

Abstract
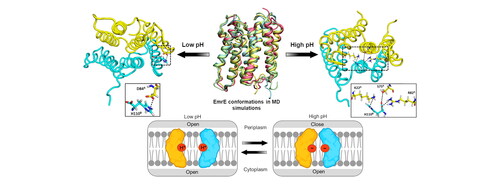
EmrE is a small multidrug resistance (SMR) pump of antiparallel topology that confers resistance to a broad range of polyaromatic cations in Escherichia coli. Atomic-level understanding of conformational changes for the selectivity of substrate and transport of a diverse array of drugs through the smallest known efflux pumps is crucial to multi-drug resistance. Therefore, the present study aims to provide insights into conformational changes during the transport through EmrE transporter at different pH. Molecular dynamics simulations have been carried out on the complete structure of EmrE in the absence of substrate. Computational analyses such as secondary structure, principal component, dynamic cross-correlation matrix, and hydrogen bond calculations have been performed. Analysis of MD trajectories in this study revealed pH-dependent interactions that influenced the structural dynamics of EmrE. Notably, at high pH, Glu14 and Tyr60 in both monomers formed electrostatic interactions, while these interactions decreased significantly at a low pH. Interestingly, a kink at helix 3 (H3) and dual open conformation of EmrE at low pH were also observed in contrast to a closed state discerned towards the periplasmic side at high pH. Significant interactions between C-terminal residues and residues at the edge of H1 & Loop1 and H3 & Loop3 were identified, suggesting their role in stabilizing the closed conformation of EmrE at the periplasmic end under high pH conditions. The present study enhances our understanding of EmrE’s conformational changes, shedding light on the pH-dependent mechanisms that are likely to impact its function in multi-drug resistance.
Communicated by Ramaswamy H. Sarma
Authors’ contributions
MK and BS conceived and designed the study. MK interpreted, analyzed MD simulation data and wrote the manuscript. PA and SC gave suggestions for the analysis of data and helped in reviewing the manuscript. BS supervised, guided the whole work, and reviewed the manuscript.
Acknowledgments
MK is grateful to the University Grants Commission (UGC) for her research fellowship. SC acknowledges the Council of Scientific and Industrial Research (CSIR) in New Delhi, India, for providing research fellowship. PA expresses gratitude to the Indian Council of Medical Research (ICMR) for the ICMR Research Associate fellowship. Authors are thankful to Dr. Gurprit Sekhon for his assistance in analyzing Molecular Dynamics simulation data.
Disclosure statement
No potential conflict of interest was reported by the author(s).