
Abstract
Pseudomonas aeruginosa is an opportunistic pathogen prone to developing drug-resistance and is a major cause of infection for burn patients and patients suffering from cystic fibrosis or are hospitalized in intensive care units. One of the virulence factors of this bacterium is the lipase enzyme that degrades the extracellular matrix of the host tissue and promotes invasion. Bromhexine is a mucolytic drug and has recently been reported to function as a competitive inhibitor of lipase with an IC50 value of 49 µM. In the present study, an attempt was made to identify stronger inhibitors from the ChEMBL database of bioactive compounds, as compared to the reference compound Bromhexine. Following docking and MD simulations, four hit compounds (N1-N4) were selected that showed promising binding modes and low RMSD values indicative of stable protein-ligand complexes. From subsequent binding pose metadynamics (BPMD) simulations, two of these (N2 and N4) stood out as more potent than Bromhexine, displaying stable interactions with residues in the catalytic site of the enzyme. Biological investigations were performed for all four compounds. Among them, the same two hit compounds were found to be the most effective binders with IC50 values of 22.1 and 27.5 µM, respectively; i.e. roughly twice as efficient as the reference Bromhexine. Taken together, our results show that these hits can be promising new candidates to use as leads for the development of drugs targeting the P. aeruginosa lipase enzyme.
Communicated by Ramaswamy H. Sarma
1. Introduction
Pseudomonas aeruginosa is an opportunistic, aerobic, Gram-negative, rod-shaped bacterium and is an important factor in nosocomial infections, particularly in patients with compromised host defense mechanisms (Lyczak et al., Citation2000; Talebi et al., Citation2018). It is also considered the second leading cause of pneumonia and the third leading cause of urinary tract infection (Flockton et al., Citation2019; Gellatly & Hancock, Citation2013). Several virulence factors such as flagellum, lipopolysaccharide, type IV pili, type III secretion system, exotoxin A, alginate, quorum sensing, antibiotic-resistant biofilm formation, and extracellular enzymes may cause pathogenicity that facilitates adhesion and disconnects the signaling pathways in host cells (Rocha et al., Citation2019; Le Berre et al., Citation2011). In this regard, some extracellular enzymes such as lipase break down the extracellular matrix in the tissue of host patients and cause biofilm on the skin surface (Gholami et al., Citation2022). Hence, the inhibition of lipase in P.aeruginosa can reduce biofilm formation and subsequent tissue damage (Talebi et al., Citation2018). The main natural function of lipases is to catalyze the hydrolysis of long-chain triglycerides into monoacylglycerol (Angkawidjaja et al., Citation2006) (), with several potential areas of application. Thus, exploring the interactions involving the catalytic triad residues of lipase will be helpful to further understand the main functionality and will provide means to modulate its activity.
Figure 1. (A) Hydrolysis of triglycerides. (B) Catalytic mechanism of lipolysis in Pseudomonas aeruginosa (adapted from Jegannathan et al., Citation2008 and Röttig et al., Citation2010).
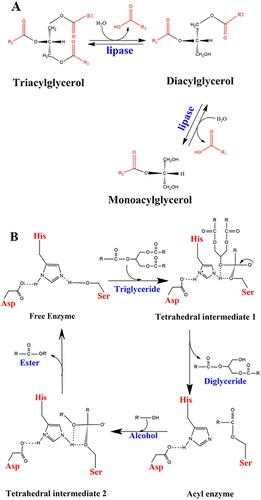
The three amino acids in the catalytic triad, Ser-His-Asp, act as a charge-relay system (). The histidine forms hydrogen bonds with the aspartic acid on the one side, and the serine sidechain on the other. In the first step of the reaction, the proton of the serine hydroxyl group is transferred to Nε of His, forming an oxyanion. This enables a nucleophilic attack by the serine. In the second step, tetrahedral intermediate 1 is produced when the serine oxyanion attacks a carbonyl carbon on the substrate. In the third step, the proton on the histidine is transferred to the diglyceride and the electrons on the oxyanion are pushed back to the carbonyl carbon. For the completion of the transesterification, the serine ester reacts with an alcohol and the histidine nitrogen removes the hydrogen from the OH group, giving an alkyl oxide anion as tetrahedral intermediate 2.
Transfer of the proton from His to the serine oxygen and rupture of the O-C bond gives the free fatty acid (ester). During the whole procedure, the proton shuttle between the aspartic acid and histidine helps to promote the proton transfers to/from Nε of His to push the reaction forward (Jegannathan et al., Citation2008; Nardini et al., Citation2000).
Inhibition of lipase may not be useful in industrial applications (Rosenau & Jaeger, Citation2000) but is important in the medical field (Tielen et al., Citation2013). In this context, Bromhexine is a mucolytic agent which is prescribed in the treatment of respiratory disorders, for patients who suffer from excessive mucus (Pospıšilová et al., Citation2001). In addition, the use of Bromhexine as a prophylactic medication was considered to prevent viral entry for SARS-CoV-2 during the pandemic (Ansarin et al., Citation2020; Depfenhart et al., Citation2020). In our previous studies, kinetic analyses (IC50 = 49 µM and Ki = 20 µM) and fluorescence measurements of Bromhexine revealed that the binding of the drug to lipase could induce structural changes to the enzyme and inhibit it in a competitive manner (Gholami et al., Citation2022). Another study showed that the muscarinic receptor antagonist Dicyclomine used to reduce the symptoms of stomach and intestinal cramping (Vikram & Gupta, Citation2019), could also be considered a promising candidate against lipase from P.aeruginosa (Talebi et al., Citation2018). Docking analyses demonstrated that the drug attached tightly to the binding pocket by forming several hydrogen bonds and hydrophobic interactions. Hanes-Woolf plots indicated that the drug inhibited lipase activity through a competitive mechanism, with IC50 and Ki values reported to be 60 µM and 30 µM respectively (Talebi et al., Citation2018). Investigation of the antibacterial activity of sulfonamide compounds as potential tools against quorum sensing (QS) and biofilm formation was reported by another research group. Studies showed that the compounds decreased the virulence expression, which in turn can reduce the risk of developing drug-resistant strains. Molecular docking analyses were performed and provided promising binders for inhibiting the production of QS (Mizdal et al., Citation2018).
In light of the above findings, we herein compare the inhibitory effect of Bromhexine on P.aeruginosa lipase with that of some bioactive agents from the ChEMBL database, with the aim to discover more potent and stronger candidates against lipase and contribute to the improvement of existing treatments for this highly antibiotic-resistant bacterium. In this regard, the use of computational methods for virtual screening along with docking analyses and molecular dynamics simulations has proven to be a fast and valuable approach to the discovery of novel compounds. The in vitro inhibitory concentrations (IC50 values) of selected hits and kinetic parameters for the two most promising candidates towards the enzyme are reported, confirming the in silico findings.
2. Materials and methods
2.1. In silico studies
2.1.1. Protein preparation
The lipase protein in the open structure (PDB ID:1EX9) (Nardini et al., Citation2000), containing co-crystallized octyl-phosphinic acid 1,2-bis-octylcarbamoyloxy-ethyl ester, was downloaded from the protein data bank (PDB) (http://www.rcsb.org). The protein was prepared and refined using the Protein Preparation Wizard in Maestro (Schrödinger 2021–4, www.schrodinger.com).
Bond orders were assigned during the preprocessing stage of the crystal structure, and after retrieving missing loops or side chains, all water molecules located beyond 3.0 Å from the ligand were deleted from the system. Protein hydrogen bond assignments were optimized and protonation states at pH 7 determined using PROPKA (Madhavi Sastry et al., Citation2013). Finally, a restrained minimization with the OPLS4 force field (Roos et al., Citation2019) was performed with an RMSD convergence threshold for heavy atoms of 3.0 Å.
2.1.2. Ligand identification, preparation and docking
Bioactive compounds were searched within the ChEMBL database (https://www.ebi.ac.uk/chembl). Based on structure similarity with Bromhexine, 11 compounds having up to 40% identity were selected, downloaded from the ChEMBL database, and transferred into Maestro Schrödinger using LigPrep. The Epik module (Greenwood et al., Citation2010) was used to determine possible ionization states at physiological pH 7.0 ± 2.0, and the OPLS4 force field (Roos et al., Citation2019) was selected for the optimization.
In order to perform the molecular docking and predict the interactions of the protein-ligand complexes, the Schrödinger Induced Fit Docking (IFD) methodology was used (Farid et al., Citation2006; Sherman et al., Citation2006). All compounds were docked toward the active site of the holoenzyme of the lipase protein. The grid box for the docking was defined from the centroid of the binding site. During the initial docking procedure, the van der Waals scaling factor was set at 0.5 for both receptor and ligand. The Prime refinement step was used on sidechains of residues within 5 Å of the ligand. No constraints were applied, and all remaining parameters were set to default.
2.1.3. Molecular dynamics (MD) simulations
The stabilities of the ligand-protein complexes were investigated through 200 ns MD simulations in triplicate, using an NPT ensemble and the OPLS4 force field, with the Desmond MD engine (Bowers et al., Citation2006) implemented in Schrödinger (Schrödinger 2021-4, www.schrodinger.com). The TIP3P force field (Jorgensen et al., Citation1983) was used to model water molecules. Periodic boundary conditions were applied with a 10 Å water buffer around the protein in a cuboid simulation box. The appropriate number of counterions (i.e. Na+/Cl-) were added to neutralize the system and to obtain a salt concentration of 150 mM to mimic physiological conditions. The temperature 300 K and pressure 1 atm were controlled using the Nose–Hoover thermostat (1ps relaxation time) and the Martyna–Tobias–Klein barostat (Martyna et al., Citation1992) (isotropic coupling), respectively. Electrostatic forces were treated using particle-mesh Ewald (PME) (Essmann et al., Citation1995) summation with a cut-off of 9 Å both for electrostatics and van der Waals interactions. The relaxation protocol consisted of several steps as follows: (1) NVT Brownian Dynamics with restraints on solute heavy atoms at T = 10 K for 100 ps, (2) NVT MD simulation at T = 10 K with restraints on solute heavy atoms for 12 ps, (3) NPT MD simulation at T = 10 K with restraints on solute heavy atoms for 12 ps, (4) NPT MD simulation at T = 300 K with restraints on solute heavy atoms for 12 ps, and (5) NPT MD simulation at T = 300 K without restraints for 24 ps.
2.1.4. Clustering
From the MD trajectories, clustering of the resulting structures was performed to group similar molecular conformations into distinct sets such that the structures in each cluster are more similar to each other compared to structures in any other cluster. This gives a refined view of how a given molecule is sampling the conformational space and allows direct characterization of the separate conformational sub-states visited during the MD simulation.
To obtain representative structures for the Binding Pose Metadynamics simulations (BPMD), Desmond Trajectory clustering in Maestro (Schrödinger 2021–4, www.schrodinger.com) was performed, setting the initial number of clusters to 3. After running the clustering tool, the lowest energy structures from the most populated clusters were selected for the BPMD calculations.
2.1.5. Binding Pose Metadynamics (BPMD)
Binding Pose Metadynamics simulations were performed using a set biasing force, to explore how stable ligands are in the binding pocket of the receptor. Weaker ligands will experience higher fluctuations with larger RMSD values in comparison with the stably bound ones (Fusani et al., Citation2020).
MD clustering based on the previous stage was done to remove any bad contacts and select the most appropriate starting structures, followed by BPMD simulations as implemented in Schrödinger Maestro version 2021-4. 10 independent metadynamics simulations of 10 ns each were performed for each system, using as Collective Variable (CV) the Root-Mean-Square Deviation (RMSD) of the ligand heavy atoms relative to their starting position.
2.2. In vitro studies
2.2.1. Culture medium and cell harvesting
The most preferable culture for producing lipase in P.aeruginosa PAO1 is salt medium containing 2.5 g Na2HPO4, 2.5 g KH2PO4, 1 g NH4NO3, 0.2 g MgSO4, and 0.01 g CaCl2 per 1 L of distilled water, and adjusting the pH to 7.0. All chemicals were obtained from commercial sources and were of the highest quality available. As carbon source, olive oil was used with a final concentration of 1%. The source of the bacterium was provided by CCUG (culture collection university of Gothenburg). The medium was aerated in a rotary shaker at 30 °C for 96 h. The cells were precipitated at 7000 g during 1 min of centrifugation and the supernatant used for the enzyme assay.
2.2.2. Enzyme assay
Stock solution: All four compounds (: N1-N4) were purchased from Enamine. A detailed list of purchased compounds, including purity and quantity, is available in the supplementary information (Table S1). The compounds were dissolved in dimethyl sulphoxide (DMSO) with a purity of 99.9% to final concentration of 10 mM, through careful pipetting. The enzyme assay was performed in the presence of the different compounds with concentration ranges of 10–200 µM.
Table 1. Docking score values and free energies of binding (MMGBSA) for each compound, obtained from the virtual screening.
Work solution: Para-nitrophenyl palmitate (pNPP) was purchased from Sigma Aldrich (Merck KGaA, Darmstadt, Germany). As lipase substrate, 1 mg of pNPP was dissolved in 1 ml of 2-propanol containing 1% Triton X-100. Enzyme assay buffer was prepared by mixing pNPP solution with 0.1 M Tris buffer at pH 8. The control reaction was initiated by adding 50 µl of the supernatant to 2 ml of the working buffer in a test tube. The catalytic activity of lipase was monitored by continuously following the increased absorption at 410 nm for 5 min by a UV-Visible spectrophotometer, which was sufficient for the enzyme to reach the substrate depletion plateau. Different concentrations of each ligand sample (10–200 µM) was added to the work solution and the enzymatic activity measured with the same protocol as described above. The experiments were performed in triplicate for each concentration. The IC50 values were calculated using GraphPad Prism 9 for all compounds, followed by calculation of kinetic parameters of the enzyme and type of inhibition for the two most promising hit compounds.
3. Results and discussion
3.1. Sequence alignment
In order to identify conserved amino acids, sequence alignment was made for a selection of prokaryotic and eukaryotic lipase enzymes (Figure S1). Knowledge of the structures of the enzyme and conserved residues in the active site is essential for compound selectivity and determination of binding modes of the lipase inhibitors.
The FASTA sequence of triacylglycerol lipase protein for Pseudomonas aeruginosa PAO1 was retrieved from UniProt (ID code: P26876) and used as reference in the alignment. The same protein from different species as outlined below were also selected and the sequence similarity and identity percentages were compared to the P. aeruginosa reference.
Triacylglycerol lipase protein from Pseudomonas cepacia (Burkholderia cepacia) (P22088)
Triacylglycerol lipase protein from Burkholderia plantarii (P0DUB8),
Triacylglycerol lipase protein from Pseudarthrobacter phenanthrenivorans (Arthrobacter phenanthrenivorans) (P0DUB9),
Lipase 1 from Candida rugosa (Diutina rugosa) (P20261),
Gastric triacylglycerol lipase from Homo sapiens (Human) (P07098),
Hepatic triacylglycerol lipase from Homo sapiens (Human) (P11150),
Pancreatic triacylglycerol lipase from Homo sapiens (Human) (P16233),
Pancreatic triacylglycerol lipase from Rattus norvegicus (Rat) (P27657).
The sequence identity between the reference and P. cepacia, B. plantarii, and P. phenanthrenivorans is 36%, 37%, and 38%, respectively, and the corresponding sequence similarity 55%, 54%, and 55%. The sequence identity between our reference protein and that of yeast and pancreatic lipase in human and rat is only 13%, 9%, and 12%, respectively, which indicates that the enzyme is less conserved with eukaryotic species. However, a closer look at the aligned sequences (Figure S1) reveals that the identity of the active site Ser residue is conserved in all organisms, indicating the importance of this amino acid. As described in the introduction, the catalytic triad residues in P. aeruginosa lipase are Ser-His-Asp. Studies have shown that Ser is situated in the nucleophile position and is crucial for the first step of hydrolysis (Jegannathan et al., Citation2008; Stefanucci et al., Citation2019). As a result, if inhibitors bind or block access to this residue, formation of the tetrahedral intermediate that occurs during the acylation step () will be disrupted and the reaction stopped at the initial hydrolysis step. The His and Asp residues of the catalytic triad (Figure S1, blue boxes) are conserved in all prokaryotic microorganisms but differ in human, rat, and yeast (eukaryotic organisms).
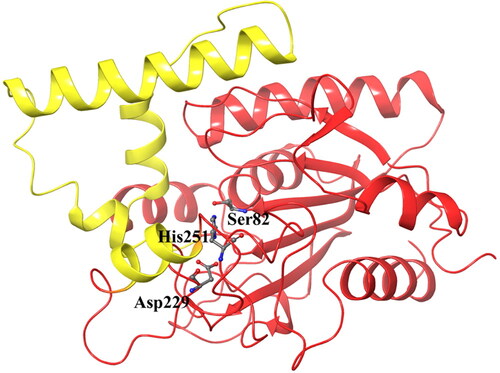
The P. aeruginosa lipase is a globular protein with an approximate radius of 20 Å. It is divided into two units: a core domain with typical features of an α/β hydrolase fold topology, and a cap domain containing four α-helixes that form the active site cleft. The catalytic triad residues Ser82, Asp229, and His251 () are located in the canonical positions in the α/β hydrolase fold (Nardini et al., Citation2000).
Figure 2. The structure of Pseudomonas aeruginosa lipase with the catalytic triad in stick model and the four alpha-helices of the cap domain indicated in yellow.
3.2. In silico evaluation of selected compounds
3.2.1. Identification of potential hit compounds
Previous studies have shown the effect of Bromhexine and Ambroxol on the activity of P.aeruginosa. For instance, alkaline phosphatases detoxify lipopolysaccharide (LPS) by dephosphorylation. Ambroxol and Bromhexine, as mocolytic and bronchial expectorants, were suggested to accelerate the secretion of pulmonary surfactant particles as their pharmacological action. In this context, Bromhexine was found to be less effective than Ambroxol as an LPS detoxificant (Koyama et al., Citation2004).
The efficiency of Ambroxol for the production of pulmonary surfactants was investigated further in rat pneumonia induced by P. aeruginosa. The rats were infected by injecting ATCC27853 intratracheally, after which changes to lung structure were investigated by electronic microscope. The study showed that Ambroxol improved the levels of surfactants and could protect rats from pneumonia. Since Ambroxol is relatively economical and the usage is simple, it was recommended to use this in order to reduce and remove lung infections (Gao et al., Citation2011).
A major problem regarding P. aeruginisa is that it is resistant to a wide range of antibiotics. In this context, affecting an extracellular hydrolytic enzyme such as lipase which is produced and secreted during biofilm formation, with small molecule inhibitors, can thus affect bacterial growth and biofilm formation without leading to resistance development (Frimmersdorf et al., Citation2010).
For example, the anti-quorum sensing and anti-biofilm efficacy of aromatic aldehyde, 5-hydroxymethylfurfural (5-HMF) against P. aeruginosa PAO1 was investigated by Rajkumari et al. (Citation2019). Their in silico studies revealed a competitive manner for this compound and a strong affinity for the binding of two proteins (LasR and RhlR) which are involved in quorum sensing.
Another important factor that produces quorum-sensing in P. aeruginosa virulence determinants is LasI. The inhibition of the LasI protein is therefore an attractive and promising drug target. The compound (z)- 5-octylidenethiazolidine-2, 4-dione (TZD-C8) was shown to be a strong inhibitor of biofilm formation. In silico docking prediction showed that the compound had high affinity for the LasI binding pocket, in which the TZD-C8 compound formed hydrogen bonds with residues Arg30 and Ile107 (Lidor et al., Citation2015).
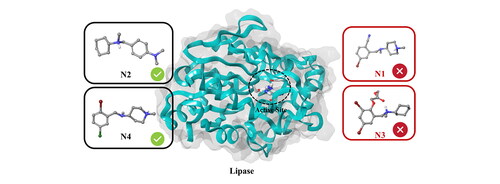
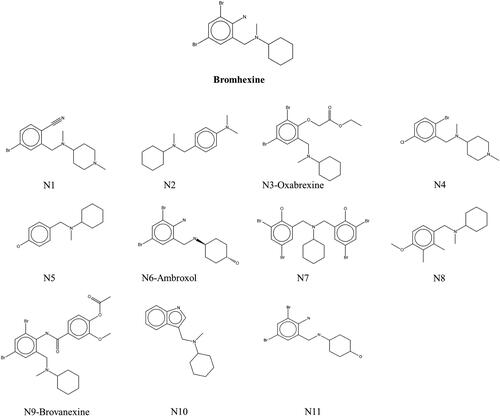
Since Bromhexine was previously identified as an inhibitor to P. aeruginosa lipase enzyme (Gholami et al., Citation2022), we used this as a template for finding structural analogs using a similarity search in the ChEMBL database based on 40% structural match with our reference. This led to the identification of compounds N1–N11 (), including Oxabrexine (N3), Ambroxol (N6) and Brovanexine (N9), carrying a similar structural scaffolds as Bromhexine.
Figure 3. Chemical structures of Bromhexine and the compounds identified in this work.
3.2.2. Molecular docking and molecular dynamics simulations
The active site of the lipase holoenzyme (PDB ID: 1EX9) was selected and used in induced fit docking of the ligand set identified from the ChEMBL database (), with Bromhexine as reference inhibitor. For simplicity, the compounds are labeled N1 to N11.
Following docking, 200 ns MD simulations were performed in triplicate on the different complexes to explore the stability and accuracy of the protein-ligand complexes obtained from the docking studies. Calculations of free energies of binding were performed on the lowest energy structures after clustering of the MD simulation trajectories. shows the docking scores and free energies of binding obtained using MMGBSA.
The binding site positions along with atomic contacts between lipase and compounds N1-N4 or Bromhexine are shown as 2D and 3D interaction diagrams in ; for the remaining compounds N5-N11 the 2D interaction diagrams are given in Supplementary Figure S2.
Figure 4. Interaction patterns of the best docked poses. 2D interaction diagrams (left) and atomic contacts (right) for (A) Bromhexine and the four compounds (B) N1, (C) N2, (D) N3, and (E) N4. Different types of interactions are represented in 3D views by color-dashed lines: blue, yellow, and purple denote π–π stacking, hydrogen bonds, and halogen bonds, respectively.

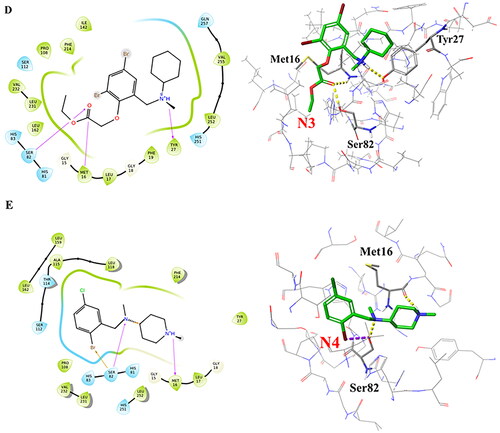
Bromhexine was docked into the active site of lipase as reference inhibitor (). It forms a halogen bond to Ser82 and a hydrogen bond with His251, which are two of the key amino acid residues in the catalytic triad. Based on our previous studies, the binding of the drug into the active site of the enzyme follows competitive inhibition (Gholami et al., Citation2022), which is confirmed by analysis of the docking pose. Based on , we can conclude that the ligand will inhibit the nucleophilic attack in the first step of the transesterification reaction when histidine becomes protonated by the serine OH, in order to generate the reactive oxyanion. Compound N1 () forms a similar halogen bond with Ser82 and a hydrogen bond with Met16. Studies show that Met16 is highly conserved in families I.1 and I.2 of bacterial lipases and is one of the residues that interact with most inhibitors (Nardini et al., Citation2000).
Compound N2 forms a hydrogen bond and a π–π stacking interaction with the catalytic residue His251. Based on the results from , the second highest free energy of binding (after N9) is noted for this ligand. Three hydrogen bonds are observed between compound N3 and amino acids Ser82, Met16, and Tyr27. For compound N4, our docking results indicate the formation of hydrogen bonds with Ser82 and Met16, and a halogen bond with Ser82. These interactions are similar as for compound N1, and it can thus be predicted based on the docking interactions and MMGBSA data () that N4 could have the potential to be a stronger inhibitor than N1 or Bromhexine. The interactions between compounds N5-N11 with lipase, based on the best docking poses, are displayed in Supplementary Figure S2. Among those seven hit compounds, only Ambroxol (N6) and N11 formed π–π stacking interaction or a hydrogen bond, respectively, with one of the catalytic residues (His251). For the rest of the compounds, no interactions with the main catalytic residues were seen. For compounds N5, N7, N8, N10 and N11, the MMGBSA values () do not indicate stronger binding than Bromhexine. Compound N9, which displayed the highest docking score and MMGBSA value (), had as only interaction a hydrogen bond with Leu17 which is not involved in the active site. As a result, N9 may not be a good candidate in comparison with ligands N1-N4. However, for further insights of potential interactions, analysis of RMSD values for compounds N6 and N9 are detailed below and compared with ligands N1-N4 and Bromhexine.
In order to verify the interactions between the ligands and lipase, MD simulations were performed for all complexes. A comparison of the RMSD values for Bromhexine and the 11 identified compounds are shown in . Analyses of the MD results show that the three inhibitors N1, N2, and N4 preserve their binding interactions and are tightly bound to the catalytic site. No significant fluctuations were observed throughout the simulation periods, implying that the binding of the ligands to the active site of lipase are very stable and strong (). In spite of the fact that the analyses of the replica (Figure S3) shows that compound N2 displays some variations, for example at t = 120–140 ns in replica 1, and t = 170–180 ns in replica 2, the ligand remained very stably bound to the binding pocket. Our observations show that it is primarily the benzene ring of the ligand that is involved in these changes due to its partial exposure to the solvent. Analysis of the MD trajectories for compound N3 shows that the ligand remains stable in the binding pocket after an initial large movement/adjustment as noted by the large jump in RMSD at t = 30 ns ( and S3), and then remained in the binding cavity; however, our observations show that the solvent exposed cyclohexane ring undergoes fluctuations during the entire simulation. In addition, the higher RMSD value and the lower value from the MMGBSA calculations () indicate that this ligand does not interact as strongly as the three ligands N1, N2 and N4 or Bromhexine. The reference inhibitor Bromhexine remains very stably bound to the active site cavity of the protein with RMSD values below 1 Å (, black curve), except for a significant fluctuation at t = 150–175 ns after which it returns to the original conformation. From the replicas (Figure S3) we also note some fluctuations during the later parts of the simulations. However, in none of the triplicate simulations did the ligand detach from the binding cavity. Instead, the interaction between the ligand and the protein induced conformational changes to the protein, as described further below.
Figure 5. Root-mean-square deviation (RMSD) values for the 11 hits and the reference compound during one of the 200 ns MD simulations. (A) RMSD values for the 4 best hit compounds N1-N4, (B) RMSD values for the 7 hit compounds N5-N11, (C) Root mean square fluctuations (RMSF) of the lipase protein during the 200 ns MD simulations in complex with Bromhexine or ligands N1-N4. Red arrows correspond to the location of catalytic residues. Black arrows indicate which parts of the protein that undergoes more movements during the simulation (see text for details).
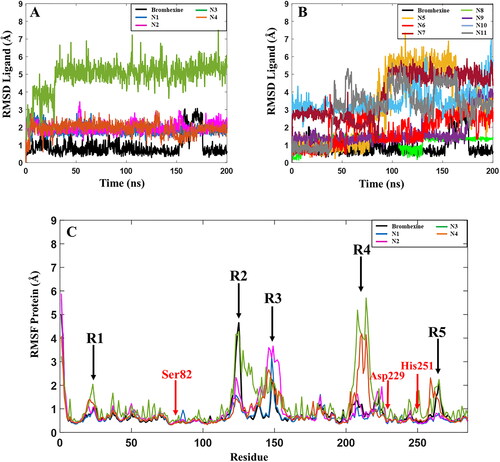
Based on the RMSD plots for N5-N11, presented in , these compounds show high fluctuations in their active site interactions, indicative of low stability of the docked complexes. Although the highest docking score and MMGBSA value is predicted for compound N9 among the full set of compounds (), a sudden increase in RMSD is observed after 170 ns of the MD simulations indicating that the ligand is unstable in the active site of the enzyme. In fact, each replica of compound N9 displayed significant RMSD variations (Figure S3), indicating that this compound is not stable enough in the cavity of the protein in comparison with Bromhexine and ligands N1-N4. The same patterns were observed for compounds N5, N10 and N11 (Figure S3), with the ligands detaching from the binding cavity in replica 2. Considering compound N6, despite the fact that the MMGBSA score is higher than Bromhexine (-47.5 kcal/mol), noticeable fluctuations are seen during all the trajectories. In replica 1 (Figure S3) this compound moves away from the cavity for 80 ns of the trajectory. For compounds N7 and N8 (Figure S3, Table S2), considerable changes seen in each replica, and the average results from the three simulations, clearly show that these compounds are not promising candidates in comparison with our reference. Based on these findings, and in comparison to the reference inhibitor, we conclude that compounds N5-N11 may not be considered as potential candidates as inhibitors against P. aerugiosa lipase.
Table 2. 50% inhibition concentration of the lipase inhibitors.
In addition, we analyzed the protein root-mean-square-fluctuations (RMSF) of the lipase, showing which residues or parts of the protein that fluctuate the most during the MD simulation trajectory (). A close look at the RMSF graph for lipase with Bromhexine bound (black curve) shows that apart from regions R2 and R3 () this complex is highly stable during the entire simulation. The locations of the three main residues Ser82, Asp229 and His251 in the catalytic pocket are indicated by red arrows. One of the regions with the highest variation is related to residues 261–268 (black arrow, R5), which forms part of a loop close to the N-terminal and is not involved in the active site of the protein. The region showing the largest movement is R2, containing residue 118–130. These are part of a key α-helix close to the active site. As discussed above, the RMSD of Bromhexine changes in particular after t = 160 ns, and analysis of the RMSF in each replica (Figure S3) also shows that the protein undergoes some structural changes for residues 118–151 during the MD trajectories. For the complexes with compounds N1, N2 and N4, there are no sudden movements near the active site of the protein. Two peaks in the RMSF are noted for compound N4, R1 and R5, concerning amino acids in the terminal parts of the protein, which do not interfere with the active site. R4 (complexes with compounds N3 and N4) includes residues 220–231 being part of a loop far from the active site of the enzyme. Region R3 (all systems) contains residues 139–151 and are part of the key α-helix, indicating that the interaction of the ligand with this part has a significant conformational impact on the protein. For compound N3, the RMSF values are overall higher than for the other studied ligands throughout the entire trajectories, including fluctuations near the active sites of the protein. All the above again confirms that N3 shows less stability in binding to lipase in comparison with Bromhexine and the other three compounds. The data related to the conformational changes of the protein as outlined above were confirmed in all triplicate studies for each ligand (Figure S3, Table S2). Taken together, based on the MMGBSA results from and RMSD values for compounds N2 and N4 (), we propose that these inhibitors may be more potent binders to P. aeruginosa lipase in comparison with Bromhexine.
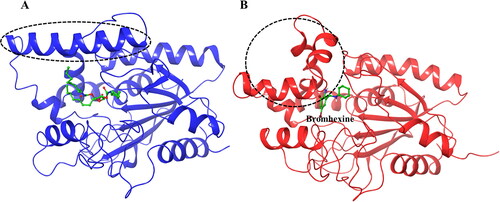
As noted above, some structural changes of the lipase protein in the amino acid sequence 118–151 were noted, in particular for the complex containing Bromhexine. For a better insight into these changes the original crystal structure (PDB ID:1EX9), containing phosphonate trioctyl as ligand (Nardini et al., Citation2000), was used as comparison (). As can be seen from the figure, the binding cavity is very wide in order to accommodate the large substrate. However, upon docking with Bromhexine, and following 200 ns MD simulations (), the cavity has narrowed to interact stronger with the new ligand. We see a clear bending of the key α-helix mentioned above, thereby closing the entrance to the active site of the protein and creating a smaller space to accommodate the ligand in the cavity. As described in connection with , similar structural changes were indicated also for the other four ligands but with some differences. After MD simulations the α-helix was not as strongly bent in the case of the complex with ligand N4, and for N2 and N3 the changes were relatively small in comparison to that seen for Bromhexine.
Figure 6. Structural changes in the lipase protein. (A) Lipase structure (PDB ID 1EX9) containing phosphonate trioctyl as ligand. (B) Lipase structure containing Bromhexine after 200 ns MD simulation. The changes in the conformation of the key α-helix is indicated with the dashed circle.
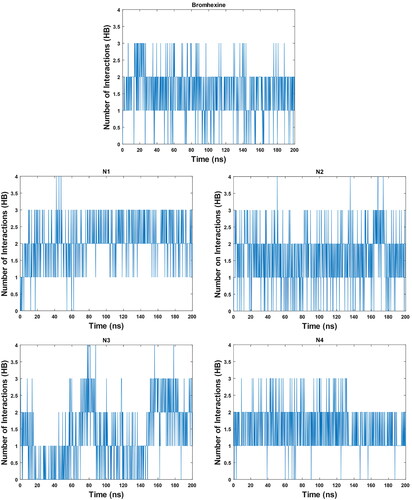
3.2.2. Hydrogen bonds
To further analyse the robustness of the formed complexes, the number of hydrogen bonded interactions were calculated (). The average number of hydrogen bonds for Bromhexine is 1.55 and for compounds N1-N4 these are 2.04, 2.12, 1.39 and 1.67, respectively. The results for compounds N1, N2 and N4 again confirm these as promising binders, and it is also clear that compound N3 interacts less strongly in comparison with the other inhibitors.
Figure 7. Graph of hydrogen bonds for Bromhexine and compounds N1-N4 during a 200 ns MD simulation.
3.2.3. Binding Pose Metadynamics (BPMD) simulations
On order to explore the presence of different MD conformations during a simulation and for quickly understanding the resulting sets, clustering into structurally similar groups is the most suitable technique. From the MD trajectories, clustering of the resulting structures was performed to group similar molecular conformations into distinct sets such that the structures in each cluster, based on RMSD analyses, are more similar to each other compared to structures in any other cluster. If a receptor conformation belongs to a cluster that interacts favorably with a specific ligand, one could assume that other conformations within the same cluster will behave similarly. High stability hence also implies a higher populated cluster ((Phillips et al., Citation2011; Paris et al., Citation2015).
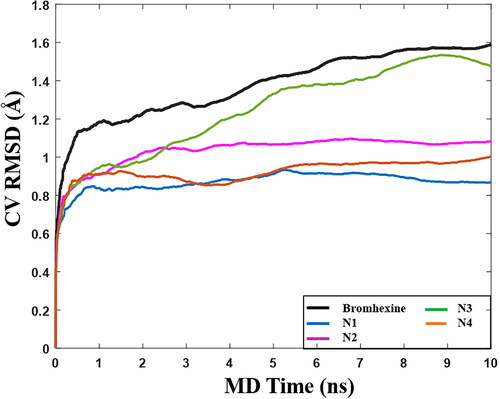
After clustering of conformations from the MD simulations, we selected Bromhexine and the four best compounds (N1-N4) for BPMD simulations. The pose stability was calculated based on the PoseScore, i.e. the RMSD of the ligand with respect to the initial heavy atom coordinates. The threshold value for ligand stability in the binding pocket is a PoseScore ≤ 2 Å (Fusani et al., Citation2020).
shows that when Bromhexine is located in the lipase cavity, the calculated value of PoseScore is 1.57 Å. For compounds N1, N2, and N4, the obtained PoseScores were 0.873 Å, 1.07 Å, and 0.976 Å, respectively, indicative of very stable binding. For compound N3, the obtained PoseScore is 1.51 Å, similar to Bromhexine. Hence all ligands are able to bind into the active site of lipase, although for N3 the highest observed RMSD values () and lower MMGBSA score (), indicate lower ligand stability within the active site in comparison with the other ligands and the reference. Following the above results, in vitro inhibitory concentrations (IC50) of the top-ranked compounds were evaluated.
Figure 8. Graph of RMSD estimate averaged over 10 BPMD trials for Bromhexine and compounds N1-N4 binding to Lipase, vs simulation time.
3.3. In vitro evaluation of selected compounds
The IC50 values of lipase inhibitory activity were determined for the four best compounds N1-N4 using a UV-Visible spectrophotometer at room temperature, with Bromhexine as reference. The control sample contained 2 ml 0.1 M Tris buffer at pH 8, to which was added 50 µl of lipase supernatant and 100 µl DMSO. The enzyme activity was followed by measuring absorption at 410 nm for 5 min. For analyzing the selected compounds, different concentrations for each inhibitor ranging from 10–200 µM were used, following the same protocol as described above. The IC50 scores for the four compounds N1-N4 and Bromhexine are given in .
As shown in , compound N2 shows the lowest IC50 of all tested compounds; less than half of that of our reference Bromhexine (49 µM). Another compound with lower IC50 value than Bromhexine is N4 with IC50 = 27.5 µM, and is thus also proposed as a possible better inhibitor against lipase compared to Bromhexine. The other two compounds (N1 and N3) showed worse inhibition scores and displayed lower stability than Bromhexine. We then calculated the kinetic parameters of the enzyme and type of inhibition for the two potential hits (N2 and N4). The results showed that, in the presence of compound N2, the Vmax of the enzyme was constant (Vmax = 2.65 mmol min−1 mg protein−1), while Km was varying, depending on concentration of the compound. The Ki value was determined to be about 19 μM. A competitive type of inhibition was thus observed for the enzyme in the presence of inhibitor N2.
Regarding compound N4, Vmax of the enzyme varies in different concentration of the compound, and the Ki and Km values were obtained as 26 and 100 μM, respectively. N4 thus displays non-competitive inhibition. Overall, the experimental data agrees perfectly with the in silico data and analysis. The result suggests that compounds N2 is more potent in comparison with our reference compound Bromhexine (IC50= 49 μM and Ki= 20 μM); however, both of these inhibitors have the same inhibition patterns and blocks the active site of the lipase protein, as confirmed by . Considering compound N4, the Ki value is close to that of Bromhexine, but the IC50 value and the type of inhibition again indicates the efficacy of this inhibitor for the lipase protein and can be a good starting point for further lead optimization.
4. Conclusions and perspective
In the current study, we have performed docking and molecular dynamics simulation analyses to identify bioactive compounds that form favorable interactions at the active site of the P. aeruginosa lipase enzyme, as possible candidate molecules for inhibitor optimization. The compounds were compared with Bromhexine as reference, which was previously identified as a novel lipase inhibitor (Gholami et al., Citation2022). In vitro results confirmed the in silico predictions, showing better IC50 values for compounds N2 (4-{[cyclohexyl(methyl)amino]methyl}-N,N-dimethylaniline and N4 (N-[(2-bromo-5-chlorophenyl)methyl]-N,1-dimethylpiperidin-4-amine). We also investigated the type of inhibition for the two potential hit compounds N2 and N4 and found that these display competitive and non-competitive inhibition, respectively. We conclude that these new compounds could be used as precursors in the development of effective drug molecules targeting the secreted lipase enzyme, thus impacting on the pathogen’s virulence without inducing the development of resistance. However, further experiments are needed to reveal possible toxic effects and physiochemical properties for each of the compounds.
Authors’ contributions
All authors designed the initial study and contributed with the analysis of data. AG performed all calculations and in vitro experiments. The manuscript was written and revised through the contributions of all authors. All authors have given approval to the final version of the manuscript.
Supplemental Material
Download PDF (2.9 MB)Acknowledgments
The authors acknowledge funding from the Swedish Research Council (VR), grant number 2019-3684 (L.A.E.), and generous allocation of computing time at the supercomputing centers C3SE and NSC by the Swedish National Infrastructure for Computing (SNIC), in part funded by the Swedish Research Council through grant agreement no 2018-05973.
Data availability
All docked structures, MD simulation trajectories, and BPMD data are available as free download from Zenodo.org, DOI: 10.5281/zenodo.7003747.
Disclosure statement
The authors declare no conflicts of interest.
Additional information
Funding
References
- Angkawidjaja, C., Kanaya, S., & Family, I. (2006). 3 lipase: Bacterial lipases secreted by the type I secretion system. Cellular and Molecular Life Sciences : CMLS, 63(23), 2804–2817. https://doi.org/10.1007/s00018-006-6172-x
- Ansarin, K., Tolouian, R., Ardalan, M., Taghizadieh, A., Varshochi, M., Teimouri, S., Vaezi, T., Valizadeh, H., Saleh, P., Safiri, S., & Chapman, K. R. (2020). Effect of bromhexine on clinical outcomes and mortality in COVID-19 patients: A randomized clinical trial. BioImpacts: BI, 10(4), 209–215. https://doi.org/10.34172/bi.2020.27
- Bowers, K. J., Chow, D. E., Xu, H., et al. Scalable algorithms for. (2006). molecular dynamics simulations on commodity clusters [Paper presentation]. In SC’06: Proceedings of [Paper presentation].The 2006 ACM/IEEE Conference on Supercomputing. IEEE,; 43. https://doi.org/10.1145/1188455.1188544
- Depfenhart, M., de Villiers, D., Lemperle, G., Meyer, M., & Di Somma, S. (2020). Potential new treatment strategies for COVID-19: Is there a role for bromhexine as add-on therapy? Internal and Emergency Medicine, 15(5), 801–812. https://doi.org/10.1007/s11739-020-02383-3
- Essmann, U., Perera, L., Berkowitz, M. L., Darden, T., Lee, H., & Pedersen, L. G. (1995). A smooth particle mesh Ewald method. Journal of Chemical Physics. 103(19), 8577–8593. https://doi.org/10.1063/1.470117
- Farid, R., Day, T., Friesner, R. A., & Pearlstein, R. A. (2006). New insights about HERG blockade obtained from protein modeling, potential energy mapping, and docking studies. Bioorganic & Medicinal Chemistry, 14(9), 3160–3173. https://doi.org/10.1016/j.bmc.2005.12.032
- Flockton, T. R., Schnorbus, L., Araujo, A., Adams, J., Hammel, M., & Perez, L. J. (2019). Inhibition of Pseudomonas aeruginosa biofilm formation with surface modified polymeric nanoparticles. Pathogens, 8(2), 55. https://doi.org/10.3390/pathogens8020055
- Frimmersdorf, E., Horatzek, S., Pelnikevich, A., Wiehlmann, L., & Schomburg, D. (2010). How Pseudomonas aeruginosa adapts to various environments: A metabolomic approach. Environmental Microbiology, 12(6), 1734–1747. https://doi.org/10.1111/j.1462-2920.2010.02253.x
- Fusani, L., Palmer, D. S., Somers, D. O., & Wall, I. D. (2020). Exploring ligand stability in protein crystal structures using binding pose metadynamics. Journal of Chemical Information and Modeling, 60(3), 1528–1539. https://doi.org/10.1021/acs.jcim.9b00843
- Gao, X., Huang, Y., Han, Y., Bai, C., Xue., & Wang, G. (2011). The protective effects of Ambroxol in Pseudomonas aeruginosa-induced pneumonia in rats. Archives of Medical Science: AMS, 7(3), 405–413. https://doi.org/10.5114/aoms.2011.23403
- Gellatly, S. L., & Hancock, R. E. W. (2013). Pseudomonas aeruginosa: New insights into pathogenesis and host defenses. Pathogens and Disease, 67(3), 159–173. https://doi.org/10.1111/2049-632X.12033
- Gholami, A., Minai-Tehrani, D., & Eftekhar, F. (2022). Bromhexine and its inhibitory effect on lipase–kinetics and structural study. Archives of Physiology and Biochemistry, 128(6), 1687–1692. https://doi.org/10.1080/13813455.2020.1788606
- Greenwood, J. R., Calkins, D., Sullivan, A. P., & Shelley, J. C. (2010). Towards the comprehensive, rapid, and accurate prediction of the favorable tautomeric states of drug-like molecules in aqueous solution. Journal of Computer-Aided Molecular Design, 24(6-7), 591–604. https://doi.org/10.1007/s10822-010-9349-1
- Jegannathan, K. R., Abang, S., Poncelet, D., Chan, E. S., & Ravindra, P. (2008). Production of biodiesel using immobilized lipase—A critical review. Critical Reviews in Biotechnology, 28(4), 253–264. https://doi.org/10.1080/07388550802428392
- Jorgensen, W. L., Chandrasekhar, J., Madura, J. D., Impey, R. W., & Klein, M. L. (1983). Comparison of simple potential functions for simulating liquid water. Journal of Chemical Physics. 79(2), 926–935. https://doi.org/10.1063/1.445869
- Koyama, I., Matsunaga, T., Harada, T., Kikuno, A., Hokari, S., & Komoda, T. (2004). Ambroxol reduces LPS toxicity mediated by induction of alkaline phosphatases in rat lung. Clinical Biochemistry, 37(8), 688–693. https://doi.org/10.1016/j.clinbiochem.2004.02.004
- Le Berre, R., Nguyen, S., Nowak, E., Kipnis, E., Pierre, M., Quenee, L., Ader, F., Lancel, S., Courcol, R., Guery, B. P., & Faure, K, Pyopneumagen Group. (2011). Relative contribution of three main virulence factors in Pseudomonas aeruginosa pneumonia. Critical Care Medicine, 39(9), 2113–2120. https://doi.org/10.1097/CCM.0b013e31821e899f
- Lidor, O., Al-Quntar, A., Pesci, E. C., & Steinberg, D. (2015). Mechanistic analysis of a synthetic inhibitor of the Pseudomonas aeruginosa LasI quorum-sensing signal synthase. Scientific Reports, 5(1), 1–13. https://doi.org/10.1038/srep16569
- Lyczak, J. B., Cannon, C. L., & Pier, G. B. (2000). Establishment of Pseudomonas aeruginosa infection: Lessons from a versatile opportunist. Microbes and Infection, 2(9), 1051–1060. https://doi.org/10.1016/s1286-4579(00)01259-4
- Madhavi Sastry, G., Adzhigirey, M., Day, T., Annabhimoju, R., & Sherman, W. (2013). Protein and ligand preparation: Parameters, protocols, and influence on virtual screening enrichments. Journal of Computer-Aided Molecular Design, 27(3), 221–234. https://doi.org/10.1007/s10822-013-9644-8
- Martyna, G. J., Klein, M. L., & Tuckerman, M. (1992). Nosé–Hoover chains: The canonical ensemble via continuous dynamics. Journal of Chemical Physics. 97(4), 2635–2643. https://doi.org/10.1063/1.463940
- Mizdal, C. R., Stefanello, S. T., Nogara, P. A., Soares, F. A. A., de Lourenço Marques, L., & de Campos, M. M. A. (2018). Molecular docking, and anti-biofilm activity of gold-complexed sulfonamides on Pseudomonas aeruginosa. Microbial Pathogenesis, 125, 393–400. https://doi.org/10.1016/j.micpath.2018.10.004
- Nardini, M., Lang, D. A., Liebeton, K., Jaeger, K. E., & Dijkstra, B. W. (2000). Crystal structure of Pseudomonas aeruginosa lipase in the open conformation: The prototype for family I. 1 of bacterial lipases. The Journal of Biological Chemistry, 275(40), 31219–31225. https://doi.org/10.1074/jbc.M003903200
- Paris, R. D., Quevedo, C. V., Ruiz, D. D., Souza, O. d., & Barros, R. C. (2015). Clustering molecular dynamics trajectories for optimizing docking experiments. Computational Intelligence and Neuroscience, 2015, 1–9. https://doi.org/10.1155/2015/916240
- Phillips, J. L., Colvin, M. E., & Newsam, S. (2011). Validating clustering of molecular dynamics simulations using polymer models. BMC Bioinformatics, 12(1), 1–23. https://doi.org/10.1186/1471-2105-12-445
- Pospıšilová, M., Polášek, M., & Jokl, V. (2001). Determination of ambroxol or bromhexine in pharmaceuticals by capillary isotachophoresis. Journal of Pharmaceutical and Biomedical Analysis, 24(3), 421–428. https://doi.org/10.1016/s0731-7085(00)00448-9
- Rajkumari, J., Borkotoky, S., Reddy, D., Mohanty, S. K., Kumavath, R., Murali, A., Suchiang, K., & Busi, S. (2019). Anti-quorum sensing and anti-biofilm activity of 5-hydroxymethylfurfural against Pseudomonas aeruginosa PAO1: Insights from in vitro, in vivo and in silico studies. Microbiological Research, 226, 19–26. https://doi.org/10.1016/j.micres.2019.05.001
- Rocha, A. J., de Oliveira Barsottini, M. R., Rocha, R. R., Laurindo, M. V., de Moraes, F. L. L., & da Rocha, S. L. (2019). Pseudomonas aeruginosa: Virulence factors and antibiotic resistance genes. Brazilian Arch Biol Technol, 62, 1–15.
- Roos, K., Wu, C., Damm, W., Reboul, M., Stevenson, J. M., Lu, C., Dahlgren, M. K., Mondal, S., Chen, W., Wang, L., Abel, R., Friesner, R. A., & Harder, E. D. (2019). OPLS3e: Extending force field coverage for drug-like small molecules. Journal of Chemical Theory and Computation, 15(3), 1863–1874. https://doi.org/10.1021/acs.jctc.8b01026
- Rosenau, F., & Jaeger, K. E. (2000). Bacterial lipases from Pseudomonas: Regulation of gene expression and mechanisms of secretion. Biochimie, 82(11), 1023–1032. https://doi.org/10.1016/s0300-9084(00)01182-2
- Röttig, A., Wenning, L., Bröker, D., & Steinbüchel, A. (2010). Fatty acid alkyl esters: Perspectives for production of alternative biofuels. Applied Microbiology and Biotechnology, 85(6), 1713–1733. https://doi.org/10.1007/s00253-009-2383-z
- Sherman, W., Day, T., Jacobson, M. P., Friesner, R. A., & Farid, R. (2006). Novel procedure for modeling ligand/receptor induced fit effects. Journal of Medicinal Chemistry, 49(2), 534–553. https://doi.org/10.1021/jm050540c
- Stefanucci, A., Dimmito, M. P., Zengin, G., Luisi, G., Mirzaie, S., Novellino, E., & Mollica, A. (2019). Discovery of novel amide tripeptides as pancreatic lipase inhibitors by virtual screening. New Journal of Chemistry, 43(7), 3208–3217. https://doi.org/10.1039/C8NJ05884A
- Talebi, M., Minai-Tehrani, D., Fazilati, M., & Minai-Tehrani, A. (2018). Inhibitory action of dicyclomine on lipase activity, kinetics and molecular study. International Journal of Biological Macromolecules, 107(Pt B), 2422–2428. https://doi.org/10.1016/j.ijbiomac.2017.10.123
- Tielen, P., Kuhn, H., Rosenau, F., Jaeger, K. E., Flemming, H. C., & Wingender, J. (2013). Interaction between extracellular lipase LipA and the polysaccharide alginate of Pseudomonas aeruginosa. BMC Microbiology, 13(1), 159. https://doi.org/10.1186/1471-2180-13-159
- Vikram, V., & Gupta, S. (2019). Problematic dicyclomine use: A case report and narrative review. Asian Journal of Psychiatry, 48, 101891.