

Abstract
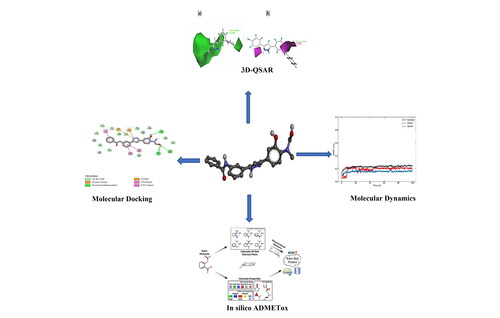
The efficacy of 40 synthesized variants of 3,5-diaryl-1H-pyrazole and spiropyrazoline’ derivatives as acetylcholinesterase inhibitors is verified using a quantitative three-dimensional structure-activity relationship (3D-QSAR) by comparative molecular field analysis (CoMFA) and molecular similarity index analysis (CoMSIA) models. In this research, different field models proved that CoMSIA/SE model is the best model with high predictive power compared to several models (Qved2 = O.65; R2 = 0.980; R2test = 0.727). Also, contour maps produced by CoMSIA/SE model have been employed to prove the key structural needs of the activity. Consequently, six new compounds have been generated. Among these compounds, M4 and M5 were the most active but remained toxic and had poor absorption capacities. While the M1, M2, M3 and M6 remained highly active while respecting ADMET's characteristics. Molecular docking results showed compound M2 better with acetylcholinesterase than compound 22. The interactions are classical hydrogen bonding with residues TYR:124, TYR:72, and SER:293, which play a critical role in the biological activity as AChE inhibitors. MD results confirmed the docking results and showed that compound M2 had satisfactory stability with (ΔGbinding = −151.225 KJ/mol) in the active site of AChE receptor compared with compound 22 (ΔGbinding = −133.375 KJ/mol). In addition, both compounds had good stability regarding RMSD, Rg, and RMSF. The previous results show that the newly designed compound M2 is more active in the active site of AChE receptor than compound 22.
Communicated by Ramaswamy H. Sarma
Low acetylcholine levels and amyloid deposits (AB) are among the leading causes of Alzheimer's disease.
Five new derivatives were designed as potential Alzheimer's disease inhibitors using a three-dimensional quantitative activity relationship (3D-QSAR).
Molecular docking and molecular dynamics results showed that the proposed compound M3 remained stable at the receptor's active site.
The inhibitor activity can be improved by replacing the R groups with hydrogen bond acceptors or larger groups or atoms.
Highlights
Disclosure statement
No potential conflict of interest was reported by the author(s).