC2 Sec7 activity by Ca2+ via integration of molecular modeling and experimental data
Previous experimental work has shown that apoCM can bind to the IQ domain of wild type IQSEC2 and that increasing the Ca2+ concentration results in an increase in Sec7 activity (Myers et al., Citation2012; Brown et al., Citation2016, Rogers et al., Citation2019). This has led to the hypothesis that apoCM is inhibiting the Sec7 domain. Myers et al. (Myers et al., Citation2012) first demonstrated that the binding of Ca2+ to apoCM results in the dissociation of apoCM from the IQ domain of IQSEC2. Combining molecular modeling with molecular dynamics simulations (sections 2.4 and 2.6) we have generated complexes formed by CaCM and apoCM with a helical fragment of the IQ domain (Figure S5). The calculated relative thermodynamic stabilities of apoCM and CaCM complexes by the MMPBSA.py method (Miller et al., Citation2012) are presented in Tables S1 and S2. The Coulomb charges of the IQ domain, apoCM, and CaCM are +6, −24, and −16, respectively. The four Ca2+ chelated by CM dramatically reduce the ability of CaCM to bind to the IQ domain. The MMPBSA.py method usually overestimates the absolute values of binding energies, so only the trend in the relative binding energy values should serve as the basis for comparison between the calculated and experimental determined interactions. The calculated difference in binding energies between CaCM-IQ and apoCM-IQ complexes is (-36.3 kcal/mol)—(-88.6 kcal/mol) = 52.3 kcal/mol, demonstrating that the apoCM-IQ complex is markedly more stable than the CaCM-IQ complex. Therefore, the CaCM-IQ complex concentration should be negligible when intracellular Ca2+ is increased. This conclusion perfectly corresponds with experimental observations (Myers et al., Citation2012; Brown et al., Citation2016, Rogers et al., Citation2019).
How does the Ca2+-mediated dissociation of CM from the IQ domain increase Sec7 activity? Complexation of apoCM with the IQ domain fragment of the model of wild type IQSEC2 was constructed by the superimposition of amino acid residues 348–360 of both the IQ domain of IQSEC2 (section 2.10) and the IQ domain of the modeled apoCM-IQ domain complex (sections 2.4 and 2.6). As discussed in Section 2.10 when generating this complex in order to remove inter-residual clashes between residues the Φ torsional angle between glycine residues 342 and 343, serving as a hinge (Figure S12), was changed leading to a large-scale conformational movement of the IQSEC2 N-terminal fragment. The principal difference between 3D structures of free wild type IQSEC2 and its complex with apoCM bound to its IQ domain may be seen in and . When apoCM is bound to the IQ domain, the N-terminal segment of wild type IQSEC2 moves to a position that hinders the access of the Sec7 domain to ARF6 ().
Figure 3. 3D structures in two projections of apoCM-IQ complexes for wild type and mutant IQSEC2 proteins demonstrating that the binding of apoCM to wild type IQSEC2 leads to an interaction of the N terminal domain with the Sec7 domain thereby inhibiting the accessibility to the latter for ARF6. Colors: Sec 7 domain – red, N-terminal fragment – yellow, IQ domain – magenta, apoCM – green, the rest of the residues are grey. The apoCM-IQ complexes are displayed in molecular surface style. All other fragments of IQSEC2 are presented in ribbon style. The table summarizes calculated binding energies of apoCM-IQ complexes.
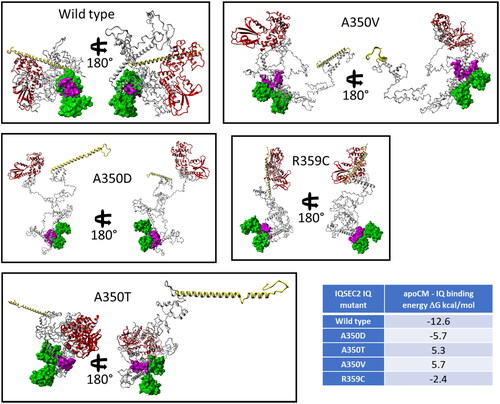
Attempts to form a stable complex between apoCM and IQ domain IQSEC2 mutants constructed by the method used for the wild type IQ domain was not possible due to multiple nonremovable conformational clashes between the protein backbones of apoCM and parts of the mutant IQSEC2 proteins (section 2.10 and Figures S14–S17). To resolve this problem, we docked apoCM to the IQ domain of the IQSEC2 mutants using the protein-protein docking method used in the HADDOCK web server (Honorato et al., Citation2021; van Zundert et al., Citation2016). The geometry of all apoCM-IQ complexes including that for wild type IQSEC2 and the IQ domain mutants (the latter after the docking) were optimized by YASARA Structure under the AMBER ff14SB force field (Maier et al., Citation2015) in a periodic cell filled with explicit water molecules and physiological concentration of Na + and Cl- ions as described in section 2.10. presents in two projections the 3D structures of wild type IQSEC2 and IQ domain mutants with apoCM bound to the IQ domain. There is a principal structural difference between the wild type and all of the mutant apoCM-IQ complexes. In the former case the IQ is squeezed between ‘Pac-Man’s jaws’ (Pac-Man,2022) of apoCM. In contrast, the mutant IQ domain is positioned behind the Pac-Man’s jaws. Consequently, the apoCM-IQ binding energies, calculated by the FoldX web server (Schymkowitz et al., Citation2005), predicted the formation of a thermodynamically stable wild type IQ complex. In contrast, the apoCM-IQ complex for the IQ mutants were predicted to have negligible stability or to be impossible to form (Table in ). These theoretical predictions are confirmed by our biological experiments (). shows a schematic representation of the mechanism regulating the ability of the Sec7 domain of wild type IQSEC2 and IQ domain mutants to bind to ARF6.
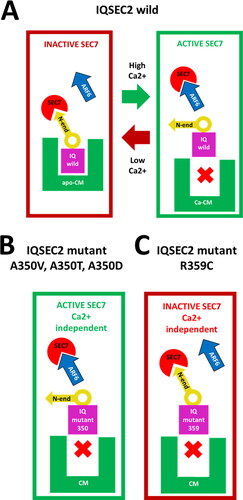
Figure 4. Schematic representation of the regulation of the interaction of the Sec7 domain with ARF6. A. Wild type IQSEC2. Under low Ca2+ concentration conditions, apoCM bound to the IQ domain initiates movement of a large-scale conformational flap of IQSEC2 amino acid residues 1-342 on a hinge formed by glycine 342 and glycine 343. The movement of this flap causes the N-terminal fragment of IQSEC2 (residues 1-71) to sterically inhibit access of ARF6 to Sec7 to form a catalytically competent ARF6-Sec7 complex. High Ca2+ concentration results in the dissociation of apoCM from the IQ domain which causes a change in the position of the N-terminal helical fragment of IQSEC2 so that it no longer inhibits access of the Sec7 region to ARF6. B. A350 mutant IQSEC2. Calmodulin is unable to bind to A350 mutant IQ domains under any Ca2+ concentration conditions, so access of ARF6 to the Sec7 domain is always permitted. C. R359C mutant IQSEC2. Calmodulin is unable to bind to the R359C IQ domain mutant under any Ca2+ concentration. The intrinsic structural feature of this mutant is that access of ARF6 to the Sec7 domain to form catalytically competent ARF6-Sec7 complex is blocked by the N-terminal helical fragment of IQSEC2 regardless of the Ca2+ concentration.
3.4. The effect of the 3D structure of the binary GTPase-GEF complex on the nucleotide exchange process
Numerous experimental studies have led to the conclusion that nucleotide exchange factors (GEFs) increase the dissociation rate of GDP from trimeric GTPase-GDP-GEF complexes, forming the resulting stable binary nucleotide-free GTPase-GEF complex (Vetter & Wittinghofer, Citation2001; Cherfils et al., Citation1998; Renault et al., Citation2003; Jackson & Casanova, Citation2000; Goldberg, Citation1998; Béraud-Dufour et al., Citation1998; Gray et al., Citation2020; Klebe et al., Citation1995). The GTPase-GEF complex formation is a key stage in the maintenance of the GDP/GTP nucleotide exchange process. Indeed, the next stage in the cycle is rebinding of GTP nucleotide to the nucleotide binding domain, which requires the appropriate geometrical structure of the GTPase-GEF complex and an increase in its concentration (dependent on its thermodynamic stability) in the cell. If at least one of the mentioned factors is not satisfied the cyclic process of nucleotide exchange will be interrupted.
In this section we analyze the 3D structure and stability of the binary nucleotide-free ARF6-Sec7 (IQSEC2) complex as a function of the IQ domain sequence and apoCM binding. Paying attention to the high conformational flexibility of the ARF6-Sec7 complex (Vetter & Wittinghofer, Citation2001; Cherfils et al., Citation1998; Renault et al., Citation2003; Jackson & Casanova, Citation2000; Goldberg, Citation1998; Béraud-Dufour et al., Citation1998; Gray et al., Citation2020; Klebe et al., Citation1995), we have applied an accelerated molecular dynamic (aMD) (Hamelberg et al., Citation2004; Gedeon et al., Citation2015) simulation protocol allowing enhanced conformational sampling (section 2.6). We have demonstrated in sections 3.2 and 3.3 that for wild type IQSEC2 (in the absence of apoCM) and for the A350 mutant IQSEC2 proteins, the Sec7 domain can freely bind to ARF6 ( and ). Therefore, the aMD simulated ARF6 (residues 14–171)-Sec7 (residues 749–1094) complex (Figure S6B) may serve as a unified model for the analysis of its structural features common for wild type IQSEC2 without apoCM bound and the A350 IQSEC2 mutants. To construct the complex of ARF6 (residues 14–171) with wild type IQSEC2 (residues 749–1094) and its A350 mutants we superposed their Sec7 domain by means of YASARA Structure software with the corresponding Sec7 fragment of the MD equilibrated ARF6-Sec7 complex (section 2.6). Subsequently, the Sec7 fragment of the ARF6-Sec7 complex was deleted. Finally, the geometries of the generated ARF6-IQSEC2 complexes were fully optimized under a ff14SB force field (Maier et al., Citation2015) by YASARA Structure software in periodic simulation cells filled with explicit water molecules and Na + and Cl- ions in physiological concentrations.
Analysis by YASARA Structure of intermolecular residue binding interactions on the interface surface between ARF6 and the Sec7 domain of wild type IQSEC2 without ApoCM bound () and the A350D IQSEC2 mutant (Figure S18) identified three oppositely charged amino acid pairs that dominate the stability of the complexes. They are K69(L)-D879(R), R15(L)-D894(R) and K26(L)-E849(R). There is an additional fourth pair D22(L)-K852(R) responsible for the greater stabilization of ARF6 complexes with the A350T and A350V mutants, (Figures S19 and S20, respectively). L and R refer here to ARF6 (Ligand) and Sec7 (Receptor) respectively. The K69(L)-D879(R) pair forms a strong ionic hydrogen bond between positively and negatively charged residues. The charged functional groups of the remaining pairs are too distant for hydrogen-bond formation; nevertheless, their electrostatic binding interaction contributes to the complex stability.
Figure 5. 3D structure of ARF6-Sec 7 complexes (see details in Section 3.4) generated by Raptor X or Alpha Fold. Complexes are presented in two projections. Free energies of binding ΔG are calculated by the FoldX server. The three amino acid pairs that dominate the stability of the ARF6-IQSEC2 wild type complex are presented in the right side of figure. Interactions between corresponding functional groups of amino acid pairs are connected by arrows and labeled by distance values in Å. Color scheme: ARF6 – blue, Sec7– red, the remainder of IQSEC2 is grey. L and R refer to ARF6 (Ligand) and Sec 7 (Receptor) respectively. Residues dominating stability of the ARF6-Sec7 complexes are element colored. A. 3D structure predicted by the Raptor X server of ARF6 (molecule L) complexed with wild type IQSEC2 (molecule R). B. 3D structure of ARF6 (molecule L)-IQSEC2 (molecule R) wild type complex where the IQSEC2 3D structure (amino acid residues 1-1094) was previously predicted by the AlphaFold server. There are numerous conformational clashes between ARF6 and structural fragments of IQSEC2 which do not permit the Sec7 domain to bind with ARF6 when using the structure of IQSEC2 generated by the AlphaFold server.
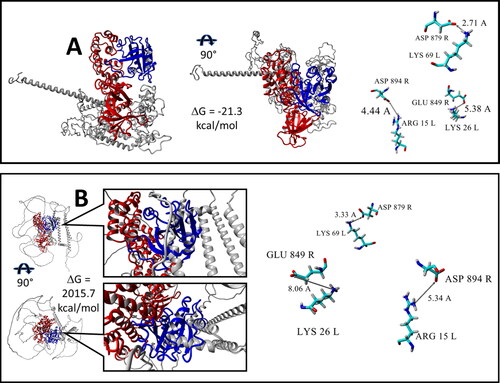
We have also examined the most popular and widely used method to model protein structure, AlphaFold,(Jumper et al., Citation2021) to predict the 3D structure of wild type IQSEC2. AlphaFold is currently available in the UniProt database, allowing us to acquire from this source the AlphaFold predicted 3D structure of wild type full length IQSEC2 (1488 amino acid residues). As we are only modeling in this study the N-terminal 1094 amino acids of IQSEC2 we deleted the C terminal end of IQSEC2 obtained by AlphaFold. We then constructed, by the protocol described above for IQSEC2 generated by RaptorX, the complex of ARF6 with AlphaFold generated IQSEC2 (). There were numerous conformational clashes between ARF6 and structural fragments of IQSEC2 when using the AlphaFold model which could not be removed by any geometry optimization procedure. As calculated by FoldX there was an enormously high positive value of the binding free energy ΔG for the AlphaFold generated complex clearly indicating that it cannot be formed. Therefore, the AlphaFold algorithm does not appear to be relevant for the 3D structure prediction of the intrinsically disordered protein IQSEC2. This lack of validity of a structure produced by AlphaFold is in agreement with the previously described poor ability of AlphaFold to predict the 3D structure of other intrinsically disordered proteins (Azzaz et al., Citation2022; Ruff & Pappu, Citation2021).
It should be noted that the functional role of the Sec7 domain of the GEF in the ARF-GDP-GEF complex is to trigger sterically and electrostatically the expulsion of the GDP nucleotide by insertion of Sec7 residues into the nucleotide binding site of ARF6 (Goldberg et al., 1998). Specifically, it is the so called ‘glutamic finger’—a specific Glu residue of the Sec7 domain corresponding to E156 for ARNO (Cherfils et al., Citation1998; Renault et al., Citation2003; Béraud-Dufour et al., Citation1998) and E97 for Gea2 (Goldberg et al., 1998). The analogous role of the glutamic finger is played by E849 of the Sec7 domain of IQSEC2 (Gray et al., Citation2020). In contrast, as demonstrated above, in the absence of nucleotide, the Sec7 domain of IQSEC2 stabilizes the nucleotide-free ARF6-Sec7 complex (binding free energy ΔG = −17.4 kcal/mol). The E849 residue of the Sec7 domain also contributes to the complex stabilization by an electrostatic interaction with K26 of ARF6.
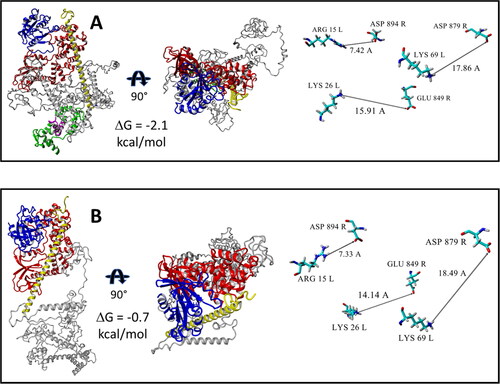
Protein-protein docking of previously MD equilibrated structures of ARF6 to wild type IQSEC2 and the R359C mutant, where the former was bound to apoCM, was assessed by the HADDOCK server (Honorato et al., Citation2021; van Zundert et al., Citation2016) using the default parameters of the server (the default parameters of the HADDOCK 2.4 server are provided as a SI file). The geometries of the generated complexes were optimized by YASARA software under the AMBER ff14SB force field (Maier et al., Citation2015) in a periodic cell filled with explicit water molecules and physiological concentration of Na + and Cl- ions. The N-terminal fragment (1–71 residues) of IQSEC2 interferes with optimal binding of ARF6 to the Sec7 domain for both apoCM bound wild type IQSEC2 as well as for mutant R359C IQSEC2 (sections 1 and 2, and ). As a result, both ARF6-Sec7 complexes are unstable; the corresponding binding free energies ΔG are −2.1 kcal/mol and −0.7 kcal/mol respectively. In addition, the geometries of both ARF6-Sec 7 complexes are distorted. Indeed, the amino acid residues that were found to stabilize the interaction between ARF6 and Sec7 when apoCM was not bound to wild type IQSEC2 () are extremely distant from each other () when apoCM is bound to wild type IQSEC2 or in the R359C IQSEC2 mutant. As a result, GTP cannot be successfully bound to the corrupted ARF6-Sec7 structure, and the cyclic nucleotide exchange process is abolished.
Figure 6. 3D structures demonstrating destabilization of the interaction between ARF6 and Sec7 domain when wild type IQSEC2 is bound to apoCM or for R359C IQSEC2 (see details in Section 3.4). Complexes are presented in two projections. Binding free energies ΔG were calculated by FoldX server. The amino acid residues which stabilize the ARF6-Sec7 complex when wild type IQSEC2 is not bound to apoCM (as demonstrated in ) in contrast as shown here are very distant from one another both when wild type IQSEC2 is bound to apoCM and for the R359C IQSEC2 mutant. Interactions between functional groups of Sec7 and ARF6 domains in the demonstrated complexes are connected by arrows and labeled by distance values in Å. Color scheme: ARF6 – blue, Sec7 – red, N-terminal fragment – yellow, the rest of the fragments of IQSEC2 are grey. A. 3D structure of the ARF6 (mol L)-IQSEC2 (mol R) wild type complex, where the IQ domain (magenta) is bound to apoCM (green). B. 3D structure of ARF6 (mol L)-IQSEC2 (mol R) R359C mutant complex.
4. Limitations of this study
4.1. The hinge region
We have observed clashes between residues of apoCM and the wild type IQ domain of IQSEC2 after their superimposition (see section 3.2). To remove the bumps, we chose to focus on a specific gly-gly region as a crucial hinge due its close proximity (residues 342–343) to the IQ region (residues 348–378). As discussed above the IQ region serves as the binding site for the apoCM protein resulting in the allosteric modulation of Sec7 activity. A hinge located adjacent to this binding site is a reasonable hypothesis. This may be experimentally tested in future studies by mutating this hinge and testing whether this would result in the expected loss of the ability of the IQ region to allosterically regulate the Sec7 activity of IQSEC2.
4.2. The R359C conundrum
We have demonstrated major structural differences in the orientation of the N-terminal region of IQSEC2 relative to the IQSEC2 Sec7 domain for IQSEC2 mutations at amino acid residues 350 and 359 both of which cannot bind apoCM. These structural differences are consistent with experimentally acquired data. However, we presently do not have an explanation for these differences. Perhaps this may be due to differences in proximity to the putative hinge region (342–343). Future experiments are needed to clarify this unusual finding, such as mutagenesis studies or possibly acquiring crystallographic data.
5. Conclusions and implications of this study
Mutations in the IQSEC2 gene are associated with epilepsy, autism, and intellectual disability. The intention of this work was to present a model at a molecular level for how mutations in the IQ domain of IQSEC2 may cause disease. The IQ domain of IQSEC2 is known to bind apoCM and results in inhibition of its GEF activity. Ca2+ influx relieves this inhibition and leads to activation of ARF6. Moreover, it was found that mutations in the IQ domain can lead to permanently elevated GEF activity in A350 mutants, or to its GEF catalytic incompetence as in the case of the R359C mutant. The structural basis for this discrepancy had not been described to date. Applying various molecular modeling techniques including molecular dynamics simulations, we have found that one explanation for this phenomenon lies in the mechanism of GDP/GTP nucleotide exchange stimulated by the GEF catalytic activity of the Sec7 domain of IQSEC2. Specifically, an N-terminal fragment of IQSEC2, whose position around a hinge region is dependent on whether apoCM is bound to the IQ domain, is responsible for determining access of the Sec7 domain to ARF6.
The understanding of the mechanism by which IQSEC2 is normally regulated may allow for discovery efforts to improve the lives of children with IQSEC2 mutations as there are currently no treatments. Specifically, work by Mehta et al., Citation2021 has demonstrated that IQSEC2 mediated disease might be rescued by replacement with a wild type copy of the IQSEC2 gene (i.e. gene therapy). The Federal Drug Administration has recently approved gene therapy for several single gene defects using adeno-associated virus (AAV). The problem is that applying AAV mediated therapy to a specific disease is restricted by the size of the gene that can be used within the carrying capacity of the AAV virus. The IQSEC2 gene is too large to fit within the space permitted by the AAV virus. We are currently working on developing an IQSEC2 minigene, based in part on the molecular model presented here, that will preserve regions (functional domains) important for the biological function of IQSEC2 and delete regions that appear to only serve a linker function between these functional domains. We have developed platforms to test the efficacy of this IQSEC2 minigene in AAV in both neuronal cultures and in mice with IQSEC2 mutations. The molecular model described here has provided a starting point from which we are making IQSEC2 minigenes but the final minigene will only be able to be validated by experimental data.
Authors’ contributions
All authors have read the manuscript and have contributed to its content. MS designed the molecular modeling and wrote and edited the manuscript. RW, DF, CZ, NL, AA and VU all contributed to the editing of the manuscript and in interpreting results of experiments and modeling. AL proposed the experiments, interpreted the data and wrote the paper.
Supplemental Material
Download Zip (8.6 MB)Disclosure statement
The authors declare that they have no conflicts of interest with the contents of this article.
Additional information
Funding
References
- Azzaz, F., Yahi, N., Chahinian, H., & Fantini, J. (2022). The epigenetic dimension of protein structure is an intrinsic weakness of the alphafold program. Biomolecules, 12(10), 527. https://doi.org/10.3390/biom12101527.
- Bahler, M., & Rhoads, A. (2002). Calmodulin signaling via the IQ motif. FEBS Letters, 513(1), 107–113. https://doi.org/10.1016/S0014-5793(01)03239-2
- Béraud-Dufour, S., Robineau, S., Chardin, P., Paris, S., Chabre, M., Cherfils, J., & Antonny, B. (1998). A glutamic finger in the guanine nucleotide exchange factor ARNO displaces Mg2+ and the beta-phosphate to destabilize GDP on ARF1. The EMBO Journal, 17(13), 3651–3659. https://doi.org/10.1093/emboj/17.13.3651
- Brown, J. C., Petersen, A., Zhong, L., Himelright, L., Murphy, J. A., Walikonis, R. S., & Gerges, N. Z. (2016). Bidirectional regulation of synaptic transmission by BRAG1/IQSEC2 and its requirement in long-term depression. Nature Communications, 7, 11080. https://doi.org/10.1038/ncomms11080
- Carrascoza, F., Zaric, S., & Silaghi-Dumitrescu, R. (2014). Computational study of protein secondary structure elements: Ramachandran plots revisited. Journal of Molecular Graphics & Modelling, 50, 125–133. https://doi.org/10.1016/j.jmgm.2014.04.001
- Case, D. A., Aktulga, H. M., Belfon, K., Ben-Shalom, I. Y., Brozell, S. R., Cerutti, D. S., I. I. I., Cheatham, T. E., Cruzeiro, V. W. D., Darden, T. A., Duke, R. E., Giambasu, G., Gilson, M. K., Gohlke, H., Goetz, A. W., Harris, R., Izadi, S., Izmailov, S. A., Jin, C., Kasavajhala, … S., Kollman, P. A. (2020). Amber 2020, University of California, San Francisco.
- Cherfils, J., Ménétrey, J., Mathieu, M., Le Bras, G., Robineau, S., Béraud-Dufour, S., Antonny, B., & Chardin, P. (1998). Structure of the Sec 7 domain of the Arf exchange factor ARNO. Nature, 392(6671), 101–105. https://doi.org/10.1038/32210
- Choi, M.-H., Yang, J. O., Min, J. S., Lee, J. J., Jun, S. Y., Lee, Y.-J., Yoon, J.-Y., Jeon, S.-J., Byeon, I., Kang, J.-W., & Kim, N.-S. (2020). A novel X-linked Lennox Gastaut syndrome and mild intellectual disability in three generations of a Korean family. Genetic Testing and Molecular Biomarkers, 24(1), 54–58. https://doi.org/10.1089/gtmb.2019.0177
- Frishman, D., & Argos, P. (1995). Knowledge-based protein secondary structure assignment. Proteins, 23(4), 566–579. https://doi.org/10.1002/prot.340230412
- Gedeon, P. C., Thomas, J. R., & Madura, J. D. (2015). Accelerated molecular dynamics and protein conformational change: A theoretical and practical guide using a membrane embedded model neurotransmitter transporter. Methods in Molecular Biology (Clifton, N.J.), 1215, 253–287. https://doi.org/10.1007/978-1-4939-1465-4_12
- Goldberg, J. (1998). Structural basis for activation of ARF GTPase: Mechanisms of guanine nucleotide exchange and GTP–myristoyl switching. Cell, 95(2), 237–248. https://doi.org/10.1016/s0092-8674(00)81754-7
- Gray, J. L., von Delft, F., & Brennan, P. E. (2020). Targeting the small GTPase superfamily through their regulatory proteins. Angewandte Chemie (International ed. in English), 59(16), 6342–6366. https://doi.org/10.1002/anie.201900585
- Hamelberg, D., Mongan, J., & McCammon, J. A. (2004). Accelerated molecular dynamics: A promising and efficient simulation method for biomolecules. The Journal of Chemical Physics, 120(24), 11919–11929. https://doi.org/10.1063/1.1755656
- Honig, B., & Nicholls, A. (1995). Classical electrostatics in biology and chemistry. Science (New York, N.Y.), 268(5214), 1144–1149. https://doi.org/10.1126/science.7761829
- Honorato, R. V., Koukos, P. I., Jimenez-Garcia, B., Tsaregorodtsev, A., Verlato, M., Giachetti, A., Rosato, A., & Bonvin, A. (2021). Structural biology in the clouds: The WeNMR-EOSC ecosystem. Frontiers in Molecular Biosciences, 8, 729513. https://doi.org/10.1126/science.7761829
- Jackson, C. L., & Casanova, J. E. (2000). Turning on ARF: The Sec 7 family of guanine-nucleotide-exchange factors. Trends in Cell Biology, 10(2), 60–67. https://doi.org/10.1016/s0962-8924(99)01699-2
- Jumper, J., Evans, R., Pritzel, A., Green, T., Figurnov, M., Ronneberger, O., Tunyasuvunakool, K., Bates, R., Žídek, A., Potapenko, A., Bridgland, A., Meyer, C., Kohl, S. A. A., Ballard, A. J., Cowie, A., Romera-Paredes, B., Nikolov, S., Jain, R., Adler, J., … Hassabis, D. (2021). Highly accurate protein structure prediction with AlphaFold. Nature, 596(7873), 583–589. https://doi.org/10.1038/s41586-021-03819-2
- Klebe, C., Prinz, H., Wittinghofer, A., & Goody, R. S. (1995). The kinetic mechanism of Ran—nucleotide exchange catalyzed by RCC1. Biochemistry, 34(39), 12543–12552. https://doi.org/10.1021/bi00039a008
- Krieger, E., & Vriend, G. (2015). New ways to boost molecular dynamics simulations. Journal of Computational Chemistry, 36(13), 996–1007. https://doi.org/10.1002/jcc.23899
- Krieger, E., & Vriend, G. (2014). YASARA view - molecular graphics for all devices - from smartphones to workstations. Bioinformatics (Oxford, England), 30(20), 2981–2982. https://doi.org/10.1093/bioinformatics/btu426
- Maier, J. A., Martinez, C., Kasavajhala, K., Wickstrom, L., Hauser, K. E., & Simmerling, C. (2015). ff14SB: Improving the accuracy of protein side chain and backbone parameters from ff99SB. Journal of Chemical Theory and Computation, 11(8), 3696–3713. https://doi.org/10.1021/acs.jctc.5b00255
- Mehta, A., Shirai, Y., Kouyama-Suzuki, E., Zhou, M., Yoshizawa, T., Yanagawa, T., Mori, T., & Tabuchi, K. (2021). IQSEC2 deficiency results in abnormal social behaviors relevant to autism by affecting functions of neural circuits in the medial prefrontal cortex. Cells, 10(10), 2724. https://doi.org/10.3390/cells10102724
- Mignot, C., McMahon, A. C., Bar, C., Campeau, P. M., Davidson, C., Buratti, J., Nava, C., Jacquemont, M.-L., Tallot, M., Milh, M., Edery, P., Marzin, P., Barcia, G., Barnerias, C., Besmond, C., Bienvenu, T., Bruel, A.-L., Brunga, L., Ceulemans, B., … Depienne, C. (2019). IQSEC2 related encephalopathy in males and females: A comparative study including 37 novel patients. Genetics in Medicine: Official Journal of the American College of Medical Genetics, 21(4), 837–849. https://doi.org/10.1038/s41436-018-0268-1
- Miller, B. R., 3rd, McGee, T. D., Jr, Swails, J. M., Homeyer, N., Gohlke, H., & Roitberg, A. E. (2012). MMPBSA.py: An efficient program for end-state free energy calculations. Journal of Chemical Theory and Computation, 8(9), 3314–3321. https://doi.org/10.1021/ct300418h
- Murphy, J. A., Jense, O. N., & Walikonis, R. S. (2006). BRAG1, a Sec 7 domain containing protein, is a component of the postsynaptic density of excitatory synapses. Brain Research, 1120(1), 35–45. https://doi.org/10.1016/j.brainres.2006.08.096
- Myers, K. R., Wang, G., Sheng, Y., Conger, K. K., Casanova, J. E., & Zhu, J. J. (2012). ARF6-GEF BRAG1 regulates JNK-mediated synaptic removal of GluA1-containing AMPA receptors: A new mechanism for nonsyndromic X-linked mental disorder. The Journal of Neuroscience: The Official Journal of the Society for Neuroscience, 32(34), 11716–11726. https://doi.org/10.1523/JNEUROSCI.1942-12.2012
- Oates, M. E., Romero, P., Ishida, T., Ghalwash, M., Mizianty, M. J., Xue, B., Dosztányi, Z., Uversky, V. N., Obradovic, Z., Kurgan, L., Dunker, A. K., & Gough, J. (2013). D2P2: Database of disordered protein predictions. Nucleic Acids Research, 41(Database issue), D508–D516. https://doi.org/10.1093/nar/gks1226
- Pac-Man. n.d. Wikipedia. https://en.wikipedia.org/wiki/Pac-Man
- Rajagopalan, K., Mooney, S. M., Parekh, N., Getzenberg, R. H., & Kulkarni, P. (2011). A majority of the cancer/testis antigens are intrinsically disordered proteins. Journal of Cellular Biochemistry, 112(11), 256–3267. https://doi.org/10.1002/jcb.23252
- Renault, L., Guibert, B., & Cherfils, J. (2003). Structural snapshots of the mechanism and inhibition of a guanine nucleotide exchange factor. Nature, 426(6966), 25–530. https://doi.org/10.1038/nature02197
- Roe, D. R., & Cheatham, T. E. (2013). PTRAJ and CPPTRAJ: Software for processing and analysis of molecular dynamics trajectory data. Journal of Chemical Theory and Computation, 9(7), 084–3095. https://doi.org/10.1021/ct400341p
- Rogers, E. J., Jada, R., Schragenheim-Rozales, K., Sah, M., Cortes, M., Florence, M., Levy, N. S., Moss, R., Walikonis, R. S., Palty, R., Shalgi, R., Lichtman, D., Kavushansky, A., Gerges, N. Z., Kahn, I., Umanah, G. K. E., & Levy, A. P. (2019). An IQSEC2 mutation associated with intellectual disability and autism results in decreased surface AMPA receptors. Frontiers in Molecular Neuroscience, 12, 3. https://doi.org/10.3389/fnmol.2019.00043.
- Ruff, K. M., & Pappu, R. V. (2021). AlphaFold and implications for intrinsically disordered proteins. Journal of Molecular Biology, 433(20), 167208–167208. https://doi.org/10.1016/j.jmb.2021.167208
- Sah, M., Shore, A. N., Petri, S., Kanber, A., Yang, M., Weston, M. C., & Frankel, W. N. (2020). Altered excitatory transmission onto hippocampal interneurons in the IQSEC2 mouse model of X linked neurodevelopmental disease. Neurobiology of Disease, 137, 04758. https://doi.org/10.1016/j.nbd.2020.104758.
- Sakagami, H., Sanda, M., Fukaya, M., Miyazaki, T., Sukegawa, J., Yanagisawa, T., Miyazaki, T., Sukegawa, J., Yanagisawa, T., Suzuki, T., Fukunaga, K., Watanabe, M., & Kondo, H. (2008). IQ-ARFGEF/BRAG1 is a guanine nucleotide exchange factor for ARF6 that interacts with PSD-95 at postsynaptic density of excitatory synapses. Neuroscience Research, 60(2), 199–212. https://doi.org/10.1016/j.neures.2007.10.013
- Schymkowitz, J., Borg, J., Stricher, F., Nys, R., Rousseau, F., & Serrano, L. (2005). The FoldX web server: An online force field. Nucleic Acids Research, 33(Web Server issue), W382–W388. https://doi.org/10.1093/nar/gki387
- Shao, J., Tanner, S. W., Thompson, N., & Cheatham, T. E. (2007). Clustering molecular dynamics trajectories: 1. Characterizing the performance of different clustering algorithms. Journal of Chemical Theory and Computation, 3(6), 2312–2334. https://doi.org/10.1021/ct700119m
- Shoubridge, C., Harvey, R. J., & Dudding-Byth, T. (2019). IQSEC2 mutation update and review of the female-specific phenotype spectrum including intellectual disability and epilepsy. Human Mutation, 40(1), 5–24. https://doi.org/10.1002/humu.23670
- Shoubridge, C., Tarpey, P. S., Abidi, F., Ramsden, S. L., Rujirabanjerd, S., Murphy, J. A., Boyle, J., Shaw, M., Gardner, A., Proos, A., Puusepp, H., Raymond, F. L., Schwartz, C. E., Stevenson, R. E., Turner, G., Field, M., Walikonis, R. S., Harvey, R. J., Hackett, A., … Gécz, J. (2010). Mutations in the guanine nucleotide exchange factor gene IQSEC2 cause nonsyndromic intellectual disability. Nature Genetics, 42(6), 486–488. https://doi.org/10.1038/ng.588
- van Zundert, G. C. P., Rodrigues, J., Trellet, M., Schmitz, C., Kastritis, P. L., Karaca, E., Melquiond, A., S. J., van Dijk, M., de Vries, S., J., A., & M. J. J., Bonvin. (2016). The HADDOCK2.2 webserver: User-friendly integrative modeling of biomolecular complexes. Journal of Molecular Biology, 428(4), 720–725. https://doi.org/10.1016/j.jmb.2015.09.01
- Vetter, I. R., & Wittinghofer, A. (2001). The guanine nucleotide-binding switch in three dimensions. Science (New York, N.Y.), 294(5545), 299–1304. https://doi.org/10.1126/science
- Wiederstein, M., & Sippl, M. J. (2007). ProSA-web: Interactive web service for the recognition of errors in three-dimensional structures of proteins. Nucleic Acids Research, 35(Web Server issue), 407–W410. https://doi.org/10.1093/nar/gkm290.
- Xu, J., McPartlon, M., & Li, J. (2021). Improved protein structure prediction by deep learning irrespective of co-evolution information. Nature Machine Intelligence, 3, 01–609. https://doi.org/10.1038/s42256-021-00348-5
- Zerem, A., Haginoya, K., Lev, D., Blumkin, L., Kivity, S., Linder, I., Shoubridge, C., Palmer, E. E., Field, M., Boyle, J., Chitayat, D., Gaillard, W. D., Kossoff, E. H., Willems, M., Genevieve, D., Tran-Mau-Them, F., Epstein, O., Heyman, E., Dugan, S., … Lerman-Sagie, T. (2016). The molecular and phenotypic spectrum of IQSEC2-related epilepsy. Epilepsia, 57(11), 1858–1869. https://doi.org/10.1111/epi.13560
- Zhang, Y., Kong, W., Gao, Y., Liu, X., Gao, K., Xie, H., Wu, Y., Zhang, Y., Wang, J., Gao, F., Wu, X., & Jiang, Y. (2015). Gene mutation analysis in 253 chinese children with unexplained epilepsy and intellectual/developmental disabilities. PLoS One, 10(11), e0141782. https://doi.org/10.1371/journal.pone.0141782
- Zipper, R., Baine, S. D., Genizi, J., Maoz, H., Levy, N. S., & Levy, A. P. (2017). Developmental progression of intellectual disability, autism and epilepsy in a child with an IQSEC2 gene mutation. Clinical Case Reports, 5(10), 1639–1643. https://doi.org/10.1002/ccr3.1139