
Abstract
Hypertrophic cardiomyopathy (HCM) is the most common inherited heart disease, with the prevalence of about 1/500. During the last two decades, the knowledge of the etiology, pathogenesis, risk stratification and prevention of sudden death in HCM has substantially advanced. Most often, HCM is familial and caused by mutations in sarcomere genes, inherited in an autosomal dominant manner. In Finland, genetic background of HCM is unique, with a few founder mutations in cardiac sarcomere genes accounting for a considerable proportion of the disease. Pathogenic mechanisms induced by disease-causing mutations are still poorly understood, although alterations in intracellular calcium handling and inefficient generation of contractile force in myocytes are considered key features in triggering the hypertrophic response. Clinical features of the disease are highly variable from no symptoms to the spectrum of exertional dyspnea, angina, palpitations, syncope and sudden death. In the current patient care, implantable cardioverter defibrillators (ICDs) are successfully used to prevent sudden cardiac death in high risk subjects. Targeted genetic testing is recommended to confirm the diagnosis in patients with HCM and to identify family members with the disease. Future research is needed to elucidate key cellular mechanisms leading to HCM, which may allow specific prevention and treatment of the disease.
Hypertrophic cardiomyopathy, most often caused by defects in sarcomere genes, is the most common inherited heart disease, and a common cause of sudden cardiac death (SCD) in athletes and young subjects.
Cardiac imaging, ECG and genetic testing are pivotal in the diagnosis of the disease in patients and first-degree relatives.
Implantable cardioverter defibrillators in patients with high risk for SCD and tailored pharmacotherapy are efficient tools in patient care, but so far, exact mechanisms leading to cardiac hypertrophy in HCM are only partially understood, and there is no curative treatment for the disease.
Key messages
Definition, diagnosis, differential diagnosis and etiology
Hypertrophic cardiomyopathy (HCM) is a myocardial disease characterized by left ventricular hypertrophy [LVH, left ventricular maximal wall thickness (LVMWT) ≥ 15 mm] not caused by hypertension, valvular disease, or coronary artery disease (Citation1). In subjects with a definitive HCM-causing mutation, or in first-degree relatives of subjects with definite HCM, LVMWT ≥13 mm is sufficient for diagnosis (Citation1). The diagnosis of LVH is most often made by echocardiography, although also other cardiac imaging modalities, including cardiac magnetic resonance imaging (CMRI) and computed tomography (CT) may demonstrate LVH suggesting HCM (Citation2). The most common cause for LVH in the general population, however, is not HCM but hypertensive heart disease, in which pressure overload induces thickening of LV (Citation2). In hypertensive heart disease, LVH is usually moderate the LVMWT not exceeding 18 mm (Citation3). Aortic stenosis also causes pressure overload and is a common cause for considerable LVH (Citation1).
HCM is familial in about half of the cases, and is most often caused by mutations in sarcomere genes, although significant phenocopies exist (Citation2,Citation4,Citation5). HCM caused by sarcomere gene defects is more often familial and the risk for sudden cardiac death (SCD) greater than in HCM attributable to other causes (Citation4). In about 5–10% of adult cases, and 25% of children diagnosed with HCM, a rare metabolic or neuromuscular disorder, mitochondrial cardiomyopathy, or malformation syndrome is found (Citation2,Citation4). In adult patients with infiltrative or inflammatory diseases such as amyloidosis or sarcoidosis, LVH quite like that induced by sarcomere gene defects may be found (Citation2).
Genetics
HCM is the most common inherited heart disease, with the prevalence of about 1/500 (Citation2). So far, over 1500 different HCM-causing mutations mainly in sarcomere genes have been identified (Citation2,Citation4,Citation5) (). Rarely, a mutation in sarcomere adjacent Z lines or calcium handling genes causes HCM. In about 60% of subjects with familial HCM, a mutation in sarcomere genes is found (Citation2). With current next generation sequencing techniques and targeted genetic panels available, the percentage of patients with identifiable genetic diagnosis will most probably increase (Citation6). Worldwide, mutations in myosin-binding protein C (MYBPC3) and myosin heavy chain (MYH7) genes account for the majority of cases with HCM (Citation2,Citation4,Citation5) (). Most causative mutations are missense mutations, changing one amino acid in the sarcomere protein, but truncating mutations are common in the MYBPC3 gene (Citation5). Very often, each family has its own disease-causing mutation, but founder mutations mainly in MYBPC3 have been detected in Finland, Holland, Iceland, Italy, Japan and South Africa (Citation7–10).
Table 1. Genes associated with HCM and estimated prevalence of respective mutations in non-Finnish patient populations.
In Finland, three mutations in sarcomere genes, MYBPC3-Q1061X, TPM1-D175N, and MYH7-R1053Q, account for about 23% of all cases with HCM ((Citation7–9,Citation11). Two of these (founder mutation MYBPC3-Q1061X and MYH7-R1053Q) have been originally reported in Finnish subjects with cardiomyopathy (Citation7,Citation11,Citation12) and are very rare outside of Finland. The founder mutation TPM1-D175N is prevalent in Finland but uncommon in non-Finnish patients, reported to occur in a few families in the USA, Europe and Japan (Citation9,Citation4,Citation5). In addition to aforementioned three common mutations, a few other gene defects in MYBPC3, TPM1, and MYH7 but not in troponin T, I, C, α-actin, or myosin essential and regulatory light chain genes have been found in Finnish patients with HCM (Citation7–9,Citation11,Citation13–15). It is likely, however, that with the next generation sequencing techniques numerous new mutations will be in identified in Finnish patients with HCM (Citation6).
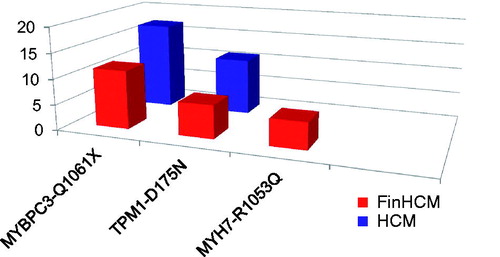
Figure 1. Prevalence of the most common HCM-causing mutations in the FinHCM study population of 306 probands from whole Finland (red bars), and in the HCM study population of 35 probands from eastern Finland (blue bars).
HCM caused by sarcomere gene defects is inherited in an autosomal dominant manner (Citation2,Citation5). Thus, the probability inheriting a mutation and risk of the disease is 50% in first-degree relatives (Citation5). About 5% of patients carry more than one gene defect in sarcomere genes, often resulting in a more severe phenotype with onset in childhood (Citation4). Homozygosity often leads to death or severe cardiomyopathy in infancy (Citation10). Some rare forms of HCM, such as Fabry disease, have X-linked inheritance, or are inherited maternally, such as some mitochondrial cardiomyopathies (Citation2). In new-born children and infants, HCM is commonly associated with rare genetic metabolic or neuromuscular disorders, or with genetic syndromes with variable modes of inheritance (Citation2,Citation4).
Cardiac phenotype
Cardiac phenotype in HCM is defined by LVH (Citation1). LVH in HCM, however, is not present at birth, but usually develops during adolescence and does not progress after 20–30 years of age (Citation16). Moreover, many subjects with confirmed HCM-causing mutations have no LVH (phenotype negative mutation carriers) (Citation5). Particularly, subjects with mutations in MYBPC3 often do not have LVH, or may develop it later in life (Citation16). Of subjects with the Finnish founder mutation MYBPC3-Q1061X, 20–30% does not have HCM phenotype (Citation8,Citation9). In subjects with mutations of other HCM-causing genes, phenotype negative carriers are not as common. In subjects with the Finnish founder mutation TPM1-D175N, 10–20% do not have LVH (Citation8,Citation9), and even some adult subjects with MYH7-R1053Q do not have manifest LVH (JKU, unpublished observations). When present, LVH in HCM is highly variable, LVMWT ranging from 15 to over 60 mm, but cases with extreme hypertrophy (≥ 35 mm) are rare (Citation2). In Finnish patients, the LVMWT is usually moderate, with mean of about 20 mm (Citation9). Hypertrophy is typically asymmetric, LVMWT locating in the interventricular septum (), but may be present also in other parts of LV, for example, in the free wall or apex. LV mass is not necessarily increased (Citation5), but with growing number of hypertrophied LV segments, LV mass ultimately exceeds normal limits. Hypertrophied LV segments typically show increased echodensity and are hypokinetic, contrasting non-hypertrophied segments of LV, which are usually hyperdynamic.
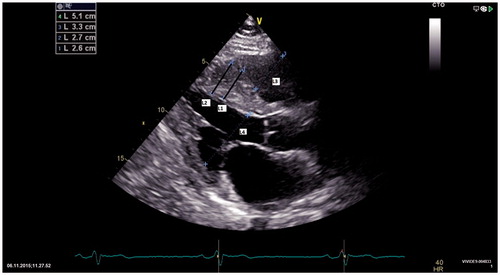
Figure 2. Two dimensional echocardiography in a patient with HCM. Interventricular septum is thickened, about 2.6–2.7 cm (bars L1 and L2), shown in bold black in the figure) and granular. RV and LV end-diastolic dimensions are normal (bars L3 and L4).
Diastolic dysfunction of LV is an early phenomenon in HCM, and some studies, but not all, have shown diastolic abnormalities in phenotype negative mutation carriers before development of LVH (Citation17–21). Diastolic function is further worsened by progressive LVH, myocardial fibrosis and left ventricular outflow tract (LVOT) obstruction. Although systolic function measured by ejection fraction is usually normal, or even hyperdynamic, subtle systolic dysfunction is present, even in phenotype negative mutation carriers (Citation20,Citation21). Systolic dysfunction and the degree of LVH are interrelated, with the number of hypokinetic segments correlating with LV mass (Citation22). LV is usually of normal size, or even smaller than in control subjects (Citation23). In about 3% of cases, however, LV dilatation and definite systolic dysfunction develop (Citation5). Patients with this end-stage HCM have particularly poor prognosis (Citation5).
Myocardial fibrosis demonstrated by gadolinium late enhancement CMRI is a common feature in HCM, present in about 65% of cases (Citation2). Fibrosis is intramyocardial and usually located in the hypertrophied septum and right ventricular insertion area. Marked myocardial fibrosis in CMRI (≥15% of the LV mass) is related to the risk of SCD, LV dilatation and systolic heart failure (Citation5).
LVOT obstruction, caused by hypertrophied basal septum, systolic anterior motion of the anterior mitral valve leaflet, and hyperdynamic LV contraction, is present in about 25% of patients with HCM (Citation16). In 306 unselected Finnish patients, 2% had a history of myectomy, and 7.5% had increased LVOT flow velocity (Citation9). LVOT obstruction is dynamic, sometimes present only in exercise. LVOT obstruction is often associated with more severe clinical course of HCM, and if untreated, increases the risk of SCD and heart failure (Citation5).
Mitral valve leaflets are often elongated in HCM and variable mitral regurgitation may be present, particularly in those with LVOT obstruction (Citation16). Abnormalities in small intramyocardial arteries are usually present in subjects with HCM, although epicardial coronary arteries are normal. Thickening of the intimal and medial layers of arterial wall is variable, and may contribute to ischemia and consequent fibrosis in hypertrophied myocardium (Citation23,Citation24).
Ultrastructure
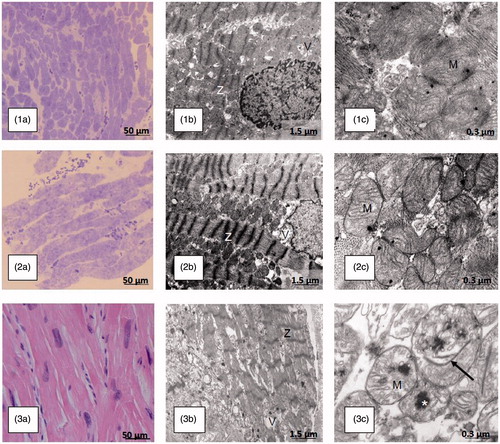
HCM is characterized by cardiomyocyte hypertrophy and disarray, and at the ultrastructural level, by sarcomere disorganization. As shown in (Naukkarinen A and Kuusisto J, unpublished), ultrastructural changes in the myocardium of patients with HCM appear to reflect the severity of clinical phenotype. Only mild changes in myocytes were visible in a myocardial biopsy specimen of a Finnish founder mutation TPM1-D175N carrier without LVH (patient 1). Increasing hypertrophy, disarray, and disorganization of Z-lines were found in a patient with definite HCM caused by the same mutation (patient 2). Even more pathological ultrastructural features including loss of myofibril integrity were found in a patient with a rare MYH7-R453C mutation and severe HCM phenotype (patient 3). Changes in mitochondrial structure shown by electron microscopy were prominent only in patient 3 with the MYH7 mutation and severe HCM phenotype.
Figure 3. Light and electron microscopic findings of myocardial biopsies in patients with hypertrophic cardiomyopathy-causing mutations in genes encoding sarcomere proteins reflect the severity of macroscopic disease and arrhythmia vulnerability. Patients 1 and 2, a 17-year-old girl and her 19-year-old male first cousin, respectively, are carriers of the HCM-causing Finnish founder mutation in the α-tropomyosin gene (TPM1-D175N). Patient 1 had no signs or symptoms of HCM and was considered a healthy mutation-carrier. The LVMWT in CMRI was 10 mm. Patient 2 had a LVMWT of 24 mm in CMRI, severe symptoms, and several risk factors for SCD. Subsequently, an ICD was implanted. Patient 3, a 14-year-old boy, died of electric VT storm, irrespective of a functional ICD, after falling in the ski slope. On the last echocardiography, the LVMWT was 29 mm; LV dimensions were 54/37 mm and ejection fraction was 57%. The patient had a previous history of ventricular tachycardia and several aborted SCDs but no overt heart failure. In the postmortem genetic analysis, a rare disease-causing mutation in the β-myosin heavy chain gene was found (MYH7-R453C). Part labels 1a, 2a and 3a show growing hypertrophy of myocytes (light microscopy, bar 50 μm) in the endomyocardial biopsies (1a: normal muscle/mild hypertrophy, 2a: moderate hypertrophy) and the postmortem heart specimen (3a: marked hypertrophy) in patients 1, 2 and 3, respectively. Part labels 1b, 2b and 3b show increasing derangement of myocyte ultrastructure, including myofibril disarray, abnormalities of Z-lines (Z), and vacuoles (V) [electron microscopy (EM), bar 1.5 μm) in patients 1–3. Part label 3b displays partial loss of myofibril integrity in patient 3. In patients 1 and 2 (Part labels 1c and 2c), no unequivocal changes are evident in mitochondria (M) (EM, bar 0.3 μm). Part label 3c shows swollen mitochondria with anomalous cristae, internal rod-like structures (arrow) and large dark calcium deposits (asterisk) (EM, bar 0.3 μm) in the severely affected patient 3.
Histological myocardial fibrosis in HCM is interstitial, located mainly adjacent to intramyocardial vessels (Citation24). Factors resulting in the development of fibrosis are poorly defined, but may be related, at least partly, to LVH, as disease-causing mutation carriers without LVH in general do not have myocardial fibrosis (Citation5). Also, myocardial ischemia and active low-grade inflammation have been shown to be related to myocardial fibrosis (Citation24,Citation25).
Pathogenesis
Most gene defects causing HCM are missense mutations of the sarcomere genes, changing one amino acid in the encoded protein, and resulting in a “poison polypeptide”, which is incorporated in the sarcomere (Citation5). Gene defects in MYBPC3, however, often result in truncated proteins. MYBPC3 mutations are the most common cause for HCM, accounting for 40–50% of all cases (Citation2,Citation4,Citation5,Citation10). Of all MYBPC3 mutations, about 60% lead to the synthesis of truncated protein. Consequently, approximately 30% of all HCM-causing mutations represent this kind of gene defect (Citation10). Truncated sarcomere proteins are removed from the myocardium, which results in decreased content of the protein (haploinsufficiency) (Citation10). Possibly, mutations leading to haploinsufficiency versus missense mutations resulting in poison polypeptides may induce distinct pathogenic responses in the sarcomere.
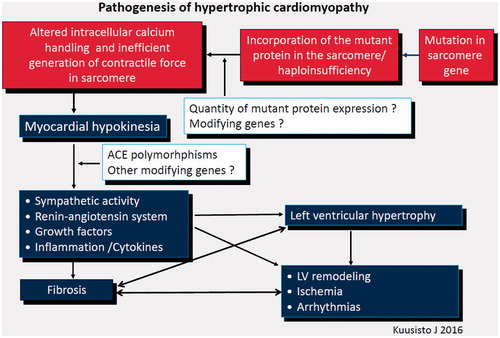
Pathogenic changes induced by HCM-causing mutations in myocytes are poorly understood. Alterations in intracellular calcium handling, sarcomere function, and the generation of contractile force in myocytes are associated with cardiomyocyte energy depletion or imbalance (Citation26–29), which is considered to be the key feature in triggering the hypertrophic response, cell death and fibrosis (). Modifying genes, of which only ACE gene I/D polymorphisms is well established (Citation28), are suggested to influence the development of hypertrophy. When present, LVH, in turn, leads to other features of HCM, including ischemia, advancing LV remodeling, fibrosis and arrhythmias, all contributing to the vicious cycle of pathophysiological manifestations. As indicated by a considerable number of carriers of disease-causing mutations who do not develop LVH or other macroscopic cardiac manifestations of the disease, pathogenesis of HCM is not entirely determined by the disease-causing mutation but is an active process, influenced by sympathetic activity, renin–aldosterone–angiotensin system, growth factors, and inflammation (Citation24,Citation30,Citation31), which are possibly modifiable. In our study on patients with TPM1-D175N, LVH in CMRI was related to cardiac adrenergic activity (Citation30). There are some experimental and animal studies suggesting that losartan, diltiazem, simvastatin, nebivolol and ranolazine might prevent LVH in HCM, but so far, large scale human studies are lacking (Citation26,Citation31–34). A recent study shows that in patients with unequivocal LVH, losartan is not beneficial with respect to LVH (Citation35).
Figure 4. Pathogenesis of HCM. Hypothesis.
Prognosis and natural history
Prognosis and modes of death
Natural history of HCM is highly variable. Most subjects with HCM have a prognosis similar to that of the general population. According to recent reports on unselected patient populations, the mortality rate is about 0.5–1.5%/year (Citation5). The prognosis and clinical course of HCM are not unequivocally related to the causative mutation, and vary even in subjects from the same family with the same causative mutation. In patients with Finnish founder mutations MYBPC3-Q1061X and TPM1-D175N, clinical course is usually, but not always, favorable (Citation8,Citation9). Mutations of MYH7 usually cause more severe phenotype. In carriers of MYH7-R1053Q, dilatation and systolic dysfunction of LV often develop (Citation11,Citation12).
About 20–30% of the patients with HCM are at an increased risk for SCD (Citation5,Citation36). SCD is usually attributable to ventricular tachycardia degenerating to ventricular fibrillation (Citation5). Although SCD may occur at any age, it is more common in the young (Citation5). HCM is a common cause SCD in athletes and young subjects. According to a recent meta-analysis, HCM was the cause of SCD in young (≤35 years) in 10.3% of cases, whereas SCDs with structurally normal hearts were more common (26.7%) (Citation37). In athletes, there was no significant difference between HCM and structurally normal hearts as a cause of SCD. However, postmortem diagnosis of HCM may be challenging (Citation37), as for example, LV mass is not increased in a considerable number of patients (Citation5). Consequently, the real prevalence of HCM in young victims of SCD may be higher than aforementioned numbers.
Other significant modes of death in HCM are heart failure and embolic stroke (Citation5). Overt heart failure may develop as a consequence of LVOT obstruction, LV dilatation and systolic dysfunction, or as a consequence of marked hypertrophy and subsequent diastolic dysfunction and restriction. In our own study on 306 Finnish subjects with HCM, only 4% had a history of systolic congestive heart failure (Citation9). In Europe, about 7% of the patients on the heart transplantation waiting list have HCM (Citation2). Embolic stroke is very common in subjects with atrial fibrillation and HCM, and contributes to increased mortality in HCM (Citation5).
Risk factors of SCD in HCM
Patients with aborted SCD or sustained monomorphic ventricular tachycardia have a very high risk of recurrence of the malignant arrhythmia, and implantation of cardioverter defibrillator (ICD) is recommended (level of evidence IB) (Citation2).
Other risk factors of SCD in patients with HCM are marked LVH (LVMWT ≥30 mm), family history of SCD caused by HCM, unexplained syncope, presence of non-sustained ventricular tachycardia (NSVT), and abnormal blood pressure response in clinical exercise test (Citation2). Recently, additional risk factors have been identified, including increased size of the left atrium, LVOT obstruction, myocardial ischemia, extensive myocardial fibrosis in CMRI, overt heart failure, and increased levels of proBNP and TnT (Citation2,Citation5). The risk of SCD in HCM increases with the number of risk factors, and patients without any risk factors are usually at low risk (Citation2,Citation5).
NSVT is present in about 20-30% of patients with HCM and is a significant risk factor of SCD (Citation2). Outbursts of NSVT are often asymptomatic and detected only on 24- hour ambulatory ECG (Citation2) but may also cause palpitations and presyncope. In our own study on 306 Finnish patients with HCM, NSVT on Holter ECG registration was found in 20% of patients, 3% had a history of sustained VT and 5% a history of ventricular fibrillation, respectively (Citation9).
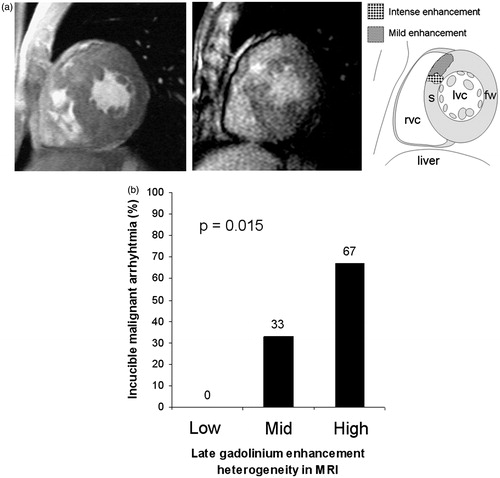
In our study on patients with the Finnish founder mutation TPM1-D175N, one third had inducible malignant ventricular arrhythmias in electrophysiological testing (EPS), indicating arrhythmia vulnerability (Citation36). All inducible patients had LVMWT over 20 mm and two or more risk factors for HCM-associated SCD. In contrast, all patients without inducible ventricular arrhythmias had LVMWT under 20 mm and at most 1–2 traditional risk factors for SCD (Citation36). In the same patients with TPM1-D175N, no ventricular arrhythmias were induced in patients with low late enhancement heterogeneity in CMRI (, Sipola P and Kuusisto J, unpublished). In contrast, patients with moderate or high late enhancement heterogeneity had inducibility rates of 33% and 67%, respectively. Consequently, LVH and myocardial fibrosis may be the prerequisite for malignant ventricular arrhythmias in HCM. In agreement with this concept, phenotype negative mutation carriers, as a rule, are not considered to be at risk for SCD (Citation2).
Figure 5. (a) CMRI from a patient with HCM caused by the TPM1-D175N mutation. Left panel shows marked septal LVH in the patient. The middle panel shows septal gadolinium late-enhancement (GLE) heterogeneity, as clarified by the right panel (s = septum, fw = free wall, lvc = left ventricle, rvc = right ventricle). (b) Association of inducibility of malignant ventricular arrhythmias in EPS with CMRI-derived myocardial fibrosis in patients with HCM caused by the TPM1-D175N mutation. Patients with low GLE heterogeneity in CMRI had no inducible malignant ventricular arrhythmias, whereas those with mid or high level of GLE had increasing rate of inducibility.
Clinical course
In many patients with HCM, the clinical course is stable with no adverse events or LV remodeling, and patients have normal life-expectancy (Citation5). In a considerable number of patients with HCM, however, the natural history of the disease is defined by progressive symptoms of exertional dyspnea, angina, arrhythmias, and in some, also heart failure and death, reflecting advancing cardiac remodeling and arrhythmia vulnerability.
Many of the factors associated with the risk of SCD appear to increase also the risk for adverse remodeling of LV and atrial fibrillation. Definite LVH and/or LVOT obstruction is usually present in subjects with adverse outcomes (Citation2,Citation5). Increased left atrium size, diastolic dysfunction, LVOT obstruction and mitral regurgitation increase the risk of atrial fibrillation, which, according to recent review, is present in about one quarter of subjects with HCM (Citation2,Citation5). In our own study on Finnish subjects with HCM, the prevalence atrial fibrillation was 25% (Citation8). LVOT obstruction and marked myocardial fibrosis are risk factors for LV dilatation and systolic dysfunction (Citation5).
Increased levels of proBNP and TnT predict heart failure and adverse cardiac events in patients with HCM (Citation2,Citation38). In our own study, in patients with HCM caused by the TPM1-D175N mutation, increased levels of NT-proBNP were associated with the LV volume index, LV mass and proportion of hypokinetic LV segments, reflecting adverse LV remodeling (Citation39).
Clinical features
Symptoms
Patients with HCM who have mild LVH commonly have no cardiac symptoms (Citation2). In general, HCM-causing mutation carriers without LVH are also asymptomatic. In patients with moderate to marked LVH and/or LVOT obstruction, definite symptoms usually develop. The most common symptom is exertional dyspnea, assumed to be caused by LVH, ischemia, or LVOT obstruction (Citation16). Physical exercise capacity is often reduced but may be normal even in subjects with marked LVH (Citation9). About one fifth of the patients have anginal chest pain. In our own study on 306 Finnish patients with HCM, prevalence of dyspnea was 43%, with NYHA class 1-2 in 85% of patients (Citation9). Chest pain was reported in 14% of cases (Citation9). Many patients have palpitations, which may reflect atrial fibrillation, supraventricular or ventricular ectopic beats, NSVT, or hypercontractile myocardium. Unexplained syncope is a malignant symptom particularly in the young, and is often caused by prolonged ventricular arrhythmias (Citation2). In our own study on Finnish patients with HCM, the prevalence of syncope was 27% (Citation9).
Clinical examination
Cardiovascular examination is often within normal limits (Citation2,Citation16). S4 gallop, caused by LVH, may be audible on cardiac auscultation. Systolic murmur associated with hyperdynamic LVOT flow or obstruction, or mitral regurgitation, may be heard. In the obstructive form of the disease, trill, or multiplied apical thrust or carotid pulse may be palpable (Citation2). Signs of overt heart failure are uncommon. In HCM attributable to causes other than sarcomere gene defects, specific features, such as angiokeratomata in Fabry disease may be encountered.
Diagnostic assessment
HCM is usually suspected because of cardiac symptoms (see above), abnormal ECG, or currently more often, family history of HCM. According to the quite recently published guidelines on diagnosis and management of HCH by the European Society of Cardiology, when HCM is suspected, evaluation by a cardiologist with expertise in cardiomyopathies is recommended (Citation2). HCM is diagnosed if LVMWT equal or over 15 mm, not solely explained by loading conditions, is detected by any imaging technique (Citation2). Echocardiography, however, is the most commonly used cardiac imaging modality in the diagnosis of HCM. In patients with a definitive HCM-causing mutation, or in first-degree relatives of patients with definite HCM, LVMWT ≥13 mm suffices for diagnosis.
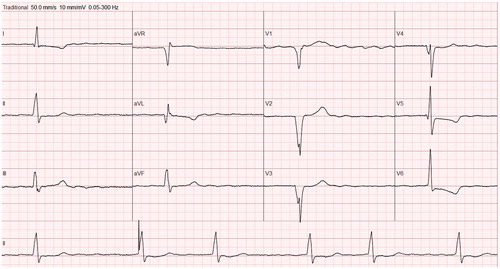
ECG should be part of the diagnostics both in patients and relatives. ECG is almost always abnormal, showing variable features of LVH, pathological Q waves, ST-depression, T wave abnormalities, wide QRS (), right or left bundle branch block, or left atrial enlargement. ECG abnormalities may precede LVH in carriers of causal mutations (Citation2).
Figure 6. Twelve-lead ECG from a patient with HCM. Note pathological Q waves (leads V1–V4 and aVL), ST-depression and inverted T waves (leads I and V5–V6), somewhat wide QRS (121 ms) with notching (leads III, aVL, aVF, V2–V3). The patient has atrial fibrillation and an implanted pacemaker (see rhythm section in the lower part of the figure).
CMRI, if available, should be considered in all patients with definite or uncertain cardiomyopathy (Citation2). Three-dimensional quality and acoustic window-independent views of CMRI make it the golden standard in the evaluation of myocardial diseases. LV mass and dimensions, apical and anterolateral hypertrophy, aneurysms and thrombi are most reliably measured by CMRI. Furthermore, late gadolinium enhancement in CMRI indicates myocardial fibrosis, which cannot be recognized by other cardiac imaging modalities. Cardiac CT may be considered in subjects in whom CMRI is contraindicated, as in patients with pace makers not compatible with CMRI.
Coronary angiography is recommended if coronary artery disease is suspected. Endomyocardial biopsy should be taken if the etiology of HCM is indefinite, specific pathology such as myocardial sarcoidosis or storage disease is suspected, or the course of the disease is atypical, such as progressive hypertrophy.
In the initial assessment, laboratory tests including hemoglobin, fasting glucose, and renal and thyroid function tests are recommended (Citation2). Creatinine phosphokinase, liver transaminases and plasma lactate may be elevated in metabolic and mitrochondrial diseases and should be measured if the specific etiology of HCM is not evident. Elevated brain natriuretic peptide (NT-proBNP) and troponin T are associated with higher risk of cardiovascular events, heart failure and death, and should be assessed (Citation2).
Genetic testing
Probands
According to the current guidelines, targeted genetic testing is recommended in patients with HCM (level of evidence IB) (Citation2). Genetic testing confirms the exact diagnosis, may reveal a rare cause for HCM, such as Fabry disease, and enables the cascade genetic screening of relatives. Next-generation sequencing techniques have developed rapidly, and currently targeted panels for cardiomyopathy causing genes, including sarcomere and also other more rare genes, for example, α-galactosidase A, are available for clinicians.
Relatives
In families with definite disease causing gene defects, first-degree relatives (children, siblings and parents of the proband) should be offered screening of the gene defect (Citation2). As a rule, clinical assessment is necessary only in the relatives with the disease-causing mutation. In economic decision models, this approach of cascade genetic screening has been found to be more cost-effective than clinical screening alone (Citation2). If no definite causative gene defect is found in the patient with HCM, repeated clinical assessment (history, clinical examination, ECG, echocardiography) is recommended in all first-degree relatives (Citation2).
Risk assessment of SCD
The assessment of the risk of SCD is one of the most important issues in the evaluation of patients with HCM, and should be performed at first evaluation and after that, at 1–2-year intervals (Citation2). Family history, symptoms of the patient, clinical examination, echocardiography, genetic testing, the levels of NT-proBNP and troponin T, and clinical exercise test are reviewed in the risk stratification of the patient. In addition, ambulatory 24-h ECG registration is used to detect outbursts of NSVT and atrial fibrillation. If no risk markers for SCD are found (see chapter natural history), the risk for SCD is considered low. With increasing number of risk factors there is an increased risk for SCD. When in doubt, the risk of SCD may be assessed by a recently developed Internet-based risk calculator model, in which LVMWT, left atrial diameter, age, LVOT gradient, and presence of family history of SCD, NSVT, and syncope are included (Citation2). HCM Risk-SCD calculator gives an estimate of 5-year risk of SCD. However, not all known risk factors for SCD are included in the model, and the risk for SCD may be high in patients with only one severe risk marker, such as overt heart failure (Citation5). Finally, the knowledge of mechanisms, pathogenesis and risk markers for SCD is still quite limited, and further studies are needed to enable more accurate risk stratification in HCM.
Treatment
Lifestyle
Subjects with definite HCM are advised against competitive sports and extreme physical activity (Citation2,Citation5). Healthy lifestyle including non-strenuous exercise and smoking cessation is recommended. Physically demanding, night or shift work is not recommended because of arrhythmia risk. As concomitant coronary artery disease markedly increases the risk of SCD in HCM (Citation40), careful evaluation and treatment of all cardiovascular risk factors (hypertension, dyslipidemia, diabetes, obesity) is recommended.
Medical treatment
So far, there is neither curative treatment for HCM caused by sarcomere gene defects, nor medicine which can prevent the development of LVH in mutation carriers. Modern treatment of HCM improves the outcome but is still symptomatic. In phenotype negative mutation carriers or in asymptomatic subject with mild LVH, no medical treatment is recommended. The treatment in symptomatic patients is outlined in the recent ESC guidelines on diagnosis and management of HCM (Citation2). Shortly, beta-blockers or alternatively, calcium channel blockers verapamil and diltiazem may be used in the non-obstructive form of the disease (Citation2). In the obstructive form of the disease, betablockers are prescribed, and if needed, combined with disopyramide. Inhibitors of renin–angiotensin–aldosterone system are used in subjects with LV dilatation or decreased (<50%) ejection fraction (Citation2). Permanent anticoagulation with warfarin, or new anticoagulants, is recommended in all patients with documented atrial fibrillation to prevent embolic complications, irrespective of the CHA2DS2-VASc score (Citation2). To prevent paroxysmal or persistent atrial fibrillation, only amiodarone may be used in addition to beta-blockers (Citation2).
Pacemakers, ICDs and cardiac resynchronization therapy
As interventricular septum is usually affected in HCM, patients with the disease may have variable atrioventricular block. Permanent bradycardia pacemaker implantation is indicated in the third and second degree Mobitz type II atrioventricular block, and in symptomatic sick sinus syndrome, just as in respective conditions caused by other cardiac diseases.
ICD therapy is recommended in subjects who have survived cardiac arrest and in those with sustained ventricular tachycardia, as these subjects have a very high risk of the recurrence of malignant arrhythmia (level of evidence IB) (Citation2). Preventive ICD implantation should be considered in patients with multiple risk factors for SCD, whose estimated HCM Risk-SCD calculator 5-year risk is ≥6% (level of evidence IIaB). ICD therapy may be considered if the risk calculator gives SCD estimate ≥4% but <6%, or there is another condition increasing the risk of SCD, which is not included in the risk calculator, for example heart failure (IIbB) (Citation2,Citation5).
Cardiac resynchronization therapy is required only in a small proportion of patients with HCM who have systolic heart failure and wide QRS.
EPS and catheter ablation for arrhythmias
EPS and catheter ablation may be considered in subjects with recurrent or persistent atrial fibrillation if the antiarrhythmic medication fails, and there is no severe left atrial enlargement (Citation2). EPS and ablation may be considered also in subjects with recurrent VT, and is becoming life-saving standard of care in subjects with VT storm (Citation2,Citation41).
Invasive treatment of LVOT obstruction
In patients with drug refractory LVOT obstruction (gradient ≥50 mmHg) at rest and definite NYHA Class III-IV symptoms, surgical myectomy of the LV septum, or septal alcohol ablation should be considered (Citation2). Invasive treatment may be considered also in subjects with milder NYHA Class II symptoms, who have resting or provoked gradient ≥50 mmHg, if they have significant mitral regurgitation, left atrial dilatation, or atrial fibrillation.
Surgical ventricular septal myectomy is recommended in young subjects and particularly if concomitant mitral valve surgery is required (Citation2). Surgery successfully eliminates LVOT gradients with long-term benefit and favorable survival. Septal alcohol ablation creates myocardial infarction and is associated with a higher risk of AV block and larger residual LVOT obstruction, and there is no long-term survival data. Septal alcohol ablation, performed in experienced centers, may be considered particularly in elderly subjects with severe co-morbidities, or other contraindications for surgery.
Follow-up
In patients with definite HCM, life-long follow-up in an outpatient clinic with expertise in cardiomyopathies is recommended every 1–2 years, or whenever they develop new symptoms (Citation2). ECG, transthoracic echocardiography, and ambulatory ECG should be ordered for each visit. Clinical exercise test is recommended every 2–3 years and CMRI every fifth year, and earlier, if disease progression is suggested. ProBNP and TnT are not clearly recommended in the current guidelines for each visit, but as they predict adverse events, are readily available and inexpensive, it is reasonable to include them in the follow-up routine.
In families with definite causative mutations, adult LVH negative mutation carriers require permanent follow-up (Citation2) with regular intervals of 5 years, or more often, depending on the mutation and possible non-diagnostic abnormalities on ECG or echocardiography. If the definite disease-causing mutation of the family is not found in the relative, discharge is advised, except for the relatives who have suspect symptoms or findings. If the causative mutation is not found in the proband, and the first-degree relative has no LVH, repeated follow-up is nevertheless recommended, particularly if subtle ECG or echocardiographic signs of myocardial disease are detected.
Future challenges
Future research is challenged to find the key cellular mechanisms leading to LVH in HCM, and to develop targeted therapies to alter the natural history of the disease before end-stage irreversible cardiac remodeling occurs (Citation26). Utilizing models of transgenic animals and modern molecular techniques, including induced pluripotent stem cell models, may allow detecting gene defect specific mechanisms leading to HCM and applying targeted drug therapies. In the future, modern gene therapy techniques, correcting the disease-causing gene defect, may be applicable to severe forms of the disease. More accurate identification of modifying genes and pathways leading to hypertrophic response in HCM may also allow development of more effective medical therapies to counteract LVH and associated pathophysiological vicious cycle.
Disclosure statement
No declarations of interest to report.
References
- Elliott P, Andersson B, Arbustini E, Bilinska Z, Cecchi F, Charron P, et al. Classification of the cardiomyopathies: a position statement from the European Society Of Cardiology Working Group on Myocardial and Pericardial Diseases. Eur Heart J. 2008;29:270–6.
- Elliott PM, Anastasakis A, Borger MA, Borggrefe M, Cecchi F, Charron P, et al. 2014 ESC Guidelines on diagnosis and management of hypertrophic cardiomyopathy: the Task Force for the Diagnosis and Management of Hypertrophic Cardiomyopathy of the European Society of Cardiology (ESC). Eur Heart J. 2014;35:2733–79.
- Sipola P, Magga J, Husso M, Jääskeläinen P, Peuhkurinen K, Kuusisto J. Cardiac MRI assessed left ventricular hypertrophy in differentiating hypertensive heart disease from hypertrophic cardiomyopathy attributable to a sarcomeric gene mutation. Eur Radiol. 2011;21:1383–9.
- Ho CY, Charron P, Richard P, Girolami F, Van Spaendonck-Zwarts KY, Pinto Y. Genetic advances in sarcomeric cardiomyopathies: state of the art. Cardiovasc Res. 2015;105:397–408.
- Maron BJ, Ommen SR, Semsarian C, Spirito P, Olivotto I, Maron MS. Hypertrophic cardiomyopathy: present and future, with translation into contemporary cardiovascular medicine. J Am Coll Cardiol. 2014;64:83–99. Erratum in: J Am Coll Cardiol. 2014;64:1188.
- Lopes LR, Syrris P, Guttmann OP, O’Mahony C, Tang HC, Dalageorgou C, Jenkins S, et al. Novel genotype–phenotype associations demonstrated by high-throughput sequencing in patients with hypertrophic cardiomyopathy. Heart. 2015;101:294–301.
- Jääskeläinen P, Kuusisto J, Miettinen R, Kärkkäinen P, Kärkkäinen S, Heikkinen S, et al. Mutations in the cardiac myosin-binding protein C gene are the predominant cause of familial hypertrophic cardiomyopathy in eastern Finland. J Mol Med (Berl). 2002;80:412–22.
- Jääskeläinen P, Miettinen R, Kärkkäinen P, Toivonen L, Laakso M, Kuusisto J. Genetics of hypertrophic cardiomyopathy in eastern Finland: few founder mutations with benign or intermediary phenotypes. Ann Med. 2004;36:23–32.
- Jääskeläinen P, Heliö T, Aalto-Setälä K, Kaartinen M, Ilveskoski E, Hämäläinen L, et al. Two founder mutations in the alpha-tropomyosin and the cardiac myosin-binding protein C genes are common causes of hypertrophic cardiomyopathy in the Finnish population. Ann Med. 2013;45:85–90.
- Carrier L, Mearini G, Stathopoulou K, Cuello F. Cardiac myosin-binding protein C (MYBPC3) in cardiac pathophysiology. Gene. 2015;573:188–97.
- Jääskeläinen P, Heliö T, Aalto-Setälä K, Kaartinen M, Ilveskoski E, Hämäläinen L, et al. A new common mutation in the cardiac beta-myosin heavy chain gene in Finnish patients with hypertrophic cardiomyopathy. Ann Med. 2014;46:424–9.
- Kärkkäinen S, Heliö T, Jääskeläinen P, Miettinen R, Tuomainen P, Ylitalo K, et al. Two novel mutations in the beta-myosin heavy chain gene associated with dilated cardiomyopathy. Eur J Heart Fail. 2004;6:861–8.
- Jääskeläinen P, Soranta M, Miettinen R, Saarinen L, Pihlajamäki J, Silvennoinen K, et al. The cardiac beta-myosin heavy chain gene is not the predominant gene for hypertrophic cardiomyopathy in the Finnish population. J Am Coll Cardiol. 1998;32:1709–16.
- Jääskeläinen P, Miettinen R, Silvennoinen K, Vauhkonen I, Laakso M, Kuusisto J. The cardiac troponin I gene is not associated with hypertrophic cardiomyopathy in patients from eastern Finland. J Mol Cell Cardiol. 1999;31:2031–6.
- Kärkkäinen S, Peuhkurinen K, Jääskeläinen P, Miettinen R, Kärkkäinen P, Kuusisto J, Laakso M. No variants in the cardiac actin gene in Finnish patients with dilated or hypertrophic cardiomyopathy. Am Heart J. 2002;143:E6.
- Elliott P, McKenna WJ. Hypertrophic cardiomyopathy. Lancet. 2004;363:1881–91.
- Nagueh SF, Bachinski LL, Meyer D, Hill R, Zoghbi WA, Tam JW, et al. Tissue Doppler imaging consistently detects myocardial abnormalities in patients with hypertrophic cardiomyopathy and provides a novel means for an early diagnosis before and independently of hypertrophy. Circulation. 2001;104:128–30.
- Poutanen T, Tikanoja T, Jääskeläinen P, Jokinen E, Silvast A, Laakso M, Kuusisto J. Diastolic dysfunction without left ventricular hypertrophy is an early finding in children with hypertrophic cardiomyopathy-causing mutations in the beta-myosin heavy chain, alpha-tropomyosin, and myosin-binding protein C genes. Am Heart J. 2006;151:725.e1–e9.
- Gandjbakhch E, Gackowski A, Tezenas du Montcel S, Isnard R, Hamroun A, Richard P, et al. Early identification of mutation carriers in familial hypertrophic cardiomyopathy by combined echocardiography and tissue Doppler imaging. Eur Heart J. 2010;31:1599–607.
- De S, Borowski AG, Wang H, Nye L, Xin B, Thomas JD, et al. Subclinical echocardiographic abnormalities in phenotype-negative carriers of myosin-binding protein C3 gene mutation for hypertrophic cardiomyopathy. Am Heart J. 2011;162:262–3.
- Yiu KH, Atsma DE, Delgado V, Ng ACT, Witkowski TG, Ewe SH, et al. Myocardial Structural alteration and systolic dysfunction in preclinical hypertrophic cardiomyopathy mutation carriers. PLoS One. 2012;7:e36115.
- Sipola P, Lauerma K, Jääskeläinen P, Laakso M, Peuhkurinen K, Manninen H, et al. Cine MR imaging of myocardial contractile impairment in patients with hypertrophic cardiomyopathy attributable to Asp175Asn mutation in the alpha-tropomyosin gene. Radiology. 2005;236:815–24.
- Sipola P, Lauerma K, Husso-Saastamoinen M, Kuikka JT, Vanninen E, Laitinen T, et al. First-pass MR imaging in the assessment of perfusion impairment in patients with hypertrophic cardiomyopathy and the Asp175Asn mutation of the alpha-tropomyosin gene. Radiology. 2003;226:129–37.
- Kuusisto J, Kärjä V, Sipola P, Kholová I, Peuhkurinen K, Jääskeläinen P, et al. Low-grade inflammation and the phenotypic expression of myocardial fibrosis in hypertrophic cardiomyopathy. Heart. 2012;98:1007–13.
- Basso C, Thiene G, Corrado D, Buja G, Melacini P, Nava A. Hypertrophic cardiomyopathy and sudden death in the young: pathologic evidence of myocardial ischemia. Hum Pathol. 2000;31:988–98.
- Tardiff JC, Carrier L, Bers DM, Poggesi C, Ferrantini C, Coppini R, et al. Targets for therapy in sarcomeric cardiomyopathies. Cardiovasc Res. 2015;105:457–70.
- Lopes LR, Elliott PM. A straightforward guide to the sarcomeric basis of cardiomyopathies. Heart. 2014;100:1916–23.
- Kimura A. Molecular genetics and pathogenesis of cardiomyopathy. J Hum Genet. 2016;61:41–50.
- Tuunanen H, Kuusisto J, Toikka J, Jääskeläinen P, Marjamäki P, Peuhkurinen K, et al. Myocardial perfusion, oxidative metabolism, and free fatty acid uptake in patients with hypertrophic cardiomyopathy attributable to the Asp175Asn mutation in the alpha-tropomyosin gene: a positron emission tomography study. J Nucl Cardiol. 2007;14:354–65.
- Sipola P, Vanninen E, Aronen HJ, Lauerma K, Simula S, Jääskeläinen P, et al. Cardiac adrenergic activity is associated with left ventricular hypertrophy in genetically homogeneous subjects with hypertrophic cardiomyopathy. J Nucl Med. 2003;44:487–93.
- Lim DS, Lutucuta S, Bachireddy P, Youker K, Evans A, Entman M, et al. Angiotensin II blockade reverses myocardial fibrosis in a transgenic mouse model of human hypertrophic cardiomyopathy. Circulation. 2001;103:789–91.
- Westermann D, Knollmann BC, Steendijk P, Rutschow S, Riad A, Pauschinger M, et al. Diltiazem treatment prevents diastolic heart failure in mice with familial hypertrophic cardiomyopathy. Eur J Heart Fail. 2006;8:115–21.
- Ho CY, Lakdawala NK, Cirino AL, Lipshultz SE, Sparks E, Abbasi SA, et al. Diltiazem treatment for pre-clinical hypertrophic cardiomyopathy sarcomere mutation carriers: a pilot randomized trial to modify disease expression. JACC Heart Fail. 2015;3:180–8.
- Patel R, Nagueh SF, Tsybouleva N, Abdellatif M, Lutucuta S, Kopelen HA, et al. Simvastatin induces regression of cardiac hypertrophy and fibrosis and improves cardiac function in a transgenic rabbit model of human hypertrophic cardiomyopathy. Circulation. 2001;104:317–24.
- Axelsson A, Iversen K, Vejlstrup N, Ho CY, Norsk J, Langhoff L, et al. Efficacy and safety of the angiotensin II receptor blocker losartan for hypertrophic cardiomyopathy: the INHERIT randomised, double-blind, placebo-controlled trial. Lancet Diabetes Endocrinol. 2015;3:123–31.
- Hedman A, Hartikainen J, Vanninen E, Laitinen T, Jääskeläinen P, Laakso M, et al. Inducibility of life-threatening ventricular arrhythmias is related to maximum left ventricular thickness and clinical markers of sudden cardiac death in patients with hypertrophic cardiomyopathy attributable to the Asp175Asn mutation in the alpha-tropomyosin gene. J Mol Cell Cardiol. 2004;36:91–9. Erratum in: J Mol Cell Cardiol. 2004;36:607–8.
- Ullal AJ, Abdelfattah RS, Ashley EA, Froelicher VF. Hypertrophic cardiomyopathy as a cause of sudden cardiac death in the young: a meta-analysis. Am J Med. 2016;129:486–96.e2.
- Coats CJ, Gallagher MJ, Foley M, O'Mahony C, Critoph C, Gimeno J, et al. Relation between serum N-terminal pro-brain natriuretic peptide and prognosis in patients with hypertrophic cardiomyopathy. Eur Heart J. 2013;34:2529–37.
- Magga J, Sipola P, Vuolteenaho O, Risteli J, Jääskeläinen P, Peuhkurinen K, Kuusisto J. Significance of plasma levels of N-terminal Pro-B-type natriuretic peptide on left ventricular remodeling in non-obstructive hypertrophic cardiomyopathy attributable to the Asp175Asn mutation in the alpha-tropomyosin gene. Am J Cardiol. 2008;101:1185–90.
- Sorajja P, Ommen SR, Nishimura RA, Gersh BJ, Berger PB, Tajik AJ. Adverse prognosis of patients with hypertrophic cardiomyopathy who have epicardial coronary artery disease. Circulation. 2003;108:2342–8.
- Tanawuttiwat T, Nazarian S, Calkins H. The role of catheter ablation in the management of ventricular tachycardia. Eur Heart J. 2016;37:594–609.