
Abstract
Pigment epithelium-derived factor (PEDF) could bind to vascular endothelial growth factor receptor 2 (VEGFR2) and inhibit its activation induced by VEGF. But how PEDF affects VEGFR2 pathway is still poorly understood. In this study, we elucidated the precise mechanism underlying the interaction between PEDF and VEGFR2, and subsequently corroborated our findings using a rat AMI model. PEDF prevented endocytosis of VE-cadherin induced by hypoxia, thereby protecting the endothelium integrity. A three-dimensional model of the VEGFR2-PEDF complex was constructed by protein-protein docking method. The results showed that the VEGFR2-PEDF complex was stable during the simulation. Hydrogen bonds, binding energy and binding modes were analyzed during molecular dynamics simulations, which indicated that hydrogen bonds and hydrophobic interactions were important for the recognition of VEGFR2 with PEDF. In addition, the results from exudation of fibrinogen suggested that PEDF inhibits vascular leakage in acute myocardial infarction and confirmed the critical role of key amino acids in the regulation of endothelial cell permeability. This observation is also supported by echocardiography studies showing that the 34mer peptide sustained cardiac function during acute myocardial infarction. Besides, PEDF and 34mer could inhibit the aggregation of myofiber in the heart and promoted the formation of a dense cell layer in cardiomyocytes, which suggested that PEDF and 34mer peptide protect against AMI-induced cardiac dysfunction. These results suggest that PEDF inhibits the phosphorylation of downstream proteins, thereby preventing vascular leakage, which provides a new therapeutic direction for the treatment of acute myocardial infarction.
Communicated by Ramaswamy H. Sarma
Introduction
Microvascular damage and dysfunction induced by hypoxia are considered to contribute to the no-reflow phenomenon in obstructed arteries during acute myocardial infarction (AMI) (Hausenloy et al., Citation2019; Ito, Citation2006). Ischemia and hypoxia resulting from AMI lead to gaps between adjacent endothelial cells and increase vascular permeability (Contessotto & Pandit, Citation2021). These pathological changes of microvascular endothelium render it difficult to restore blood supply even if the obstructed blood vessels are recanalized (Torp et al., Citation2022). Therefore, treatments that minimize microvascular damage and while retaining its structure and function to prevent the no-reflow phenomenon represent an unmet medical need (Hausenloy et al., Citation2019; Kloner et al., Citation2018).
Previous studies have shown that VE-cadherin is a transmembrane glycoprotein that forms adherent junctions through aggregation (Juettner et al., Citation2019). Vascular endothelial growth factor receptor 2 (VEGFR2) is a membrane receptor that is involved in regulating endothelial junctions and increasing vascular permeability (Galaup et al., Citation2012; Rahman et al., Citation2016). A protein complex of VEGFR2 with VE-cadherin stabilizes the distribution of VE-cadherin on endothelial cell membranes and protects adherent junctions (Corada et al., Citation1999). Dissociation of the VEGFR2/VE-cadherin complex results in endocytosis of VE-cadherin and destabilization of adherent junctions (LeBlanc et al., Citation2019). The structure of adherent junctions in the endothelial cells plays a crucial role in regulating vascular permeability, inhibiting extravasation of blood components and stabilizing endothelial structure. Therefore, stabilizing the VEGFR2/VE-cadherin complex maintains the adherent junctions which is crucial to minimize microvascular damage. However, the expression of VEGF is rapidly increased during AMI, prompting the endocytosis of VE-cadherin into a clathrin-containing endosomal compartment (Liu et al., Citation2020). This process is initiated by the activation of the p-VE-cadherin Y685 by VEGFR-2 through the Src-dependent phosphorylation (Weis et al., Citation2004), which suggests that inhibition of VEGF binding to VEGFR may be an effective way to prevent vascular leakage (Gavard & Gutkind, Citation2006; Johnston et al., Citation2015).
Pigment epithelium-derived factor (PEDF) is a secretory glycoprotein with a highly conserved structure, known to regulate angiogenesis and development (He et al., Citation2015). It is reported that PEDF overexpression significantly reduced myocardial infarct size and improved cardiac function (Gao et al., Citation2014; Zhang, Wang, et al., Citation2015). VEGF stimulation promotes the rapid endocytosis of VE-cadherin, thereby disrupting the endothelial barrier function via the recruitment of β-arrestin2 to serine-phosphorylated VE-cadherin (Gavard & Gutkind, Citation2006). Nevertheless, the relationship between PEDF and VEGFR2 pathway is also still poorly understood, and the binding site between PEDF and VEGFR2 remains unclear.
Here we show that PEDF can inhibit the sequential phosphorylation of VEGFR2, Src and VE-cadherin. Furthermore, protein-protein docking show that the hydrogen bonds together with the hydrophobic interactions are crucial for the recognition of VEGFR2 by PEDF as well as maintaining the stability of the PEDF-VEGFR2 complex. Besides, stimulation with peptides containing key amino acids leads to the regulation of phosphorylation, and echocardiography studies also support that both PEDF and peptides can prevent vascular leakage. Thus, these results provide a new direction for the treatment of AMI.
Materials and methods
All experiments conducted in this research strictly followed the National Institutes of Health (NIH Publication, 8th Edition, 2011) guidelines on the use of laboratory animals. The Eighth Affiliated Hospital of Sun Yat-sen University Committee on Animal Care granted approval for the animal care and experimental protocols.
Animal feeding and treatment
Sprague-Dawley male rats (weighing ≈ 250 ± 10 g, at 8–10 wk of age) were purchased from the Charles River Co., Ltd, (Beijing, China). Rats were housed in a controlled environment (humidity, 50–60%). A total of three rats were housed per cage and were maintained at room temperature under a 12-h light/dark cycle, and rats were provided free access to food and water.
Preparations of lentivirus
Recombinant lentiviral vectors carrying PEDF gene (PEDF-LVs; Shanghai GeneChem Co., Ltd, Shanghai, China) were prepared as described previously (Zhang, Wang, et al., Citation2015), and the concentrated titer of virus suspension was 2 × 1012 TU/L. Recombinant lentiviral vectors carrying PEDF peptides (44–77, 201–262, 280–291, and 327–387) (34mer-LVs, 62mer-LVs, 64mer-LVs, and 61mer-LVs) were prepared as described previously (Benskey & Manfredsson, Citation2016). Briefly, Prepare the required amount of plasmid DNA (for each respective plasmid) using a standard plasmid DNA preparation method. DNA must be endotoxin-free. Prepare PEI transfection reagent. Heat 200 mL of dH2O to 80 °C in a beaker on a stirring hotplate. Add 80.75 mg of PEI to the beaker and stir with a stir bar. Stir at 80 °C until the PEI is completely dissolved. Adjust pH to 8.0 with hydrochloric acid. Adjust to 250 mL with dH2O. Filter sterilize with a 0.22 μm filter unit. On day one, human embryonic kidney 293 T cells (ATCC) were cultured in Dulbecco’s modified Eagle’s medium (Gibco) supplemented with 10% fetal bovine serum (Gibco), 100 units/mL penicillin, 100 mg/mL streptomycin (Invitrogen) and maintain in 5% CO2 at 37 °C until 90% confluency. On day 2, Prepare the transfection mixture. a transfection mix was made as the following: a solution of 500uL was first prepared consisting of 1.25 μg of shuttle plasmid pMD2.G, 3.75 μg of packaging plasmid psPAX2, 5 μg of transfer expression plasmid DNA, and 125 μl of 2 mM CaCl2 in deionized distilled water; CaCl2/DNA was then added dropwise while vortexing to equal volume of 2xHBS for a total of 1 mL. This mix was added to the dish, and the cells maintained in 5% CO2 at 37 °C. The packaged recombinant lentiviruses were harvested from the supernatant of cell cultures at 48h post-transfection.
Animal model and intra-myocardial gene delivery
Myocardial ischemia was induced by ligating the left anterior descending (LAD) coronary artery (Maczewski & Mackiewicz, Citation2008). SD rats were sedated with sodium pentobarbital (60 mg/kg) and the anesthesia was maintained with 1% isoflurane. After adequate anesthesia, the animals were intubated with a 14-gauge polyethylene catheter and ventilated with room air using a small-animal ventilator (R415, RWD Co., Ltd, Shenzhen, China). The left thoracotomy was performed through the fourth intercostal space and PEDF-LVs/34mer-LVs/62mer-LVs/64mer-LVs/61mer-LVs (2 × 107 TU) in 20 μL enhanced infection solution was delivered with a 20-μL syringe and 25-gauge needle into myocardium along the LAD coronary artery. After closing chest cavity, the animals gradually resumed spontaneous respiration and then analgesics were administered. To better illustrate the results, the rats were randomly divided into four groups: Sham, Control, Vector and PEDF/peptides. Sham operated rats underwent the surgical procedure, excluding LAD ligation. The rats were injected with 20 μL enhanced infection solution in control group. Recombinant lentiviral vectors without any genes were used in vector group. And the rats were injected with PEDF-LVs or peptides-LVs in experimental group. There were six rats in sham group and ten rats in each of the other groups.
Masson’s trichrome (MTC) staining
MTC staining as in our previous study (Zhang et al., Citation2017). Briefly, Hearts were collected in each group, then fixed in 4% paraformaldehyde. All hearts were embedded in paraffin and sliced into 4 μm sections. Subsequently, MTC stainin was porformed by kits (Fuzhou Maixin Biotech Co., Ltd., China).
Wheat germ agglutinin (WGA)
As in our previous study (Qiu et al., Citation2022), WGA (L4895, Sigma) staining were performed according to the manufacturer’s instructions. The slices of the heart from each group were placed on a slide, washed three times with PBS buffer, soaked in 0.1% Triton X-100 buffer for 10 min, and rinsed three times with PBS buffer. Then, WGA fluorescent marker was incubated at 4 °C for 1 h, and DAPI dye was incubated at room temperature for 10 min to label the cell nucleus with blue fluorescence. The left ventricle was observed under a microscope and representative pictures were taken.
Cell culture and treatment
Rat Cardiac Microvascular Endothelial Cells (Cell Biologics, #RN-6024) were used between the third and fifth passage and cultured in endothelial cell medium (ECM, ScienCell, #1001) supplemented with 5% fetal bovine serum (FBS, ScienCell, #0025), 1% endothelial cell growth supplement (ScienCell, #1052) and 1% penicillin/streptomycin solution and cultured at 37 °C in a humidified atmosphere containing 5% CO2. For hypoxia, cells were cultured in ECM without FBS supplement and in a tri-gas incubator (Heal Force, Shanghai, China) saturated with 1% O2 and 5% CO2 at 37 °C.
Western blotting
The heart tissues from animal models were harvested for extracting proteins. The equivalent amount of protein with gel-loading buffer was separated with a 10% gradient SDS-PAGE gel. The separated proteins were transferred to PVDF membrane. The membrane was blocked in 5% non-fat milk for 1 h and incubated with primary antibodies for 1 h, washed by blocking buffer, and then incubated with the secondary antibody. After washing with blocking buffer and 0.1% TBST, the protein bands were visualized by Odyssey Infrared imaging system (Li-Cor Biosciences, Lincoln, USA), and the densities of bands were determined by Image J (NIH, Bethesda, USA). β-actin is the internal control protein. The primary antibodies used in this study were anti-PEDF (DF6547, Affinity), anti-6X His tag (ab18184, Abcam), anti-VE Cadherin (ab205336, Abcam), anti-p-VE Cadherin (ab119785, Abcam), anti-VEGF Receptor 2 (ab2349, Abcam), anti-phospho-VEGF Receptor 2 (AF3281, Affinity), anti-Src (ab133283, Abcam), and anti-phospho-Src (ab185617, Abcam).
Co-immunoprecipitation
Heart tissues from animal models were lysed in immunoprecipitation lysis buffer with protease inhibitor. Cell debris was removed after centrifugation at 12,000 rpm for 15 min, and the collected supernatant was incubated with primary antibodies at 4 °C for 2 h. Protein A/G beads were added into the mixture and then rock gently overnight at 4 °C. Immunoprecipitates were washed with IP buffer at least three times. The bound proteins were eluted from the beads with SDS sample buffer and supernatants were analyzed by SDS-PAGE Western blot.
Immunofluorescence
In order to study the effect of reduced vascular leakage, fibrinogen leakage was detected by immunofluorescence. After harvest, hearts were sliced into paraffin sections (4 μm), and the sections were fixed with 4% paraformaldehyde for 15 min, programmed dewaxing and antigen retrieval, permeabilized with triton X-100 (0.1%), and blocked with solution containing 5% bovine serum before applying the primary antibody. Sections were incubated respectively with anti-CD31 (ab24590, 1:200, Abcam) and anti-fibrinogen alpha chain (ab92572, 1:200, Abcam) for 12 h in 4 °C. Secondary antibodies (A21207; 1:200; Life Technologies, Carlsbad, CA) and Alexa Fluor 488 goat anti-mouse immunoglobulin G (A11001; Life Technologies) were incubated subsequently under light-protected conditions for 1 h at room temperature. Nuclei were stained with 40,6-diamidino-2-phenylindole (KGA215–10; KeyGEN BioTECH Corp., Ltd, Nanjing, China) in the end. After final washing, coverslips were mounted on slides using 50% glycerin. Then, sections were observed under a fluorescence microscope (Olympus, Tokyo, Japan) or confocal laser scanning microscope (Olympus).
Molecular dynamics (MD) simulations
The molecular dynamics analysis was performed by Sichuan Mode Technology Co., Ltd (China). In order to obtain a stable binding conformation of VEGFR2-PEDF and VEGFR2-VEGF, the molecular dynamics (MD) simulation method was used to simulate the structure of the complex. The initial structures of MD simulation is the VEGFR2-PEDF and VEGFR2-VEGF complex structure obtained by docking, and the simulation uses the Amber 18 software package, the force field is the Amber14SB (Maier et al., Citation2015) force field, and the force field parameters of proteins are all fitted based on experimental values. The simulation temperature is 300 K, pH is 7.0, and the solvent is TIP3P water model. First, add a cubic water box with a radius of 1.0 nm around the solute, and then fill in the water solvent. Before the MD simulation, two energy optimizations were carried out separately for the system: first constraining the solute and optimizing the water solvent with the steepest descent method and the conjugate gradient method for 5000 steps; then removing the constraint force and optimizing the whole system with the steepest descent method and the conjugate gradient method for 5000 steps. The MD simulation process was also carried out in two steps: the first step was a 100 ps constrained solute MD simulation, in which the temperature of the system was gradually increased from 0 to 300 K; the second step was a 50 ns unconstrained isothermal MD simulation at a constant temperature of 300 K. During the simulation, the SHAKE algorithm was used to constrain the bond length, and the integration step was 2 fs. Each 10 ps, one conformation was collected, a total of 5000 conformations were collected, and the dynamic process of the MD simulation was monitored by VMD software package.
Echocardiography
Echocardiography was conducted under sedation by sodium pentobarbital (30 mg/kg, intraperitoneally), as described previously (Lu et al., Citation2016). Left ventricle heart function was detected by echocardiography (SiliconWave 30, KOLO medical technology Co., Ltd, China). Briefly, it was performed at the end of 4 week after induction of AMI. Two dimensional–guided M-mode echocardiography was used to determine left ventricular chamber volume at systole and diastole and contractile parameters, such as left ventricular end-diastolic dimension (LVEDD), left ventricular end-systolic dimension (LVESD), left ventricular end-diastolic volume (LVEDV), left ventricular end-systolic volume (LVESV) stroke volume (SV), and heart rate (HR). Left ventricular fractional shortening (LVFS) was calculated as: FS (%) = [(LVEDD—LVEDS)/LVEDD] × 100%. Left ventricular ejection fraction (LVEF) was calculated as: EF (%) = [(LVEDV − LVESV)/LVEDV] × 100%. Cardiac output (CO) was calculated as: SV × HR.
Statistical analysis
The data were analyzed by SPSS (version 22.0.0, SPSS Inc., Chicago, IL, United States) or Prism (version 8.3.0, GraphPad Software, LLC) and presented as the mean ± SD. A two-tailed independent sample t test was used to compare the differences between two groups. One-way ANOVA was used to compare the differences between three or more groups. p < 0.05 was considered statistically significant.
Results
PEDF inhibited the phosphorylation of downstream proteins
Animal models were established to examine the effect of PEDF on VEGF and the related downstream signaling proteins in the settings of AMI. The expression levels of PEDF and VEGF in AMI were first measured during the course of 48 h of AMI (). PEDF significantly decreased in 3 h after AMI and reached its lowest point at 48 h, while the expression of VEGF gradually increased and showed significant differences in 24 h and 48 h after AMI. Therefore, the hearts of rats suffering AMI after 24 h were used for subsequent experiments. To determine the effects of PEDF on VEGF and VEGFR2, phosphorylation of VEGFR2 and the downstream proteins was analyzed. The expression of PEDF increased approximately six-fold after transfection of LV. The total expression level of VEGFR2 remained unchanged with overexpression of PEDF, while the level of p-VEGFR2 Y949 decreased. PEDF did not affect VE-cadherin expression, but induced a significant decrease of p-VE-cadherin Y685 and p-Src (). To further detect the expression of VE-cadherin in vitro, we collected the hypoxia cells and the content of VE-cadherin was detected by western blot. By comparing the expression of the proteins in different conditions, we can conclude that hypoxia environment had little effect on expression of VE-cadherin, whereas hypoxia could affect p-VE-cadherin Y685. (Figure S1A). The observation that phosphorylation level of Src and VEGFR Y949 reduced by PEDF suggested the important role in preventing vascular leakage.
Figure 1. PEDF inhibited the phosphorylation of downstream proteins. (A) Western blot determination of PEDF/VEGF protein expression in AMI. (B) Time-course analysis of protein expression. *p < 0.05, ***p < 0.001, #p < 0.05,##p < 0.01 versus the respective sham groups, n = 3. (C) The inhibition to phosphorylation was detected by western blot when PEDF is overexpressed. The expression of proteins (D) PEDF, (E) p-VEGFR2, (F) p-Src, and (G) p-VE-cadherin were analyzed. **p < 0.01, ***p < 0.001 versus the sham group, and ##p < 0.01, ###p < 0.001 versus the control group, n = 3.
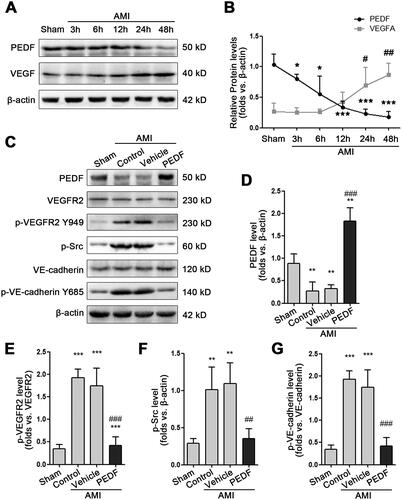
In order to confirm the effect of PEDF on endothelium integrity in vivo, we detected the fluorescence of VE-cadherin and CD31. In Sham, VE-cadherin and CD31 presented linear distribution, indicating the endothelium integrity of cells. In the control and vehicle groups, the distribution of VE-cadherin and CD31 was dispersed and the fluorescence signal suggested that endocytosis occur and thus endothelial integrity is damaged. However, VE-cadherin dispersion is inhibited by PEDF, and VE-cadherin and CD31 were still linear distribution when PEDF added. This result supports the conclusion that PEDF inhibits vascular leakage (). In addition, we examined the distribution of VE-cadherin under different experimental conditions. In normal groups, VE-cadherin is mainly concentrated on the membrane, but in the control and vehicle groups, VE-cadherin concentrated in the endochylema, but the addition of PEDF will inhibit endocytosis of VE-cadherin, thus VE-cadherin was still concentrated on the membrane (Figure S1B). We further detected the exudation of fibrinogen in the ischemic area, and a small amount of fibrinogen could be detected outside the vessel after AMI, while PEDF reduces it (). These data indicated that PEDF can maintain the distribution of VE-cadherin on the cell membrane, thereby protecting the endothelium integrity.
Figure 2. PEDF protect endothelium integrity. (A) Fluorescence imaging of VE-cadherin and CD31. (B) PEDF reduces blood vessel leakage. Fluorescence imaging of fibrinogen after incubation with PEDF.
PEDF interacts with VEGFR2
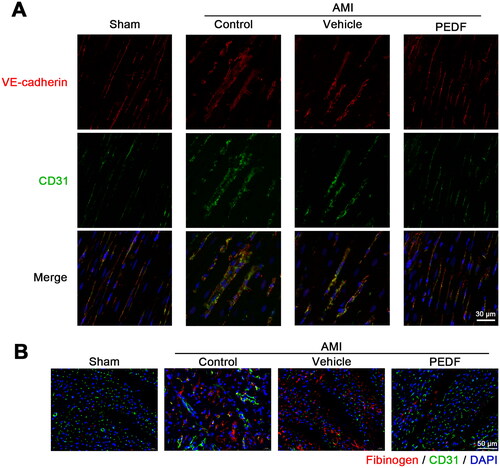
Binding of VEGF to VEGFR2 triggers phosphorylation of VE-cadherin (Weis et al., Citation2004). As shown in , PEDF does not reduce VEGF levels but inhibits p-VEGFR2 Y949. Therefore, we speculate that there is an interaction between PEDF and VEGFR2. Co-immunoprecipitation (Co-IP) was used to determine whether PEDF could compete with VEGF by binding to VEGFR2. VEGFR2 coimmunoprecipitated with VEGF when vehicle-infected myocardium cells in AMI, but PEDF was barely detected. Lentiviral infection with PEDF-LVs increased PEDF expression, and in this circumstance the interaction of VEGFR2 with VEGF was much more limited than the interaction with PEDF, indicating that PEDF may prevent the binding of VEGF to its receptor (). Moreover, PEDF did not interact with VEGF even when overexpressed in AMI (). In contrast, PEDF was able to precipitate VEGFR2 effectively even at low levels in the control groups (). Thus, these results are in agreement with the hypothesis that PEDF could bind to VEGFR2, thereby inhibiting the interaction between VEGF and VEGFR2.
Figure 3. PEDF inhibits VEGF via binding to VEGFR2. (A) Heart tissue lysate was Co-IPed with anti-VEGFR2 antibody or rabbit IgG, and then analyzed by Western blotting with anti-PEDF and anti-VEGF antibody. Densitometry analysis of PEDF (B) and VEGF (C) levels versus the sham group. (D) Heart tissue lysate was Co-IPed with anti-PEDF antibody or rabbit IgG, and then analyzed by Western blotting with anti-VEGFR2 and anti-VEGF antibody. (E) Densitometry analysis of VEGFR2 level versus the sham group. ##p < 0.01 versus the control group, n = 3.
Molecular recognition of PEDF with VEGFR2
To elucidate the protein-protein structure and interaction between PEDF and VEGFR2, a molecular docking analysis was performed. Both parameters are important to measure the stability of the system. As the sum of all the atomic deviations between the conformation and the target conformation at a certain moment, the root mean square deviation (RMSD) of the Cα atom in the VEGFR2-PEDF system changed dynamically with time, but became stable after 30 ns, with the RMSD value at 0.418 ± 0.032 nm (Figure S2a). The radius of gyration (Rg) as an indicator of the degree of looseness of the system fluctuated around an average value of 2.756 ± 0.014 nm during the MD simulation (Figure S2b). Based on the RMSD and Rg, it can be concluded that the VEGFR2-PEDF complex system reached a steady state during the MD simulation.
To determine the flexibility of amino acid residues in proteins, the root mean square fluctuation (RMSF) of amino acid residues in VEGFR2 and PEDF proteins was detected (Figure S3). Only three regions of amino acids exhibited better flexibility, including C200-Q210 in VEGFR2, K220-K225 and G370-A380 in PEDF. According to the 3D structure of the proteins, these three regions were located on the surface with segment random coils, which were subjected to strong solvent forces during MD simulation. Thus, the flexible regions of the protein in the complex structure are distributed mainly in these positions.
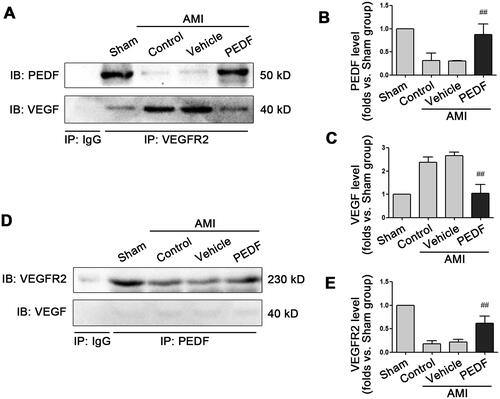
Given the important role of hydrogen bonds in maintaining the secondary structure and function of the proteins, the number of hydrogen bonds between VEGFR2 and PEDF in the simulation process was analyzed (). The number of hydrogen bonds tended to stabilize at about six after 30 ns with a stable distribution of hydrogen bonds between VEGFR2 and PEDF (Table S1), and most were distributed between charged amino acids, such as Glu331 of PEDF and Arg275 of VEGFR2. Furthermore, the binding of PEDF or VEDF to VEGFR2 was compared. The average number of hydrogen bonds between VEGFR2 and PEDF was 5.79, while the average number of hydrogen bonds between VEGFR2 and VEGF was 4.08 (). To determine the magnitude of intermolecular affinity between VEGFR2 and PEDF/VEGF, the binding energy between the 2 proteins during MD simulation was assessed. The binding energy of VEGFR2 with ligand was basically stable after 30 ns, with the average binding energies of VEGFR2-VEGF and VEGFR2-PEDF at −441.91 ± 55.35 kJ/mol and −2472.05 ± 194.16 kJ/mol, respectively (). These results imply that VEGFR2 can bind to PEDF stably during MD simulation, and the affinity between PEDF and VEGFR2 was stronger than that of VEGF with VEGFR2.
Figure 4. Molecular docking analysis. (A) The number of hydrogen bonds between VEGFR2 and VEGF (Blue line) or PEDF (Red line) during molecular dynamics simulation. (B) Binding energy between VEGFR2 and VEGF (Blue line) or PEDF (Red line) during molecular dynamics simulation. (C) Binding model of VEGFR2 docking with PEDF protein. Ribbon in green represents VEGFR2, grey represents PEDF, purple and blue linkers represent amino acid residues originating from VEGFR2 and PEDF, respectively, and green dashed lines represent hydrogen bonding interactions.
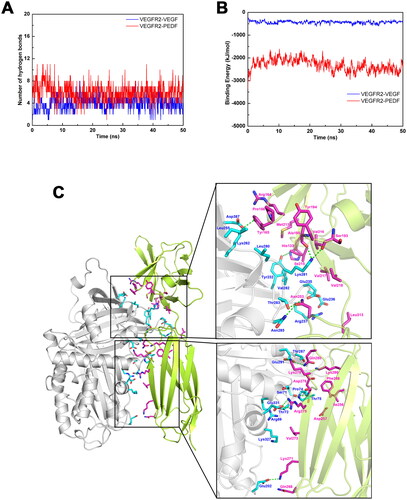
In order to further study the binding mode of VEGFR2 with PEDF, the distribution of amino acid residues at the interface between the two proteins was also analyzed using binding model of protein docking (). Charged amino acid residues in PEDF include Arg69, Ser71, Thr72, Pro74, Thr75, Asp201, Glu202, Tyr232, Glu235, Glu236, Arg237, Leu255, Lys262, Leu280, Lys281, Val283, Thr284, Asn285, Thr287, Glu291, Lys327, Glu331 and Asp387 (blue amino acids in ). In VEGFR2, the main charged amino acids are His133, Tyr137, Arg164, Tyr165, Pro166, Ser193, Tyr194, Ala195, Met213, Ile215, Val216, Val217, Val218, Asn253, Ile256, Asp257, Gln268, Lys271, Val273, Asn284, Arg275, Asp276, Lys278, Gln280, Lys286, Phe288 and Leu313 (purple amino acids in ). In this regard, it can be inferred that hydrogen bonding and electrostatic interaction are the key drivers of molecular recognition between VEGFR2 and PEDF. The presence of some hydrophobic amino acids (such as Leu and Val) in the binding interface implies that both hydrophilic and hydrophobic interactions may play an important role in the molecular recognition between VEGFR2 and PEDF (Figure S4).
The 34mer peptide prevent fibrinogen efflux during AMI
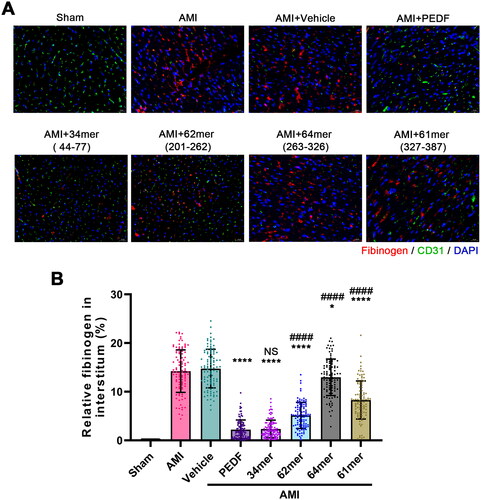
According to the protein-protein docking model, certain amino acids in PEDF play an important role in the recognition to VEGFR2. To address whether peptides containing these amino acids exhibit similar functions to PEDF, peptides 34mer (aa44-77), 62mer (aa201-262), 64mer (aa263-326), 61mer (aa327-387) were constructed to analyze the function (). The exudation of fibrinogen in the ischemic area indicated the vascular leakage. A large amount of fibrinogen was detected outside the vessels in the AMI and vehicle groups, whereas the overexpression of PEDF reduced the efflux of fibrinogen. The peptides showed varying abilities to inhibit the efflux of fibrinogen. Similar to PEDF, the 34mer-expressing cells displayed a significant inhibition of fibrinogen exudation when compared against the AMI group. The 62mer and 61mer peptides also inhibited the efflux of fibrinogen when compared against the AMI group but not as significant as compared to PEDF. 64mer barely inhibits fibrinogen efflux (). These results indicated the critical role of key amino acids similar to PEDF function in regulation of fibrinogen extrusion.
Figure 5. Peptides reduce blood vessel leakage. (A) Fluorescence imaging of fibrinogen after incubation with PEDF or peptides. (B) Relative fibrinogen in each group. *p < 0.05, ****p < 0.0001 vs the AMI group, and ####p < 0.0001 vs the AMI + PEDF group, sham group n = 6 and the other each group n = 10, ten fields of view per rat.
The 34mer peptide inhibits phosphorylation of downstream proteins
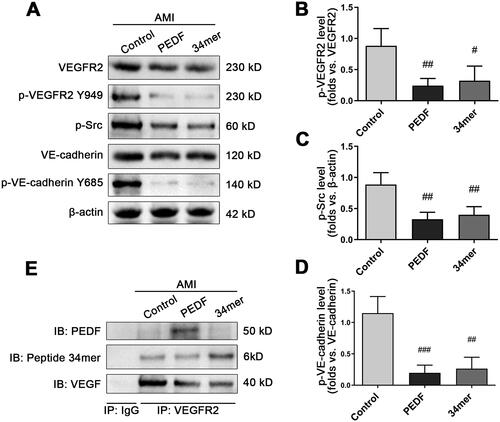
As the 34mer peptide has been demonstrated to be the most potent in preventing fibrinogen extrusion during AMI in a similar fashion to PEDF, we next explored whether the action of the 34mer peptide relies on the same underlying mechanism as the PEDF. Similar to PEDF, the expression of the 34mer peptide did not affect the expression of VEGFR2, but can potently block the VEGF-induced phosphorylation of VEGFR2 Y949. VEGF can trigger phosphorylation of VE-cadherin in control group, and the VE-cadherin Y685 signal was sharply reduced in response to PEDF and the 34mer peptide (). We then conducted co-IP experiments to examine whether the 34mer peptide interacts with VEGFR2. The results showed that PEDF and the 34mer peptide reciprocally co-IPed with VEGFR2 (). Together, these findings strongly indicate that key amino acids of PEDF, in this case the 34mer, is crucial for the function of the PEDF to prevent fibrinogen extrusion via inhibiting phosphorylation of downstream proteins
Figure 6. The 34mer peptide inhibited the phosphorylation of downstream proteins in a similar fashion to PEDF. (A) Representative Western blot image of phosphorylation of downstream proteins following PEDF or 34mer infection and overexpression during AMI. Densitometry analysis of (B) p-VEGFR2/VEGFR2, (C) p-Src/β-actin and (D) p-VE-cadherin Y685/VE-cadherin following infection with PEDF or 34mer peptide during AMI. (E) Heart tissue lysate was Co-IPed with anti-VEGFR2 antibody or rabbit IgG, and then analyzed by Western blotting with anti-PEDF, anti-34mer peptide and anti-VEGF antibody. #p < 0.05, ##p < 0.01 vs the control group, n = 3.
The 34mer peptide sustained cardiac function during AMI
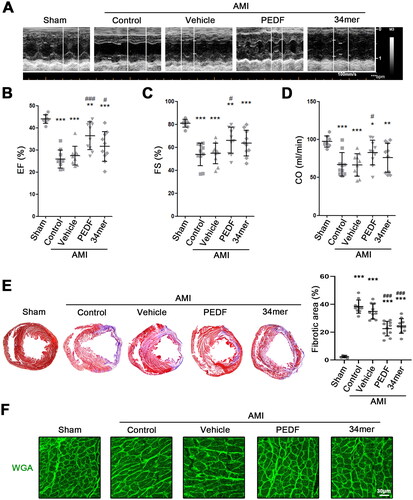
As prevention of fibrinogen extrusion and maintenance of vascular integrity sustains cardiac function during AMI, we also sought to determine whether the 34mer peptide can achieve cardioprotective benefits similar to PEDF treatment. Echocardiography was performed to assess the cardiac function in the myocardium of PEDF or the 34mer peptide-treated rats (). Rats in the sham group had normal ejection fraction (EF), fractional shortening (FS) and cardiac output (CO) (). Both control and vehicle-treated hearts, demonstrated a marked reduction in EF, FS and CO, indicating the severity of the ischemic injury. Conversely, rats treated with LVs containing PEDF demonstrated restoration of EF, FS and CO compared to the control group while the 34mer peptide restored EF when compared against the vehicle-treated group. For further pathological analysis of hearts after AMI, we performed Masson and cardiomyocyte remodeling experiments. Masson analyses showed that, in control and vehicle groups, more myofiber was observed. PEDF and 34mer could inhibit the aggregation of myofiber in the heart (). Cardiomyocyte remodeling indicated that PEDF and mer34 promoted the formation of a dense cell layer in cardiomyocytes, which suggested that PEDF and 34mer peptide protect against AMI-induced cardiac dysfunction (). Taken together, these results indicate that PEDF-overexpression protects against AMI-induced cardiac dysfunction, and confirmed that the 34mer peptide containing critical amino acids consistent with the function of PEDF.
Figure 7. The 34mer peptide sustained cardiac function during AMI. (A) Representative echocardiogram of hearts during AMI. Scatter plot of (B) ejection fraction (EF) (%), (C) fractional-shortening (FS) (%) and (D) cardiac output (CO) (ml/min) of each group. (E) MTC staining and fibrotic area analysis of hearts in each group. (F) WGA staining analysis of cardiomyocyte remodeling in each group. *p < 0.05, **p < 0.01, ***p < 0.001 versus the sham group, and #p < 0.05, ###p < 0.001 versus the control group, sham group n = 6, the other each group n = 10.
Discussion
PEDF overexpression reduces myocardial infarct size and protects myocardium against ischemia-induced necrosis after AMI (Liu et al., Citation2019), in addition to maintaining the stability of endothelial adherens junctions (AJs) and preventing the occurrence of no-reflow (Zhang et al., Citation2022). This may be due to the effect of PEDF on the phosphorylation level of VE-cadherin yet how PEDF affects the phosphorylation of downstream proteins involved in vascular permeability is still far from being fully understood. In this study, lentiviral-induced overexpression of PEDF in adult Sprague-Dawley rats was performed in myocardium along the LAD coronary artery. In the PEDF-overexpressing rats, Western blotting and Co-IP demonstrated the interaction of PEDF with VEGFR2, subsequently resulting in the decreased phosphorylation of VEGFR2, Src and VE-cadherin. PEDF represses the action of VEGF by binding to VEGFR2 via hydrogen bonding and electrostatic interaction between key amino acids. A peptide containing 34mers of the PEDF also inhibited fibrinogen extrusion, prevented the phosphorylation of downstream proteins related to vascular permeability as well as maintained cardiac function during AMI.
P-Src induces VE-cadherin phosphorylation, which leads to endocytosis of VE-cadherin and adhesion dissociation (Yuan, Huang, et al., Citation2019). PEDF inhibits p-Src, thus, the protein could reduce vascular permeability. A protein complex of VEGFR2 with VE-cadherin stabilizes the distribution of VE-cadherin on endothelial cell membrane and protects adherent junctions (Smith et al., Citation2020). However, VEGF binding to VEGFR2 causes dimerization of the receptor, which contributes to the tyrosine phosphorylation of VE-cadherin and subsequent enhanced vascular permeability (King & Hristova, Citation2019). In this study, we found that overexpression of PEDF could not affect the expression of VE-cadherin but inhibited endocytosis of VE-cadherin induced by hypoxia, which suggested that PEDF was able to protect the endothelium integrity. Our Co-IP experiments showed diminished recruitment of VEGF to VEGFR2 after exposure to high level of PEDF, while the amount of PEDF in the complex containing VEGFR2 was increased. These results strongly support the idea that PEDF treatment exerts an inhibitory control on VEGF recruitment to VEGFR2 thus protecting VEGFR2–VE-cadherin complexes from disruption and preserving vascular integrity.
Recent investigations have analyzed the interaction between VEGF and VEGFR2 by the protein-protein docking method (Leppänen et al., Citation2010; Simonovic et al., Citation2001). In our study, we observed that PEDF can bind to VEGFR2, yet the exact binding conformation between PEDF with VEGFR2 is still unknown. In this regard, we utilized the protein-protein docking method to construct a 3D model of the VEGFR2-PEDF complex which is very stable according to the RMSD and Rg values. The intermolecular interaction, such as hydrogen bonding, electrostatic interaction, hydrophilic and hydrophobic interactions not only facilitates the recognition of PEDF with VEGFR2, but also stabilizes the structure of the complex, thus affirming the stronger affinity between PEDF and VEGFR2 compared to that of VEGF with VEGFR2. During severe ischemia and anoxia, VEGF expression is rapidly increased (Chiong et al., Citation2011) causing the cardiomyocytes to lose viability and rendering reperfusion therapy less effective. Therefore, inhibition of VEGF binding to VEGFR2 via PEDF may represent a therapeutic strategy to prevent cardiomyocyte death during AMI.
Another important finding of this study is that the peptides containing key amino acids have similar functions to PEDF. Based on the molecular simulation between PEDF and VEGFR2, peptides 34mer (aa44-77), 62mer (aa201-262), 64mer (aa263-326), 61mer (aa327-387) were constructed. Data show that these peptides reciprocally co-IPed with VEGFR2 and possess the function of inhibiting fibrinogen extrusion. The mechanism of peptides exhibiting these functions is similar to PEDF in that the peptides can potently block the VEGF-induced phosphorylation of VEGFR2. In addition, similar to PEDF, the 34mer peptide also maintains cardiac function during AMI. Masson analyses showed that PEDF and 34mer could inhibit the aggregation of myofiber in the heart. Cardiomyocyte remodeling indicated that PEDF and mer34 promoted the formation of a dense cell layer in cardiomyocytes, which suggested that PEDF and 34mer peptide protect against AMI-induced cardiac dysfunction. These results confirmed the critical role of key amino acids in the regulation of endothelial cell permeability.
As in our previous studies, PEDF is a multifunctional protein that has various protective effects on ischemic myocardial cells, such as reducing apoptosis (Miao et al., Citation2018), promoting glycogen storage (Yuan, Liu et al., Citation2019), and reducing energy consumption (Qiu et al., Citation2018; Yuan, Huang, et al., Citation2019). In this study, 34mer exhibited strong anti-vascular leakage effects, but the improvement in cardiac function did not achieve the same effect as PEDF. Perhaps these cardioprotective effects against ischemia are mediated by sequences outside of the 34mer (aa44-77) region of PEDF, such as the 44mer (aa78-121) which has been reported to have direct cardioprotective effects against ischemia (Gao et al., Citation2014; Zhang, Sun, et al., Citation2015).
Conclusion
This study demonstrates that overexpression of PEDF can interact with VEGFR2, and subsequently inhibit the phosphorylation of VEGFR2, VE-cadherin and Src. The VEGFR2-PEDF complex obtained by molecular dynamics simulations is extremely stable, mediated by the presence of hydrogen bonds, hydrophilic and hydrophobic interactions between key amino acids. Synthetic peptides containing key amino acids exhibit similar function and cardioprotective properties to that of PEDF. These results put forth the implication that PEDF inhibits VEGF-induced vascular leakage during AMI and has important therapeutic values of coronary recanalization.
Ethics approval statement
All experiments conducted in this research strictly followed the National Institutes of Health (NIH Publication, 8th Edition, 2011) guidelines on the use of laboratory animals. The Eighth Affiliated Hospital of Sun Yat-sen University Committee on Animal Care granted approval for the animal care and experimental protocols.
| Abbreviations | ||
| PEDF | = | Pigment epithelium-derived factor |
| VEGFR2 | = | vascular endothelial growth factor receptor 2 |
| AMI | = | acute myocardial infarction |
| LAD | = | left anterior descending |
| LVEDD | = | left ventricular end-diastolic dimension |
| LVESD | = | left ventricular end-systolic dimension |
| LVEDV | = | left ventricular end-diastolic volume |
| LVESV | = | left ventricular end-systolic volume |
| LVFS | = | Left ventricular fractional shortening |
| RMSD | = | root mean square deviation |
| MD | = | Molecular Dynamics |
Supplemental Material
Download MS Word (1.6 MB)Disclosure statement
No potential conflict of interest was reported by the authors.
Data availability statement
The data used to support the findings of this study are available from the corresponding author upon reasonable request.
Additional information
Funding
References
- Benskey, M. J., & Manfredsson, F. P. (2016). Lentivirus production and purification. Methods in Molecular Biology (Clifton, N.J.), 1382, 107–114. https://doi.org/10.1007/978-1-4939-3271-9_8
- Chiong, M., Wang, Z. V., Pedrozo, Z., Cao, D. J., Troncoso, R., Ibacache, M., Criollo, A., Nemchenko, A., Hill, J. A., & Lavandero, S. (2011). Cardiomyocyte death: Mechanisms and translational implications. Cell Death & Disease, 2(12), e244–e244. https://doi.org/10.1038/cddis.2011.130
- Contessotto, P., & Pandit, A. (2021). Therapies to prevent post-infarction remodelling: From repair to regeneration. Biomaterials, 275, 120906. https://doi.org/10.1016/j.biomaterials.2021.120906
- Corada, M., Mariotti, M., Thurston, G., Smith, K., Kunkel, R., Brockhaus, M., Lampugnani, M. G., Martin-Padura, I., Stoppacciaro, A., Ruco, L., McDonald, D. M., Ward, P. A., & Dejana, E. (1999). Vascular endothelial-cadherin is an important determinant of microvascular integrity in vivo. Proceedings of the National Academy of Sciences of the United States of America, 96(17), 9815–9820. https://doi.org/10.1073/pnas.96.17.9815
- Galaup, A., Gomez, E., Souktani, R., Durand, M., Cazes, A., Monnot, C., Teillon, J., Le Jan, S., Bouleti, C., Briois, G., Philippe, J., Pons, S., Martin, V., Assaly, R., Bonnin, P., Ratajczak, P., Janin, A., Thurston, G., Valenzuela, D. M., … Germain, S. (2012). Protection against myocardial infarction and no-reflow through preservation of vascular integrity by angiopoietin-like 4. Circulation, 125(1), 140–149. https://doi.org/10.1161/CIRCULATIONAHA.111.049072
- Gao, X., Zhang, H., Zhuang, W., Yuan, G., Sun, T., Jiang, X., Zhou, Z., Yuan, H., Zhang, Z., & Dong, H. (2014). PEDF and PEDF-derived peptide 44mer protect cardiomyocytes against hypoxia-induced apoptosis and necroptosis via anti-oxidative effect. Scientific Reports, 4(1), 5637. https://doi.org/10.1038/srep05637
- Gavard, J., & Gutkind, J. S. (2006). VEGF controls endothelial-cell permeability by promoting the beta-arrestin-dependent endocytosis of VE-cadherin. Nature Cell Biology, 8(11), 1223–1234. https://doi.org/10.1038/ncb1486
- Hausenloy, D. J., Chilian, W., Crea, F., Davidson, S. M., Ferdinandy, P., Garcia-Dorado, D., van Royen, N., Schulz, R., & Heusch, G. (2019). The coronary circulation in acute myocardial ischaemia/reperfusion injury: A target for cardioprotection. Cardiovascular Research, 115(7), 1143–1155. https://doi.org/10.1093/cvr/cvy286
- He, X., Cheng, R., Benyajati, S., & Ma, J. X. (2015). PEDF and its roles in physiological and pathological conditions: Implication in diabetic and hypoxia-induced angiogenic diseases. Clinical Science (London, England: 1979), 128(11), 805–823. https://doi.org/10.1042/CS20130463
- Ito, H. (2006). No-reflow phenomenon and prognosis in patients with acute myocardial infarction. Nature Clinical Practice. Cardiovascular Medicine, 3(9), 499–506. https://doi.org/10.1038/ncpcardio0632
- Johnston, E. K., Francis, M. K., & Knepper, J. E. (2015). Recombinant pigment epithelium-derived factor PEDF binds vascular endothelial growth factor receptors 1 and 2. In Vitro Cellular & Developmental Biology. Animal, 51(7), 730–738. https://doi.org/10.1007/s11626-015-9884-0
- Juettner, V. V., Kruse, K., Dan, A., Vu, V. H., Khan, Y., Le, J., Leckband, D., Komarova, Y., & Malik, A. B. (2019). VE-PTP stabilizes VE-cadherin junctions and the endothelial barrier via a phosphatase-independent mechanism. The Journal of Cell Biology, 218(5), 1725–1742. https://doi.org/10.1083/jcb.201807210
- King, C., & Hristova, K. (2019). Direct measurements of VEGF-VEGFR2 binding affinities reveal the coupling between ligand binding and receptor dimerization. The Journal of Biological Chemistry, 294(23), 9064–9075. https://doi.org/10.1074/jbc.RA119.007737
- Kloner, R. A., King, K. S., & Harrington, M. G. (2018). No-reflow phenomenon in the heart and brain. American Journal of Physiology. Heart and Circulatory Physiology, 315(3), H550–H562. https://doi.org/10.1152/ajpheart.00183.2018
- LeBlanc, M. E., Saez-Torres, K. L., Cano, I., Hu, Z., Saint-Geniez, M., Ng, Y. S., & D'Amore, P. A. (2019). Glycocalyx regulation of vascular endothelial growth factor receptor 2 activity. FASEB Journal: Official Publication of the Federation of American Societies for Experimental Biology, 33(8), 9362–9373. https://doi.org/10.1096/fj.201900011R
- Leppänen, V.-M., Prota, A. E., Jeltsch, M., Anisimov, A., Kalkkinen, N., Strandin, T., Lankinen, H., Goldman, A., Ballmer-Hofer, K., & Alitalo, K. (2010). Structural determinants of growth factor binding and specificity by VEGF receptor 2. Proceedings of the National Academy of Sciences of the United States of America, 107(6), 2425–2430. https://doi.org/10.1073/pnas.0914318107
- Liu, D., Xu, H., Zhang, C., Xie, H., Yang, Q., Li, W., Tian, H., Lu, L., Xu, J.-Y., Xu, G., Liu, K., Sun, X., Xu, G.-T., & Zhang, J. (2020). Erythropoietin maintains VE-cadherin expression and barrier function in experimental diabetic retinopathy via inhibiting VEGF/VEGFR2/Src signaling pathway. Life Sciences, 259, 118273. https://doi.org/10.1016/j.lfs.2020.118273
- Liu, X., Liu, Z., Chen, J., Zhu, L., Zhang, H., Quan, X., Yuan, Y., Miao, H., Huang, B., Dong, H., & Zhang, Z. (2019). Pigment epithelium-derived factor increases native collateral blood flow to improve cardiac function and induce ventricular remodeling after acute myocardial infarction. Journal of the American Heart Association, 8(22), e013323. https://doi.org/10.1161/JAHA.119.013323
- Lu, P., Zhang, Y.-Q., Zhang, H., Li, Y.-F., Wang, X.-Y., Xu, H., Liu, Z.-W., Li, L., Dong, H.-Y., & Zhang, Z.-M. (2016). Pigment epithelium-derived factor (PEDF) improves ischemic cardiac functional reserve through decreasing hypoxic cardiomyocyte contractility through PEDF receptor (PEDF-R). Journal of the American Heart Association, 5(7), e003179. https://doi.org/10.1161/JAHA.115.003179
- Maczewski, M., & Mackiewicz, U. (2008). Effect of metoprolol and ivabradine on left ventricular remodelling and Ca2+ handling in the post-infarction rat heart. Cardiovascular Research, 79(1), 42–51. https://doi.org/10.1093/cvr/cvn057
- Maier, J. A., Martinez, C., Kasavajhala, K., Wickstrom, L., Hauser, K. E., & Simmerling, C. (2015). ff14SB: Improving the accuracy of protein side chain and backbone parameters from ff99SB. Journal of Chemical Theory and Computation, 11(8), 3696–3713. https://doi.org/10.1021/acs.jctc.5b00255
- Miao, H., Qiu, F., Huang, B., Liu, X., Zhang, H., Liu, Z., Yuan, Y., Zhao, Q., Zhang, H., Dong, H., & Zhang, Z. (2018). PKCalpha replaces AMPK to regulate mitophagy: Another PEDF role on ischaemic cardioprotection. Journal of Cellular and Molecular Medicine, 22(11), 5732–5742. https://doi.org/10.1111/jcmm.13849
- Qiu, F., Yuan, Y., Luo, W., Gong, Y. S., Zhang, Z. M., Liu, Z. M., & Gao, L. (2022). Asiatic acid alleviates ischemic myocardial injury in mice by modulating mitophagy- and glycophagy-based energy metabolism. Acta Pharmacologica Sinica, 43(6), 1395–1407. https://doi.org/10.1038/s41401-021-00763-9
- Qiu, F., Zhang, H., Yuan, Y., Liu, Z., Huang, B., Miao, H., Liu, X., Zhao, Q., Zhang, H., Dong, H., & Zhang, Z. (2018). A decrease of ATP production steered by PEDF in cardiomyocytes with oxygen-glucose deprivation is associated with an AMPK-dependent degradation pathway. International Journal of Cardiology, 257, 262–271. https://doi.org/10.1016/j.ijcard.2018.01.034
- Rahman, H. N. A., Wu, H., Dong, Y., Pasula, S., Wen, A., Sun, Y., Brophy, M. L., Tessneer, K. L., Cai, X., McManus, J., Chang, B., Kwak, S., Rahman, N. S., Xu, W., Fernandes, C., Mcdaniel, J. M., Xia, L., Smith, L., Srinivasan, R. S., & Chen, H. (2016). Selective targeting of a novel Epsin-VEGFR2 interaction promotes VEGF-mediated angiogenesis. Circulation Research, 118(6), 957–969. https://doi.org/10.1161/CIRCRESAHA.115.307679
- Simonovic, M., Gettins, P. G., & Volz, K. (2001). Crystal structure of human PEDF, a potent anti-angiogenic and neurite growth-promoting factor. Proceedings of the National Academy of Sciences of the United States of America, 98(20), 11131–11135. https://doi.org/10.1073/pnas.211268598
- Smith, R. O., Ninchoji, T., Gordon, E., André, H., Dejana, E., Vestweber, D., Kvanta, A., & Claesson-Welsh, L. (2020). Vascular permeability in retinopathy is regulated by VEGFR2 Y949 signaling to VE-cadherin. eLife, 9, e54056. https://doi.org/10.7554/eLife.54056
- Torp, M.-K., Ranheim, T., Schjalm, C., Hjorth, M., Heiestad, C. M., Dalen, K. T., Nilsson, P. H., Mollnes, T. E., Pischke, S. E., Lien, E., Vaage, J., Yndestad, A., & Stensløkken, K.-O. (2022). Intracellular complement component 3 attenuated ischemia-reperfusion injury in the isolated buffer-perfused mouse heart and is associated with improved metabolic homeostasis. Frontiers in Immunology, 13, 870811. https://doi.org/10.3389/fimmu.2022.870811
- Weis, S., Shintani, S., Weber, A., Kirchmair, R., Wood, M., Cravens, A., McSharry, H., Iwakura, A., Yoon, Y.-S., Himes, N., Burstein, D., Doukas, J., Soll, R., Losordo, D., & Cheresh, D. (2004). Src blockade stabilizes a Flk/cadherin complex, reducing edema and tissue injury following myocardial infarction. The Journal of Clinical Investigation, 113(6), 885–894. https://doi.org/10.1172/jci20702
- Yuan, Y., Huang, B., Miao, H., Liu, X., Zhang, H., Qiu, F., Liu, Z., Zhang, Y., Dong, H., & Zhang, Z. (2019). A “hibernating-like” viable state induced by lentiviral vector-mediated pigment epithelium-derived factor overexpression in rat acute ischemic myocardium. Human Gene Therapy, 30(6), 762–776. https://doi.org/10.1089/hum.2018.186
- Yuan, Y., Liu, X., Miao, H., Huang, B., Liu, Z., Chen, J., Quan, X., Zhu, L., Dong, H., & Zhang, Z. (2019). PEDF increases GLUT4-mediated glucose uptake in rat ischemic myocardium via PI3K/AKT pathway in a PEDFR-dependent manner. International Journal of Cardiology, 283, 136–143. https://doi.org/10.1016/j.ijcard.2019.02.035
- Zhang, H., Hui, H., Li, Z., Pan, J., Jiang, X., Wei, T., Cui, H., Li, L., Yuan, X., Sun, T., Liu, Z., Zhang, Z., & Dong, H. (2017). Pigment epithelium-derived factor attenuates myocardial fibrosis via inhibiting endothelial-to-mesenchymal transition in rats with acute myocardial infarction. Scientific Reports, 7(1), 41932. https://doi.org/10.1038/srep41932
- Zhang, H., Li, Z., Quan, X., Liu, X., Sun, T., Wei, T., Pan, J., Liu, Z., Wang, M., Dong, H., & Zhang, Z. (2022). Strategies to attenuate myocardial infarction and no-reflow through preservation of vascular integrity by pigment epithelium-derived factor. Human Gene Therapy, 33(5-6), 330–345. https://doi.org/10.1089/hum.2021.068
- Zhang, H., Sun, T., Jiang, X., Yu, H., Wang, M., Wei, T., Cui, H., Zhuang, W., Liu, Z., Zhang, Z., & Dong, H. (2015). PEDF and PEDF-derived peptide 44mer stimulate cardiac triglyceride degradation via ATGL. Journal of Translational Medicine, 13(1), 68. https://doi.org/10.1186/s12967-015-0432-1
- Zhang, H., Wang, Z., Feng, S.-J., Xu, L., Shi, H.-X., Chen, L.-L., Yuan, G.-D., Yan, W., Zhuang, W., Zhang, Y.-Q., Zhang, Z.-M., & Dong, H.-Y. (2015). PEDF improves cardiac function in rats with acute myocardial infarction via inhibiting vascular permeability and cardiomyocyte apoptosis. International Journal of Molecular Sciences, 16(3), 5618–5634. https://doi.org/10.3390/ijms16035618