

Abstract
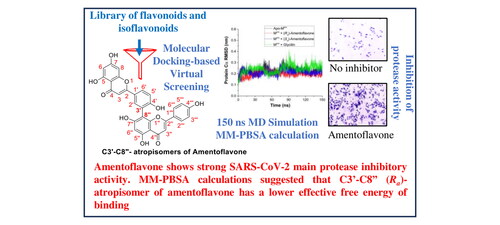
Small-molecule inhibitors of SARS-CoV-2 Mpro that block the active site pocket of the viral main protease have been considered potential therapeutics for the development of drugs against SARS-CoV-2. Here, we report the identification of amentoflavone (a biflavonoid) through docking-based virtual screening of a library comprised of 231 compounds consisting of flavonoids and isoflavonoids. The docking results were further substantiated through extensive analysis of the data obtained from all-atom 150 ns MD simulation. End-state effective free energy calculations using MM-PBSA calculations further suggested that (Ra)-amentoflavone (C3’-C8’’-atropisomer) may show a greater binding affinity towards the Mpro than (Sa)-amentoflavone. In vitro cytotoxicity assay established that amentoflavone showed a high CC50 value indicating much lower toxicity. Further, potent inhibition of the Mpro by amentoflavone was established by studying the effect on HEK293T cells treated with SARS-CoV-2 Mpro expressing plasmid.
Communicated by Ramaswamy H. Sarma
Acknowledgements
P.B. and A.M. acknowledge Mrinalini Datta Mahavidyapith for the infrastructural support. Authors thank Prof. B. Chaubey, Department of Botany, University of Calcutta, for providing the laboratory facilities for conducting in vitro studies.
Authors’ contribution
P.B.—Conceptualization, Computational studies, Data Analysis, Supervision, Manuscript writing; A.M.—In vitro studies and assisted in drafting results obtained from the in vitro experiments. All the authors have read and agreed to the final version of the manuscript.
Disclosure statement
No potential conflict of interest was reported by the author(s).