
Abstract
The Developmental Origins of Health and Disease (DOHaD) hypothesis proposes that several non-communicable diseases have their origins in prenatal life and in early childhood. This is believed to work through programming, an insult, taking place at a sensitive period of development, may have lifelong consequences, increasing and programming disease risk later in life. The Helsinki Birth Cohort Study (HBCS) has been focusing upon the importance of factors active during periods in early life and their influence on later health in 20,431 people born 1924–44. This review will focus upon findings from the HBCS over the past 20 years. Early growth patterns associated with coronary heart disease, type 2 diabetes and other health outcomes are described. The long-term health impact of maternal adiposity is also discussed. Potential underlying mechanisms explaining the associations are discussed including epigenetic factors.
Several non-communicable diseases – including coronary heart disease and type 2 diabetes – have their origins in early life.
Early life programming during sensitive periods of development may permanently program future health and disease risk.
Optimizing the health and lifestyle of women of reproductive age will have positive health consequences for their offspring.
Key messages
Introduction
The Developmental Origins of Health and Disease (DOHaD) hypothesis proposes that several non-communicable diseases (NCDs) – including coronary heart disease (CHD) and type 2 diabetes – have their origins in prenatal life and in early childhood (Citation1–4). The intrauterine milieu which is influenced by a large number of factors – including maternal factors, hormones and placental function – affects the developing fetus via a number of pathways resulting in the programming of either positive or adverse health outcomes. In 1995 David Barker wrote: “The fetal origins hypothesis states that fetal undernutrition in middle to late gestation, which leads to disproportionate fetal growth, programmes later coronary heart disease” (Citation5). Since those days, this field of research has emerged largely. Originally focusing primarily on body size at birth and the long-term consequences of being born small, it has been moving towards understanding the underlying causes of programming of health and disease as well as potential means to intervene.
The underlying principle within the DOHaD-field is that nutritional, hormonal and metabolic factors active during sensitive periods of development may permanently influence the health of the growing fetus (Citation6–8). This is believed to work through programming, which in general refers to a process where a stimulus or an insult, taking place at a sensitive period of development, may have lifelong consequences, by affecting the structure and physiology of cells and organs. In the animal world there are numerous examples of life long effects of programming during sensitive periods of development. One classical example is when a female rat is injected with testosterone propionate on the 5th day after birth. The development is normal until puberty, however the female rat fails to ovulate. Pituitary and ovarian functions are normal, but release of gonadotrophin by the hypothalamus has been permanently altered. If the same injection is given when the animal is 20 days old, it has no similar effects (Citation9).
Early programming may be advantageous from an evolutionary point of view, in order for the organism to remain plastic during development, it may also have long-term deleterious effects in modern societies. The HBCS has been focusing upon the importance of factors active during periods in early life as well as throughout the life course and their influence on health in 20,431 people born 1924–44. This review will focus upon findings from the HBCS over the past 20 years.
Helsinki Birth Cohort Study
HBCS II includes 13,345 people born between 1934 and 1944 at Helsinki University Central Hospital or at the Midwives’ Hospital in Helsinki, Finland. Information on maternal characteristics, prenatal and childhood growth has been collected from birth, child welfare clinics, and school health care records. The birth records include data on birth weight, length at birth, head circumference, placental measurements, gestational age, and maternal age, height and weight prior to delivery. Serial measurements of body size throughout childhood are available and the subjects have, on average, 17 measurements of height and weight from birth to 11 years of age. Besides data on health and growth during childhood these records also include information on socioeconomic factors including living conditions and paternal occupation. The cohort has been followed up longitudinally by register linkage to national Finnish registers, which provide information on both morbidity and mortality from a life course perspective (Citation3,Citation10–12).
Several clinical examinations originally including 2003 randomly selected individuals have provided more detailed information on metabolic and genetic aspects and their associations with growth and adult health outcomes. The clinical examinations have included 75 g oral glucose tolerance testing, measurement of blood pressure, anthropometric measurements including height, weight, waist circumference, and body composition. Blood samples have been drawn for measurements of e.g. lipids, inflammatory markers, and adipocytokines. Dietary and exercise habits, physical functioning, mental health and aging-related aspects have also been assessed.
An older study cohort born 1924–1933 has also been studied longitudinally. This cohort included 3639 men and 3447 women who were born at Helsinki University Central Hospital and who went to school in Helsinki (Citation13). Five hundred people from this cohort have been studied clinically (Citation14). This review will focus primarily on the cohort born between 1934 and 1944, although soma data from the older cohort will be presented.
Body size at birth and coronary heart disease (CHD)
Finland used to be a dark spot on the world map in relation to CHD morbidity and mortality, this was the case especially among Finnish men (Citation15). Therefore one of the original aims of the HBCS was to study whether early growth would be associated with an increased risk of CHD in Finnish men. This certainly seemed to be the case and Finnish men who were thin at birth – they had a low ponderal index – had higher death rates from CHD compared with those not born thin (Citation13).
These findings supported previous findings by Barker and colleagues in the UK (Citation1). Further studies from the HBCS have been able to replicate the original findings by showing that various markers of non-optimal prenatal growth associate with an increased risk for CHD later in life. These markers include birth weight, body mass index and ponderal index at birth (Citation3,Citation13).
There were marked differences in hazards ratios (HR) for CHD in different birth weight categories; men with a birth weight <2500 g had a HR for CHD of 3.63 (95% CI 2.02–6.51) compared with men born with a birth weight >4000 g (Citation16). Among men a 1 kg decrease in birth weight was associated with a 33% increase in mortality from CHD.
There are well-recognized gender differences, e.g. in age at onset of CHD. Interestingly there are also gender differences in relation to early programming of CHD. We have been examining whether women who develop CHD in adult life have different patterns of early growth compared with men who develop CHD. CHD among women in the HBCS was associated with low birth weight but it was more strongly associated with short length at birth. The risk for women to develop CHD increased by 10% for each cm decrease in length at birth (Citation3,Citation17,Citation18).
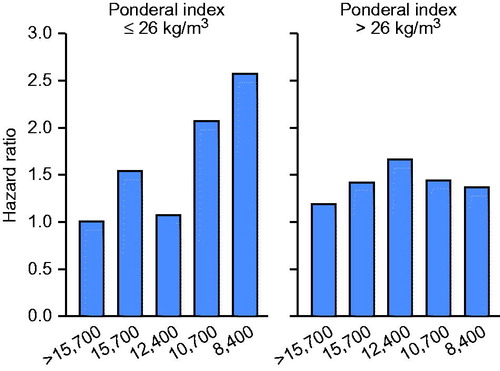
It is a well-known fact that socio-economical factors influence later risk to develop CHD (Citation19). In other words, a lower socioeconomic status (SES) has been associated with an increased risk of CHD. However, little is known whether there is an interaction between SES and prenatal growth on later risk for CHD. These findings are illustrated in showing the HR for CHD among men according to income in adult life and ponderal index at birth. Men who had lower SES in adult life had increased rates of CHD, however these effects were strongest in men who were thin at birth. Men not born thin tended to be resilient to the adverse effects of a low SES in adult life as shown in the right hand panel of (Citation20).
Figure 1. Hazard ratio for CHD among Finnish men according to yearly income (in pounds sterling) and ponderal index at birth.
Childhood growth and CHD – the mismatch concept
After the original study findings from Hertfordshire, UK, showing an inverse association between body size at birth and CHD risk in adult life several studies around the world have been able to replicate the findings (Citation1,Citation21). In the Hertfordshire study it was reported that not only a small body size at birth but also a low weight at 12 months was associated with an increased risk for CHD (Citation21).
One major aim of the HBCS was to assess whether childhood growth modifies the risk for CHD that seems to be established already in utero. Using a longitudinal study setting we have explored this question. Irrespective of body size at birth, low weight gain during infancy was associated with an increased risk of CHD. Low height, weight, and BMI at 1 and 2 years of age increased the risk for CHD among men in adult life. A low SES in childhood further increased the risk for CHD, probably because it was associated with a less optimal growth during infancy (Citation3,Citation13,Citation16,Citation17).
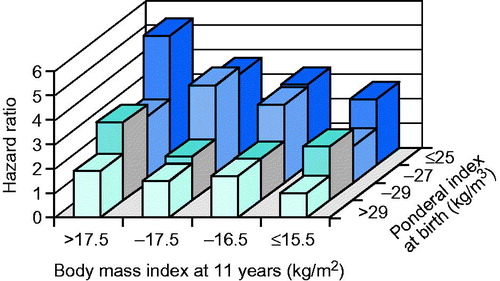
However, a rapid gain in weight and BMI later in childhood, after infancy, seemed to be associated with an increased risk of CHD. This effect was strongest among those born small or thin. This pattern of growth has very appropriately been termed “mismatch” (Citation22). Mismatch refers to the situation when one becomes relatively overweight later in childhood in relation to one’s starting point in life. This is illustrated by the HRs for coronary events among men and women in the HBCS. In simultaneous regressions, the HRs associated with 1 SD increase in BMI at 2 years were 0.76 (CI 95% 0.66–0.87) and 1.14 (CI 95% 1.00–1.31) at 11 years of age, among males, respectively. The corresponding HRs for females were 0.62 (CI 95% 0.46–0.82) and 1.35 (CI 95% 1.02–1.78), respectively (Citation3). Besides illustrating the mismatch concept these findings also illustrate that knowing body size at one time point in childhood is not very helpful in predicting later disease risk. Further the risk for coronary events in adult life was more strongly associated with the tempo of gain in adiposity in childhood that with the degree of adiposity at any particular age. This is further illustrated in showing the HR for CHD among Finnish men according to ponderal index at birth and body mass index at age 11 years.
Figure 2. Hazard ratio for CHD among Finnish men according to ponderal index at birth and body mass index at age 11 years.
Prenatal growth and type 2 diabetes
Type 2 diabetes (T2D) has traditionally been associated with adiposity and a high birth weight; offspring of women with gestational diabetes mellitus (GDM) typically have a high birth weight and they are at increased risk of T2D later in life (Citation23,Citation24). Therefore the diabetes society was largely surprised by the findings from the Hertfordshire study among men aged 64 years showing a strong inverse association between birth weight, weight at 12 months and impaired glucose regulation in adult life (Citation2). These original findings have been replicated in several cohorts around the world including the HBCS (Citation4).
In the HBCS those individuals who developed T2D were not large at birth or during infancy. On the other hand childhood obesity was extremely uncommon in those days in Finland, when the people belonging to the HBCS cohort grew up. T2D is a major risk factor for CHD and therefore it is no surprise that the early growth of those who developed T2D in adult life is rather similar to the growth of those who develop CHD. A small body size in infancy is associated with an increased risk for T2D. Further a mismatch between body size at birth and during infancy and body size later in childhood is a characteristic feature of the growth pattern predisposing to T2D. In other words a very similar pattern of growth as described in relation to CHD (Citation25,Citation26).
Adiposity rebound has been defined as the age, after infancy, when a child’s BMI is at a minimum. In children after 1–2 years of age adiposity usually decreases to a nadir at around 6 years of age before it starts to increase again. This increase in BMI has been termed the adiposity or BMI rebound (Citation27). An early age at BMI rebound has been associated with later obesity (Citation28,Citation29). Within the HBCS we have studied whether an early age at BMI rebound is associated with an increased risk of T2D (Citation30). This was indeed the case and those with the youngest age at BMI rebound had the highest cumulative incidence of T2D in adult life. The difference in cumulative incidence being almost five-fold when comparing those who had a BMI rebound before 5 years of age (8.6%) with those with a BMI rebound after 7 years of age (1.8%). We have been speculating that an early BMI rebound could be associated with a life-long setting of hormones and growth factors that facilitate the deposition of fat and thereby cause a predisposition to obesity, an unfavorable body composition and T2D.
T2D is a heterogeneous disorder characterized by several different phenotypes. Therefore it is no surprise that there is not just one single pathway of early growth leading to T2D. This has been shown among the Pima Indians already decades ago (Citation31). Among Pima Indians a U-shaped association between birth weight and later risk for T2D has been reported. The increased risk for T2D among those born with a high birth weight was largely explained by the high prevalence of gestational diabetes among the Pima Indian women. A similar U-shaped association between body size at birth and T2D has also been reported in other populations (Citation32). Therefore we wanted to study whether there are different pathways of early growth identifiable in the HBCS associated with an increased risk of T2D in later life. In fact we identified two different pathways of early growth associated with T2D in later life. One of them was the “classical” low birth weight pathway, while the other was associated with a higher birth weight and higher BMI throughout childhood. These different pathways of early growth may explain some of the heterogeneity and different phenotypes encountered in clinical practice (Citation26).
Early growth and other health outcomes
The association between a small body size at birth and elevated blood pressure in later life has been reported in numerous studies including the HBCS (Citation33–35). In general this has been considered being a result of slow fetal growth rather than premature birth. However, less is known about how childhood growth modifies the risks associated with a low birth weight. Hypertension seems to originate in slow prenatal growth followed by rapid compensatory growth in childhood. At birth those who developed hypertension were thin and short, and they gained weight slowly up to age 2 years; thereafter they grew rapidly so that at age 11 years their body size was around the average. The odds ratio for hypertension associated with each kg increase in birth weight was 0.42 (95% CI 0.32–0.56); while the corresponding odds ratio for each 10 kg increase in current adult weight was 1.85 (95% CI 1.66–2.05) (Citation33,Citation36–38).
Hypertension is one major risk factor for stroke and therefore one could assume that the early growth pattern associated with an increased risk for stroke is similar to that of hypertension. This is also the case; hazard ratios for stroke declined with increasing weight in infancy. In other words thinness in infancy was associated with an increased risk for stroke later in life while no major influence of growth later in childhood was observed (Citation11,Citation39).
Non-alcoholic fatty liver disease (NAFLD), defined by a liver fat content exceeding 5–10% has a prevalence of 20–30% in Western societies, is one of the most common causes of chronic liver disease (Citation40,Citation41). NAFLD is closely associated with obesity, the metabolic syndrome and T2D. The developmental origins of NAFLD – defined by the NAFLD liver fat score and equation – have also been studied in the HBCS in 1587 individuals. Several measurements of body size at birth and during childhood were negatively associated with NAFLD. The findings remained significant also after adjustment for adult body size and degree of adiposity. A mismatch between body size in infancy and adiposity in adult life was associated with a significantly higher risk for NAFLD; the odds ratio being 18.5 (95% CI 10.1–33.6) when comparing those belonging to the lowest BMI tertile at age 2 years and who were obese as adults to those from the same BMI tertile at 2 years but who were normal weight in adulthood. In the first group over two thirds of the subjects had NAFLD in adult life (Citation42).
Early growth and adult lifestyle
A sedentary lifestyle is one major risk factor for several NCDs including CHD and T2D (Citation23,Citation43). There have been suggestions from animal studies that physical activity patterns as well as dietary habits could be programmed already during prenatal life (Citation44,Citation45).
Within the HBCS we have been assessing the association between early growth and leisure time physical activity (LTPA) among 1967 individuals at a mean age of 62 years (Citation46). A larger body size at birth and a higher weight at 2 years predicted higher intensity of total LTPA, while higher weight and height at 2, 7 and 11 years predicted higher energy expenditure spent on total LTPA.
Cardiorespiratory fitness (CRF) is a good indicator of overall health even after adjustment for physical activity. The possible association between CRF and early growth has been focused upon in a sub-study of the HBCS including 606 people (Citation47). In this study we did not find any significant associations between body size at birth and CRF. However, childhood growth was associated with CRF in adult life, especially height was positively associated with CRF also after adjustment for adult body composition.
Not only physical activity but also dietary habits and dietary intake are associated with later health outcomes. It has in fact been proposed that one plausible mechanism explaining the association between a small body size at birth and the increased disease risk could be through early programming of lifestyle factors including dietary habits and food preferences. From animal studies there is evidence that the early environment can alter food preferences (Citation48–50). Very little human data on the potential association between body size at birth and adult food intake exist (Citation51–53).
Among 1797 people from the HBCS, studied at the mean age of 62 years, we assessed whether body size at birth was associated with food and macronutrient intake. Food intake was assessed with a validated 128 item FFQ (Citation54). Daily intake of fruit and berries was 83 g higher per 1 kg increase in birth weight; on a weekly level this is more than 500 g. Larger body size at birth was further associated with lower intake of fat, higher intake of carbohydrates and higher fiber intake in adult life. In other words prenatal growth seems to modify and program food and macronutrient intake in adult life. Therefore, one could speculate that adult dietary habits could at least partly explain the increased risk between chronic diseases in adulthood and a small body size at birth.
As previously described a small body size at birth has been associated with an increased risk for elevated blood pressure levels. A high intake of salt is a classical risk factor for hypertension (Citation55). To study whether there is a possible interaction between salt intake and birth size we performed one sub-study in the HBCS (Citation56). Among 1512 people on whom we had dietary data available from validated FFQs the relationship between salt intake and blood pressure was assessed. The well-known inverse association between birth weight and systolic blood pressure was evident in the study as a whole. Interestingly among those born with a birth weight <3050 g a higher salt intake was associated with elevated blood pressure; a 1 g higher daily intake of salt was associated with 2.5 mmHg (95% CI 0.4–4.5 mmHg) higher systolic blood pressure – until a saturation point of 10 g. These findings suggest that people born with a low birth weight might be particularly sensitive to the blood pressure elevating effect of salt. No similar association between salt intake and blood pressure was seen in those born with a birth weight >3050 g.
Early growth, mental health and cognition
Findings from epidemiological studies suggest that slower prenatal growth and growth in early childhood could have long-term consequences on mental health and cognition. Data from the HBCS suggest that a smaller body size at birth and slower growth during infancy are associated with poorer cognitive performance and may predict a more rapid age-related cognitive decline. This area has recently been reviewed in great detail and will not be further discussed here (Citation57).
Early life stress and later health outcomes
Severe early life stress (ELS) is a risk factor for several mental health outcomes both during childhood and later in life (Citation58,Citation59). However not only mental health outcomes are influenced by ELS. We have shown that early life stressors can induce physical and biological changes associated with an increased risk for T2D and cardiovascular outcomes later in life, probably as a consequence of re-programming of the HPA-axis. Within the HBCS severe ELS was defined as being sent abroad – unaccompanied by the parents – as a war child during World War II. A recent review focusing upon both epidemiological and clinical studies ranging from mental health outcomes to T2D and cardiovascular disease as well as potential underlying mechanisms has been published and therefore these findings will not be discussed in this review (Citation60).
Physical functioning and aging
As previously discussed health in adulthood is in part a consequence of development and growth taking place during sensitive periods in early life. Consequently, one could assume that physical functioning and aging-related health outcomes would also be associated with early growth.
We have been assessing physical functioning among 1999 individuals born between 1934 and 1944 belonging the HBCS, by the Short Form 36 scale (SF-36) (Citation61). The risk of lower physical functioning at the mean age of 62 years was increased in those with a birth weight <2.5 kg compared with those weighing 3.0–3.5 kg at birth (OR 2.73 95% CI 1.57–4.72). Gain in weight from birth to 2 years was associated with a decreased risk of lower physical functioning, the OR for a 1 SD increase being 0.84 (95% CI 0.75–0.94). The risk of lower physical functioning was highest in individuals with birth weight in the lowest third and BMI at 11 years in the highest third compared with those whose birth weight was in the middle third and BMI at age 11 years was in the highest third (OR 3.08, 95% CI 1.83–5.19) (Citation61).
It has not been explored previously whether early growth is associated with physical performance in old age from a life course perspective. At a mean age of 71 years, physical performance was assessed using the validated Senior Fitness Test (SFT) in 1078 individuals belonging to the HBCS (Citation62). We used multiple linear regression analysis to assess the association between the SFT physical fitness scores and individual life course measurements. Several adult characteristics were associated with physical performance including SES, lifestyle factors, and adult anthropometry. The strongest individual association between life course measurements and physical performance in old age was adult body fat percentage. Higher birth weight and length were associated with better physical performance, even after adjusting for potential confounders, seven decades later. These findings suggest that physical performance in old age is at least partly programmed already during prenatal life.
Maternal factors and offspring health
The prevalence of overweight and obesity is increasing worldwide and within the European Union about one-third of women of reproductive age are overweight, and every fifth is obese (Citation63–66). Maternal obesity is associated with immediate adverse maternal and neonatal outcomes including an increased risk of congenital defects and miscarriage. Further there is increasing evidence suggesting that maternal obesity also has long-term consequences for the offspring’s later health and wellbeing (Citation67,Citation68).
As previously discussed, early life programming has mostly been studied in relation to long-term health outcomes in relation to being born with a small body size or being born preterm. In a study from Scotland maternal obesity in pregnancy was associated with an increased risk of premature death in adult offspring (Citation69). Among 13,345 people belonging to the HBCS we focused upon overall mortality as well as morbidity from cancer, CHD, stroke and T2D among the offspring. Higher maternal pregnancy BMI was associated with an increased risk of cancer, cardiovascular disease, and T2D among the offspring. These associations were independent of socioeconomic measures in childhood and in adult life. The association with T2D was stronger in female offspring, consistent with the transmission of T2D from the mother to her daughters being stronger than the transmission to her sons. Most women in our study cohort were not obese by today’s standard, and therefore we did not focus upon an obese group but upon the trend with maternal adiposity. Nevertheless we still observed several positive associations between maternal BMI and offspring health decades later (Citation70).
Maternal BMI was positively associated with BMI in the offspring as expected (Citation71). Higher maternal BMI was associated with a less favorable body composition in the offspring in later life. There was an interaction between birth weight and maternal BMI on offspring body composition in adult life. In mothers with low BMI, a higher offspring birth weight was associated with lower body fat percentage in adult life. Among those with maternal BMI in the highest fourth, higher offspring birth weight predicted higher body fat percentage in adult life. Our findings suggest that a disadvantageous body composition is programmed in early life, as a consequence of prenatal growth and maternal factors, including adiposity. This may in part underlie the association between maternal obesity and later cardio-metabolic health in the offspring. Further, these findings support the importance of prevention of overweight and obesity in women of childbearing age.
Genetic factors
Several genes have been identified that have been associated with the disease outcomes described in this review, e.g. around 100 genes have been identified that are associated with T2D, among them the insulin gene (Citation72). Insulin is one growth factor involved in and regulating fetal growth and therefore prenatal factors could be involved in the pathogenesis of insulin resistance. Also changes in gene expression could be initiated during the prenatal phase. Further there might be an interaction between prenatal growth and genes associated with T2D. The peroxisome proliferator-activated receptor (PPAR) γ2 gene is involved in the regulation of glucose and lipid metabolism (Citation73,Citation74). We have reported that there is an interaction between PPAR γ2 genotype and early environment in relation to insulin sensitivity (Citation75). The high risk allele, i.e. the Pro12Pro allele was associated with insulin resistance but only among those born with a birth weight <3.500 g. In a similar way we have reported an interaction between body size at birth, genotype (PPARγ2) and T2D (Citation76). In a similar way, interactions between early life environment and PPAR γ2 gene genotype of lipid metabolism have been reported (Citation77). Furthermore, recently we have been studying whether the risk of T2D might be influenced by an interaction between birth weight and common variants of genes identified to be associated with T2D by GWAS studies. In this study we focused upon the TCF7L2, HHEX, PPARG, KCNJ11, SLC30A8, IGF2BP2, CDKAL1, CDKN2A/2B and JAZF1-genes, and formed a genetic risk score. Low birth weight affected the strength of the association between some common high-risk variants – HHEX, CDKN2A/2B and JAZF1 – with T2D (Citation78). More recently studies have been focusing upon genetic loci associated with both body size at birth and adult metabolism; some genetic loci have been identified, e.g. the ADCY5 gene (Citation79). It is obvious that genetic factors play a major role in the pathogenesis of several NCDs. However there seem to be strong evidence for interactions between environmental factors – including early life environment – and genes in the development of several NCDs.
Underlying mechanisms
Epidemiological findings can only report associations but not imply on causality. There are several potential underlying mechanisms explaining why a non-optimal growth might increase the risk for CHD. Dyslipidemia is one major risk factor for CHD. The liver regulates lipid metabolism, and slow growth during infancy can alter liver growth and metabolism. We have been reporting that the pattern of early growth associated with CHD is also associated with an atherogenic lipid profile in adulthood (Citation80). At a mean age of 62 years, about one fifth of the participants in HBCS used lipid lowering drugs. This group had significantly lower BMI at birth and at 2 years of age. When these subjects were excluded from further analyses a low birth BMI was still associated with higher non-HDL cholesterol and apolipoprotein B concentrations. In a similar way a lower BMI at 2 years was associated with lower HDL cholesterol and higher non-HDL cholesterol and apolipoprotein B concentrations. The early growth patterns predisposing to an adverse lipid profile were growth patterns very similar to those described to be associated with CHD and T2D (Citation80). However, these associations are not particularly strong and it has been suggested that postprandial responses might be more important than fasting values. We have shown that a small body size at birth and slow growth during infancy predict postprandial triglyceride and insulin responses in an unfavorable way (Citation81,Citation82). Interestingly there have been suggestions that postprandial regulatory hormone responses differ between those born small compared to those born with normal birth weight (Citation83).
Adiposity and more specifically visceral adiposity is a risk factor for both CHD and T2D. Therefore, another plausible underlying mechanism explaining the association between early growth and later health – including CHD and T2D – is through early programming of adiposity and body composition. The mismatch concept is relevant also in relation to development of an unfavorable body composition (Citation22). A small body size at birth as well as low weight in infancy are associated with less lean body mass in adult life (Citation12,Citation84). Muscle mass is largely responsible for glucose and insulin metabolism. If a thin newborn develops a relatively higher degree of adiposity in later childhood, this usually leads to an unfavorable body composition including a high body fat percentage, predisposing to various cardio-metabolic outcomes. We have shown in the HBCS that both among men and women birth weight is strongly associated with muscle mass in later life. The effect size is large because a 1 kg increase in birth weight among men is associated with a 4.1 kg increase in lean mass in later life; the corresponding increase in women was 2.9 kg. Further, a rapid gain in body mass index during infancy was associated with an increase in adult lean mass without excess fat accumulation, while a rapid gain in body mass index later in childhood resulted in relatively larger increases in fat mass (Citation84).
A small body size at birth is associated with at least two classical hallmarks of T2D, namely insulin resistance and an impairment in insulin secretion (Citation78). The association between body size at birth and insulin resistance could well be mediated by the well-known association between birth size and body composition described previously. Based on animal experiments it is known that a non-optimal prenatal growth is associated with a smaller pancreas, fewer beta cells and less vascularization of the beta cells (Citation85). This could in the presence of a larger body size later in life lead to an increased pressure on the pancreatic beta cells over the life course predisposing to T2D.
Interestingly both human and animal studies have shown that a small body size at birth is associated with a lower number of nephrons in the kidneys at birth and an increased risk for hypertension later in life (Citation86). It has been proposed that the link between non-optimal prenatal growth and hypertension may be through impaired nephrogenesis.
Coronary artery diameter is known to be inversely associated with mortality related to coronary artery bypass surgery. This association is believed to be responsible for the increased risk seen in women and smaller people. The inverse association between height and CHD has been proposed to be due to the fact that taller people having larger coronary vessel diameters. The explanation for this difference is not clear, but, since shorter adult height is believed to reflect, at least in part, early life circumstances this could be another plausible explanation for the association between a small body size at birth and an elevated risk for CHD (Citation87–90).
Early programming of the hypothalamic-pituitary-adrenal axis (HPA-axis) is another interesting mechanism potentially mediating the link between slow prenatal growth and later health outcomes (Citation91,Citation92). Several studies including findings from the HBCS have linked a small body size at birth with non-optimal HPA-activity in adult life. HPA-axis dysfunction again might be involved in the pathogenesis of hypertension, cardiovascular disease, T2D as well as mental and cognitive functions.
Maternal adiposity during pregnancy can lead to an adverse intrauterine environment that impacts embryonic programming and the offspring's susceptibility to developing chronic metabolic diseases later in life. Epigenetic modifications have been suggested to be a major underlying factor linking maternal adiposity and offspring health outcome (Citation93). These epigenetic changes can be mediated through DNA methylation, histone modification and non-coding RNAs. This process can even act over several generations, resulting in changes in gene function without changes in the DNA sequence. It has been shown that maternal dietary intake can cause epigenetic changes in metabolically important genes, which have been associated with offspring body composition later in childhood (Citation94). ELS has also been shown to be associated with epigenetic changes in risk genes associated with depression (Citation95). However, it is unknown whether these findings are causal. Another interesting and less studied area are the potential epigenetic effects induced by the gut microbiome.
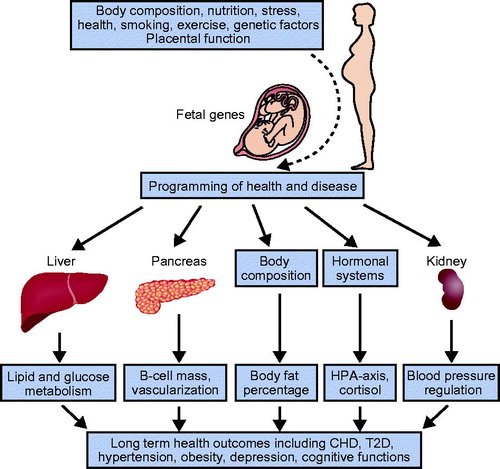
Some potential underlying mechanisms explaining the early programming of adult health and disease are schematically presented in . These include maternal factors, placental function, genetic and epigenetic mechanisms, as well as a large number of other factors.
Figure 3. Some potential underlying mechanisms explaining the early programming of adult health and disease.
End
Birth size serves as a marker of the prenatal environment and considering that body size at birth is only a snapshot of the trajectory of intrauterine growth it is fascinating that long-term health outcomes can be predicted by the body size of the baby and infant. Interestingly fetal programming can occur without any marked effects on body size at birth, as shown in the Dutch Hunger Winter study. Malnutrition – and stress – during prenatal life can have long-term effects by increasing the future risk of CHD and T2D in later life (Citation96). These programming effects depend largely upon timing of the exposure. From a public health point of view preventions aiming at influencing birth size are not feasible. Therefore we should keep in mind that most adult diseases are not programmed, rather the tendency and risk towards a disease is programmed. The early risk factors are to a large extent modifiable by various factors working during the life course. A small body size at birth is associated with an increased risk for T2D in later life. However, regular and frequent exercise is associated with lower rates of glucose intolerance among elderly men. The beneficial effect of exercise was dependent of body size at birth, being strongest among subjects born with a small body size at birth, i.e. those belonging to the high-risk group (Citation97). Another example of the beneficial effects of a healthy life is the fact that resistance training in elderly women born to obese mothers can improve their metabolic status, thus overcoming the deleterious effects of maternal adiposity. These findings stress that, irrespective of parental legacy, a healthy lifestyle can largely improve the metabolic health, among high-risk individuals (Citation98). In other words, lifestyle matters from the cradle to the grave. But ,even more import is ensuring the optimal health of women of reproductive age, since this will have long-term positive health consequences on their offspring.
Funding information
HBCS has been supported by grants from British Heart Foundation, NIH, Finska Läkaresällskapet, the Finnish Special Governmental Subsidy for Health Sciences, Academy of Finland, Samfundet Folkhälsan, Liv och Hälsa, Juho Vainio Foundation, Yrjö Jahnsson Foundation, The Diabetes Research Foundation, Finnish Foundation for Cardiovascular Research, the Signe and Ane Gyllenberg Foundation, EU FP7 (DORIAN) project number 278603, and EU Horizon 2020 (DynaHealth) project number 633595.
Disclosure statement
The author reports no conflicts of interest.
References
- Barker DJ, Osmond C. Infant mortality, childhood nutrition, and ischaemic heart disease in England and Wales. Lancet. 1986;1:1077–81.
- Hales CN, Barker DJ, Clark PM, Cox LJ, Fall C, Osmond C, et al. Fetal and infant growth and impaired glucose tolerance at age 64. BMJ. 1991;303:1019–22.
- Barker DJ, Osmond C, Forsén TJ, Kajantie E, Eriksson JG. Trajectories of growth among children who have coronary events as adults. N Engl J Med. 2005;353:1802–9.
- Whincup PH, Kaye SJ, Owen CG, Huxley R, Cook DG, Anazawa S, et al. Birth weight and risk of type 2 diabetes: a systematic review. JAMA. 2008;300:2886–97.
- Barker DJ. Fetal origins of coronary heart disease. BMJ. 1995;311:171–4.
- Lucas AA. role of nutritional programming in determining adult morbidity. Arch Dis Child. 1994;71:288–90.
- Barker DJ. Developmental origins of adult health and diseases. J Epidemiol Community Health. 2004;58:114–15.
- Warner MJ, Ozanne SE. Mechanisms involved in the developmental programming of adulthood diseases. Biochem J. 2010;427:333–47.
- Barraclough CA. Production of anovulatory, sterile rats by single injections of testosterone propionate. Endocrinology. 1961;68:62–7.
- Eriksson JG, Osmond C, Kajantie E, Forsén T, Barker DJ. Patterns of growth among children who later develop type 2 diabetes or its risk factors. Diabetologia. 2006;49:2853–8.
- Osmond C, Kajantie E, Forsén TJ, Eriksson JG, Barker DJ. Infant growth and stroke in adult life: the Helsinki Birth Cohort Study. Stroke. 2007;38:264–70.
- Ylihärsilä H, Kajantie E, Osmond C, Forsén T, Barker DJ, Eriksson JG. Body mass index during childhood and adult body composition in men and women aged 56–70 y. Am J Clin Nutr. 2008;87:1769–75.
- Eriksson JG, Forsén T, Tuomilehto J, Winter PD, Osmond C, Barker DJ. Catch-up growth in childhood and death from coronary heart disease: longitudinal study. BMJ. 1999;318:427–31.
- Eriksson J, Forsén T, Tuomilehto J, Osmond C, Barker D. Size at birth, fat-free mass and resting metabolic rate in adult life. Horm Metab Res. 2002;34:72–6.
- Keys A, Aravanis C, Blackburn HW, van Buchem FS, Buzina R, Djordjevic BD, et al. Epidemiological studies related to coronary heart disease: characteristics of men aged 40-59 in seven countries. Acta Med Scand Suppl. 1966;460:1–392.
- Eriksson JG, Forsén T, Tuomilehto J, Osmond C, Barker DJ. Early growth and coronary heart disease in later life: longitudinal study. BMJ. 2001;322:949–53.
- Forsén T, Eriksson JG, Tuomilehto J, Osmond C, Barker DJ. Growth in utero and during childhood among women who develop coronary heart disease: longitudinal study. BMJ. 1999;319:1403–7.
- Eriksson JG, Kajantie E, Thornburg K, Osmond C. Prenatal and maternal characteristics and later risk for coronary heart disease among women. Eur J Prev Cardiol. 2016;23:385–90.
- Marmot M, McDowell ME. Mortality decline and widening social inequalities. Lancet. 1986;2(8501):274–6.
- Barker DJ, Forsén T, Uutela A, Osmond C, Eriksson JG. Size at birth and resilience to effects of poor living conditions in adult life: longitudinal study. BMJ. 2001;323:1273–6.
- Barker DJ, Winter PD, Osmond C, Margetts B, Simmonds SJ. Weight in infancy and death from ischaemic heart disease. Lancet. 1989;2:577–80.
- Godfrey KM, Lillycrop KA, Burdge GC, Gluckman PD, Hanson MA. Epigenetic mechanisms and the mismatch concept of the developmental origins of health and disease. Pediatr Res. 2007;61:5R–10R.
- Zaccardi F, Webb DR, Yates T, Davies MJ. Pathophysiology of type 1 and type 2 diabetes mellitus: a 90-year perspective. Postgrad Med J. 2016;92:63–9.
- Baz B, Riveline JP, Gautier JF. Endocrinology of pregnancy: gestational diabetes mellitus: definition, aetiological and clinical aspects. Eur J Endocrinol. 2015;174:R43–51.
- Forsén T, Eriksson J, Tuomilehto J, Reunanen A, Osmond C, Barker D. The fetal and childhood growth of persons who develop type 2 diabetes. Ann Intern Med. 2000;133:176–82.
- Eriksson JG, Forsen TJ, Osmond C, Barker DJ. Pathways of infant and childhood growth that lead to type 2 diabetes. Diabetes Care. 2003;26:3006–10.
- Rolland-Cachera MF, Deheeger M, Bellisle F, Sempé M, Guilloud-Bataille M, Patois E. Adiposity rebound in children: a simple indicator for predicting obesity. Am J Clin Nutr. 1984;39:129–35.
- Rolland-Cachera MF, Deheeger M, Guilloud-Bataille M, Avons P, Patois E, Sempé M. Tracking the development of adiposity from one month of age to adulthood. Ann Hum Biol. 1987;14:219–29.
- Rolland-Cachera MF. Rate of growth in early life: a predictor of later health? Adv Exp Med Biol. 2005;569:35–9.
- Eriksson JG, Forsén T, Tuomilehto J, Osmond C, Barker DJ. Early adiposity rebound in childhood and risk of type 2 diabetes in adult life. Diabetologia. 2003;46:190–4.
- McCance DR, Pettitt DJ, Hanson RL, Jacobsson LT, Knowler WC, Bennett PH. Birth weight and non-insulin dependent diabetes: thrifty genotype, thrifty phenotype, or surviving small baby genotype? BMJ. 1994;308:942–5.
- Wei JN, Sung FC, Li CY, Chang CH, Lin RS, Lin CC, et al. Low birth weight and high birth weight infants are both at an increased risk to have type 2 diabetes among schoolchildren in Taiwan. Diabetes Care. 2003;26:343–8.
- Eriksson J, Forsén T, Tuomilehto J, Osmond C, Barker D. Fetal and childhood growth and hypertension in adult life. Hypertension. 2000;36:790–4.
- Ylihärsilä H, Eriksson JG, Forsén T, Kajantie E, Osmond C, Barker DJ. Self-perpetuating effects of birth size on blood pressure levels in elderly people. Hypertension. 2003;41:446–50.
- Bruno RM, Faconti L, Taddei S, Ghiadoni L. Birth weight and arterial hypertension. Curr Opin Cardiol. 2015;30:398–402.
- Barker DJ, Eriksson JG, Forsén T, Osmond C. Fetal origins of adult disease: strength of effects and biological basis. Int J Epidemiol. 2002;31:1235–9.
- Barker DJ, Forsén T, Eriksson JG, Osmond C. Growth and living conditions in childhood and hypertension in adult life: a longitudinal study. J Hypertens. 2002;20:1951–6.
- Eriksson JG, Forsén TJ, Kajantie E, Osmond C, Barker DJ. Childhood growth and hypertension in later life. Hypertension. 2007;49:1415–21.
- Eriksson JG, Forsén T, Tuomilehto J, Osmond C, Barker DJ. Early growth, adult income, and risk of stroke. Stroke. 2000;31:869–74.
- Bellentani S, Scaglioni F, Marino M, Bedogni G. Epidemiology of non-alcoholic fatty liver disease. Dig Dis. 2010;28:155–61.
- Puri P, Sanyal AL. Nonalcoholic fatty liver disease: definitions, risk factors, and workup. Clin Liver Dis. 2012;1:98–102.
- Sandboge S, Perälä MM, Salonen MK, Blomstedt PA, Osmond C, Kajantie E, et al. Early growth and non-alcoholic fatty liver disease in adulthood – the NAFLD liver fat score and equation applied on the Helsinki Birth Cohort Study. Ann Med. 2013;45:430–7.
- Hu FB. Diet and lifestyle influences on risk of coronary heart disease. Curr Atheroscler Rep. 2009;11:257–63.
- Vickers MH, Sloboda DM. Leptin as mediator of the effects of developmental programming. Best Pract Res Clin Endocrinol Metab. 2012;26:677–87.
- Ellis PJ, Morris TJ, Skinner BM, Sargent CA, Vickers MH, Gluckman PD, et al. Thrifty metabolic programming in rats is induced by both maternal undernutrition and postnatal leptin treatment, but masked in the presence of both: implications for models of developmental programming. BMC Genomics. 2014;15:49.
- Salonen MK, Kajantie E, Osmond C, Forsén T, Ylihärsilä H, Paile-Hyvärinen M, et al. Prenatal and childhood growth and leisure time physical activity in adult life. Eur J Public Health. 2011;21:719–24.
- Salonen MK, Kajantie E, Osmond C, Forsén T, Ylihärsilä H, Paile-Hyvärinen M, et al. Developmental origins of physical fitness: the Helsinki Birth Cohort Study. PLoS One. 2011;6:e22302.
- Bellinger L, Lilley C, Langley-Evans SC. Prenatal exposure to a maternal low-protein diet programmes a preference for high-fat foods in the young adult rat. Br J Nutr. 2004;92:513–20.
- Bellinger L, Langley-Evans SC. Fetal programming of appetite by exposure to a maternal low-protein diet in the rat. Clin Sci. 2005;109:413–20.
- Garcia AP, Palou M, Priego T, Sanchez J, Palou A, Pico C. Moderate caloric restriction during gestation results in lower arcuate nucleus NPY- and alfa-MSH-neurons and impairs hypothalamic response to fed/fasting conditions in weaned rats. Diabetes Obes Metab. 2010;12:403–13.
- Lussana F, Painter RC, Ocke MC, Buller HR, Bossuyt PM, Roseboom TJ. Prenatal exposure to the Dutch famine is associated with a preference for fatty foods and a more atherogenic lipid profile. Am J Clin Nutr. 2008;88:1648–52.
- Stein AD, Rundle A, Wada N, Goldbohm RA, Lumey LH. Associations of gestational exposure to famine with energy balance and macronutrient density of the diet at age 58 years differ according to the reference population used. J Nutr. 2009;139:1555–61.
- Barbieri MA, Portella AK, Silveira PP, Bettiol H, Agranonik M, Silva AA, et al. Severe intrauterine growth restriction is associated with higher spontaneous carbohydrate intake in young women. Pediatr Res. 2009;65:215–20.
- Perälä MM, Männistö S, Kaartinen NE, Kajantie E, Osmond C, Barker DJ, et al. Body size at birth is associated with food and nutrient intake in adulthood. PLoS One. 2012;7:e46139.
- Van Horn L. Dietary sodium and blood pressure: how low should we go? Prog Cardiovasc Dis. 2015;58:61–8.
- Perälä MM, Moltchanova E, Kaartinen NE, Männistö S, Kajantie E, Osmond C, et al. The association between salt intake and adult systolic blood pressure is modified by birth weight. Am J Clin Nutr. 2011;93:422–6.
- Räikkönen K, Pesonen AK, Roseboom TJ, Eriksson JG. Early determinants of mental health. Best Pract Res Clin Endocrinol Metab. 2012;26:599–611.
- Wiesel R. Time does not heal all wounds: quality of life and psychological distress of people who survived the holocaust as children 55 years later. J Trauma Stress. 2003;16:295–9.
- Afifi TO, Brownridge DA, Cox BJ, Sareen J. Physical punishment, childhood abuse and psychiatric disorders. Child Abuse Neglect. 2006;30:1093–103.
- Eriksson M, Räikkönen K, Eriksson JG. Early life stress and later health outcomes – findings from the Helsinki Birth Cohort Study. Am J Hum Biol. 2014;26:111–16.
- von Bonsdorff MB, Rantanen T, Sipilä S, Salonen MK, Kajantie E, Osmond C, et al. Birth size and childhood growth as determinants of physical functioning in older age: the Helsinki Birth Cohort Study. Am J Epidemiol. 2011;174:1336–44.
- Eriksson JG, Osmond C, Perälä MM, Salonen MK, Simonen M, Pohjolainen P, et al. Prenatal and childhood growth and physical performance in old age-findings from the Helsinki Birth Cohort Study 1934–1944. Age (Dordr). 2015;37:108.
- Heslehurst N, Rankin J, Wilkinson JR, Summerbell CD. A nationally representative study of maternal obesity in England, UK: trends in incidence and demographic inequalities in 619 323 births, 1989–2007. Int J Obes (Lond). 2010;34:420–8.
- Flegal KM, Carroll MD, Kit BK, Ogden CL. Prevalence of obesity and trends in the distribution of body mass index among US adults, 1999–2010. JAMA. 2012;307:491–7.
- Popkin BM, Adair LS, Ng SW. Global nutrition transition and the pandemic of obesity in developing countries. Nutr Rev. 2012;70:3–21.
- Available at: http://www.worldobesity.org/what-we-do/policy-prevention/worldobesitypublications/reports/ (accessed 11 March 2016)
- Boney CM, Verma A, Tucker R, Vohr BR. Metabolic syndrome in childhood: association with birth weight, maternal obesity, and gestational diabetes mellitus. Pediatrics. 2005;115:e290–6.
- Roman AS, Rebarber A, Fox NS, Klauser CK, Istwan N, Rhea D, et al. The effect of maternal obesity on pregnancy outcomes in women with gestational diabetes. J Matern Fetal Neonatal Med. 2011;24:723–7.
- Reynolds RM, Allan KM, Raja EA, Bhattacharya S, McNeill G, Hannaford PC, et al. Maternal obesity during pregnancy and premature mortality from cardiovascular event in adult offspring: follow-up of 1 323 275 person years. BMJ. 2013;347:f4539.
- Eriksson JG, Sandboge S, Salonen MK, Kajantie E, Osmond C. Long-term consequences of maternal overweight in pregnancy on offspring later health: findings from the Helsinki Birth Cohort Study. Ann Med. 2014;46:434–8.
- Eriksson JG, Sandboge S, Salonen M, Kajantie E, Osmond C. Maternal weight in pregnancy and offspring body composition in late adulthood: findings from the Helsinki Birth Cohort Study (HBCS). Ann Med. 2015;47:94–9.
- Pal A, McCarthy MI. The genetics of type 2 diabetes and its clinical relevance. Clin Genet. 2013;83:297–306.
- Deeb SS, Fajas L, Nemoto M, Pihlajamäki J, Mykkänen L, Kuusisto J, et al. A Pro12Ala substitution in PPARgamma2 associated with decreased receptor activity, lower body mass index and improved insulin sensitivity. Nat Genet. 1998;20:282–7.
- Altshuler D, Hirschorn JN, Klannemark M, Lindgreb CM, Vohl MC, Nemesh J, et al. The common PPARgamma Pro12Ala polymorphism is associated with decreased risk of type 2 diabetes. Nat Genet. 2000;26:76–80.
- Eriksson JG, Lindi V, Uusitupa M, Forsen TJ, Laakso M, Osmond C, et al. The effect of the Pro12Ala polymorphism of the peroxisome proliferator-activated receptor-γ2 gene on insulin sensitivity and insulin metabolism interact with size at birth. Diabetes. 2002;51:2321–4.
- Eriksson JG, Osmond C, Lindi V, Uusitupa M, Forsen T, Laakso M, et al. Interactions between peroxisome proliferator-activated receptor gene polymorphism and birth length influence risk for type 2 diabetes. Diabetes Care. 2003;26:2476–7.
- Eriksson J, Lindi V, Uusitupa M, Forsén T, Laakso M, Osmond C, et al. The effects of the Pro12Ala polymorphism of the PPARgamma-2 gene on lipid metabolism interact with body size at birth. Clin Genet. 2003;64:366–70.
- Pulizzi N, Lyssenko V, Jonsson A, Osmond C, Laakso M, Kajantie E, et al. Interaction between prenatal growth and high-risk genotypes in the development of type 2 diabetes. Diabetologia. 2009;52:825–9.
- Horikoshi M, Yaghootkar H, Mook-Kanamori DO, Sovio U, Taal HR, Hennig BJ, et al. New loci associated with birth weight identify genetic links between intrauterine growth and adult height and metabolism. Nat Genet. 2013;45:76–82.
- Kajantie E, Barker DJ, Osmond C, Forsén T, Eriksson JG. Growth before 2 years of age and serum lipids 60 years later: the Helsinki Birth Cohort Study. Int J Epidemiol. 2008;37:280–9.
- Perälä MM, Valsta LM, Kajantie E, Leiviskä J, Eriksson JG. Impact of early growth on postprandial responses in later life. PLoS One. 2011;6:e24070.
- Perälä MM, Eriksson JG. Early growth and postprandial glucose, insulin, lipid and inflammatory responses in adulthood. Curr Opin Lipidol. 2012;23:327–33.
- Perälä MM, Kajantie E, Valsta LM, Holst JJ, Leiviskä J, Eriksson JG. Early growth and postprandial appetite regulatory hormone responses. Br J Nutr. 2013;110:1591–600.
- Ylihärsilä H, Kajantie E, Osmond C, Forsén T, Barker DJ, Eriksson JG. Birth size, adult body composition and muscle strength in later life. Int J Obes (Lond). 2007;31:1392–9.
- Desai M, Crowther NJ, Ozanne SE, Lucas A, Hales CN. Adult glucose and lipid metabolism may be programmed during fetal life. Biochem Soc Trans. 1995;23:331–5.
- Luyckx VA, Bertram JF, Brenner BM, Fall C, Hoy WE, Ozanne SE, et al. Effect of fetal and child health on kidney development and long-term risk of hypertension and kidney disease. Lancet. 2013;382:273–83.
- Elezi S, Kastrati A, Neumann FJ, Hadamitzky M, Dirschinger J, Schömig A. Vessel size and long-term outcome after coronary stent placement. Circulation. 1998;98:1875–80.
- Schunkert H, Harrell L, Palacios IF. Implications of small reference vessel diameter in patients undergoing percutaneous coronary revascularization. J Am Coll Cardiol. 1999;34:40–8.
- Sheifer SE, Canos MR, Weinfurt KP, Arora UK, Mendelsohn FO, Gersh BJ, et al. Sex differences in coronary artery size assessed by intravascular ultrasound. Am Heart J. 2000;139:649–53.
- Dodge JT, Jr, Brown BG, Bolson EL, et al. Lumen diameter of normal human coronary arteries. Influence of age, sex, anatomic variation, and left ventricular hypertrophy or dilation. Circulation. 1992;86:232–46.
- Matthews SG. Early programming of the hypothalamo-pituitary-adrenal axis. Trends Endocrinol Metab. 2002;13:373–80.
- Seckl JR, Meaney MJ. Glucocorticoid programming. Ann N Y Acad Sci. 2004;1032:63–84.
- Godfrey KM, Costello PM, Lillycrop KA. The developmental environment, epigenetic biomarkers and long-term health. J Dev Orig Health Dis. 2015;6:399–406.
- Godfrey KM, Sheppard A, Gluckman PD, Lillycrop KA, Burdge GC, McLean C, et al. Epigenetic gene promoter methylation at birth is associated with child's later adiposity. Diabetes. 2011;60:1528–34.
- Khulan B, Manning JR, Dunbar DR, Seckl JR, Raikkonen K, Eriksson JG, et al. Epigenomic profiling of men exposed to early-life stress reveals DNA methylation differences in association with current mental state. Transl Psychiatry. 2014;4:e448.
- Painter RC, Roseboom TJ, Bleker OP. Prenatal exposure to the Dutch famine and disease in later life: an overview. Reprod Toxicol. 2005;20:345–52.
- Eriksson JG, Ylihärsilä H, Forsén T, Osmond C, Barker DJ. Exercise protects against glucose intolerance in individuals with a small body size at birth. Prev Med. 2004;39:164–7.
- Bucci M, Huovinen V, Guzzardi MA, Koskinen S, Raiko JR, Lipponen H, et al. Resistance training improves skeletal muscle insulin sensitivity in elderly offspring of overweight and obese mothers. Diabetologia. 2016;59:77–86.