
Abstract
Insulin-degrading enzyme (IDE) is a major enzyme responsible for insulin degradation. In addition to insulin, IDE degrades many targets including glucagon, atrial natriuretic peptide, and beta-amyloid peptide, regulates proteasomal degradation and other cell functions. IDE represents a pathophysiological link between type 2 diabetes (T2DM) and late onset Alzheimer’s disease (AD). Potent and selective modulators of IDE activity are potential drugs for therapies of both diseases. Acute treatment with a novel IDE inhibitor was recently tested in a mouse study as a therapeutic approach for the treatment of T2DM. In contrast, effective IDE activators can be used for the AD treatment. However, because of the pleiotropic IDE action, the sustained treatment with systemic IDE modulators should be carefully tested in animal studies. Development of substrate-selective IDE modulators could overcome possible adverse effects of IDE modulators associated with multiplicity of IDE targets.
Insulin-degrading enzyme (IDE) represents a pathophysiological link between type 2 diabetes (T2DM) and Alzheimer’s disease (AD).
Selective modulators of IDE activity are potential drugs for both T2DM and AD treatment.
Development of substrate-selective IDE modulators could overcome possible adverse effects of IDE modulators associated with multiplicity of IDE targets.
KEY MESSAGES
Introduction
Insulin-degrading enzyme (IDE), the 110 kDa Zn requiring metalloproteinase, is a major enzyme responsible for insulin degradation (Citation1). The ability of IDE to degrade the insulin B chain was observed over 60 years ago (Citation2), though the enzyme responsible for this cleavage was identified more recently (Citation3). IDE is localized in the cytoplasm, cell membranes, in some cell organelles (endosomes, peroxisomes, mitochondria) and secreted into the extracellular space (Citation1,Citation4–6). IDE is ubiquitously expressed, both in insulin-sensitive and in non-insulin-sensitive cells, supporting a multifunctional role for this protein, and is found in most organisms, from E. coli to mammals (Citation1,Citation3).
Molecular mechanism of substrate degradation by IDE
Major sites of IDE-mediated insulin degradation are liver and kidney, and the liver removes approximately 75% during the first portal passage (Citation7). Insulin degradation occurs also in adipocytes, fibroblasts, monocytes, lymphocytes, gastrointestinal cells, and many other tissues (Citation1). Under normal conditions, almost all insulin is degraded intracellularly via the receptor-mediated internalization mechanism of or at least by membrane-bound processes (Citation1,Citation8,Citation9). During the receptor-mediated degradation, the insulin receptor binds insulin and internalizes into endosomes. After formation, endosomes rapidly acidify, which results in dissociation of insulin from its receptor and its degradation (maximum 50% of internalized insulin is degraded). Obviously, final degradation steps occur in lysosomes. The insulin receptor is recycled to the cell surface along with degraded and intact insulin (Citation1).
Although insulin is a preferred substrate for IDE, the enzyme also cleaves a large body of other short polypeptides including glucagon, atrial natriuretic peptide, beta-amyloid peptide (Aβ), transforming growth factor alpha, and insulin-like growth factor 1 and 2 (Citation1), many of which are amyloidogenic. A study of IDE by Shen et al. (Citation10) elucidated the functional mechanisms of the substrate degradation by IDE. N and C terminal units of IDE form two halves of a proteolytic chamber containing the zinc-binding active site. In the open conformation, substrates can enter and products can leave. The bipartite catalytic site is fully formed and active only after the protease adopts the closed conformation. Therefore, IDE can cleave only peptides of up to 70 amino acids which fit inside the chamber, but not large proteins, and it does not show strong cleavage site specificity (e.g., a particular amino acid sequence). Interestingly, targeted mutations disrupting the contacts between IDE-N and IDE-C result in a 40-fold increase in catalytic activity (Citation10).
In addition to degradative activity, IDE plays a regulatory role for IDE in a number of cell functions including regulation of androgen and glucocorticoid receptors (Citation11), peroxisomal fatty acid oxidation (Citation12), antigen presentation (Citation13), cellular growth and differentiation (Citation14), and proteasomal degradation (Citation1,Citation15,Citation16).
IDE-mediated regulation of proteasomal degradation
The well-known ability of insulin to suppress cellular protein degradation is fulfilled by the inhibition of autophagic proteolysis via the amino acid-mTOR signaling pathway (Citation17) and by the suppression of the ubiquitin–proteasomal pathways where IDE is suggested to be involved (Citation1,Citation15,Citation16). The proteasome is a multi-catalytic protease, responsible for the degradation of misfolded, abnormal, oxidized or no longer needed proteins and maintenance of cellular integrity (Citation18).
The IDE role in the regulation of proteasomal function and its mechanism were investigated extensively by Duckworth and co-workers. The interaction of cytosolic IDE with the proteasome increases its proteolytic activity (Citation1,Citation15,Citation16) which was confirmed in experiments with IDE gene silencing (Citation19). During endosomal insulin degradation, intact and degraded insulin particularly enters the cytosol. Binding of insulin to cytosolic IDE causes the dissociation of the IDE–proteasome complex which obviously leads to a decrease of proteasomal activity (Citation20). Following insulin degradation, IDE reassociates with the proteasome. Notably, substrate-degradation by IDE is required for proteasome inhibition, and substrate binding alone is insufficient to induce proteasomal inhibition (Citation15,Citation21).
Interestingly, IDE-associated diseases such as type 2 diabetes (T2DM) and Alzheimer’s disease (AD) are accompanied by aberrations of the ubiquitin–proteasome systems (Citation22,Citation23). However, IDE knockout mice do not show any overt evidence of defects in proteasomal function, as demonstrated by phenotyping data available online (http://www.mousephenotype.org/data/genes/MGI:96412#section-associations). Therefore, the IDE role in the regulation of proteasomal function needs further investigations.
Physiological modulators of IDE activity
IDE action obviously undergoes a metabolic control which adapts its activity to the actual metabolic state. Physiological regulation of IDE functions is accomplished in multiple ways, both via a change of IDE expression and via modulation of the enzyme catalytic activity. Particularly, insulin treatment induces an increase of IDE activity that obviously represents a negative feedback mechanism which switches off the insulin action (Citation9). Two IDE isoforms are known to contain exon 15a or 15b, and the 15b-isoform demonstrates less catalytic efficiency for insulin and Aβ in comparison to the wild-type 15a-isoform (Citation5). The regulation of the 15a/15b-isoform ratio may be one of the mechanisms explaining the increase of IDE activity after the insulin treatment in the hepatoma cell culture (Citation9). Furthermore, the binding of some peptide substrates to one of the IDE subunits leads to allosteric regulation of IDE activity with a shift of the IDE dimer/tetramer equilibrium to the more active dimer and activation of the adjacent subunit being induced (Citation24).
Moreover, free fatty acids (Citation25) and nucleotide triphosphates (Citation26) inhibit insulin-degrading activity, whereas sex steroids increase IDE expression (Citation27). The pH values within cellular compartments also modulate IDE activity and conformation (Citation28). High glucose concentration attenuates the insulin-induced IDE activation in vitro (Citation9). Additionally, IDE-interacting proteins are able to modulate IDE activity. Recently described IDE interaction with the mitochondrial protein SIRT4 can alter IDE protease activity by ADP-ribosylation (Citation29). Two IDE-interacting proteins (14 kDa and 6 kDa) were described as inhibiting insulin-degrading activity in rodents and one of them was identified as ubiquitin (Citation30).
Development of chemical modulators of IDE activity
Since its discovery in 1949 by I. Arthur Mirsky (Citation2), IDE was considered to be an attractive target for the treatment of diabetes and other disorders associated with impaired insulin signaling. In support of this approach, Mirsky found that liver extracts containing an IDE inhibitor enhance the action of insulin when injected into rabbits (Citation31). Nevertheless, it took more than 50 years for effective chemical modulators of IDE to be developed.
A large number of chemical inhibitors of IDE activity (chelators, divalent cations, thiol-blocking agents, etc.) have been known for a long time, but most of them could not be used as therapeutic agents because they are non-selective and/or highly toxic, and demonstrate extremely low potency (Citation1,Citation16,Citation32). Moreover, a range of well-known therapeutic agents such as the antiparasitic drug suramin, antiretroviral drug nelfinavir, antibiotic bacitracin, and novel therapeutic compounds were found to regulate IDE expression and activity (Citation27,Citation32–46) (). However, the development of more potent and selective modulators of IDE activity is needed because they represent potential drugs for T2DM and AD therapies.
Table 1. Therapeutic agents regulating IDE expression and activity.
Modern drug design instruments hold the key to realize novel potent IDE inhibitors using different approaches. The first high-potency IDE inhibitors developed by Leissring et al. were based on analysis of substrate-cleavage specificity (Citation32). These peptide hydroxamate inhibitors showed a novel mode of inhibition based on stabilization of IDE's “closed”, inactive conformation. They decreased extracellular and intracellular insulin degradation potentiating insulin signaling in the cultured cells (Citation32), but were relatively large and difficult to synthesize. Further, a much more highly selective and potent IDE inhibitor, 6bK, was discovered by the screening of a DNA-templated library of cyclic peptides (Citation47). Using high-throughput compound screening, the same research group discovered small-molecule IDE activators which significantly stimulate IDE proteolytic activity, especially in the presence of ATP (Citation48). Some small-molecule compounds enhancing IDE mediated proteolysis of insulin and Aβ were discovered by computational screening using the crystal structure of IDE (Citation49). In turn, using a structure-based approach, Durham et al. designed a series of novel dual exosite-binding IDE inhibitors which do not interact with the catalytic zinc of the protease (Citation50). However, as published in the literature, only two of abovementioned IDE inhibitors were tested in vivo and demonstrated stability, pharmacokinetic properties and physiological effects, thus confirming their potential for the development of antidiabetic drugs (Citation47,Citation50). The results of these studies are described in detail below.
Notably, the regulation of IDE activity by small molecule activators and inhibitors is in several cases substrate-specific (Citation48,Citation49,Citation51,Citation52). In particular, a series of IDE inhibitors were developed by the optimization of peptide hydroxamate inhibitors, the potency of which varies as much as 60-fold in a substrate-specific manner (Citation52). Authors observed both increases and decreases in potency, depending on the inhibitor and substrate tested (Citation52). The phenomenon of substrate-specific modulation was described previously for other substances, e.g., nucleotide polyphosphates. Indeed, ATP inhibits the degradation of insulin and Aβ but activates the degradation of short fluorogenic substrates (Citation26,Citation48). Importantly, the development of substrate-selective IDE modulators could overcome the possible adverse effects of IDE modulators associated with multiplicity of IDE targets and discussed below.
IDE and type 2 diabetes
IDE role in the pathogenesis of T2DM
T2DM is a metabolic disturbance characterized by insulin resistance which may be combined with relatively reduced insulin secretion. Genetic polymorphisms within the IDE locus on the chromosome 10 demonstrated an association with increased risk of T2DM in studies using genome wide and candidate gene approaches (Citation53–56). We recently showed association of IDE polymorphisms with all aspects of impaired insulin metabolism, i.e., decreased insulin secretion, insulin sensitivity, and hepatic insulin degradation (Citation56). IDE mutation in the Goto–Kakizaki rat model causes altered cellular insulin degradation in addition to other characteristics typical of T2DM (Citation57).
IDE knockout mice are characterized by classic features of T2DM – decreased insulin degradation, hyperinsulinemia, and glucose intolerance (Citation58). Moreover, in the culture of pancreatic beta-cells, a non-specific IDE inhibitor (bacitracin) increases amyloid accumulation (Citation46) which is also a hallmark associated with T2DM. On the other side, IDE overexpression increases insulin degradation and decreases the efficiency of insulin stimulation in the insulin signaling pathway (Citation59). These data demonstrate that the regulation of the IDE level and its activity may contribute to the T2DM pathogenesis.
Unexpectedly, data on IDE activity in the biological fluids (blood cells, plasma, wound fluid, cerebrospinal fluid) of diabetic patients available in the literature are controversial and apparently depend on a range of factors including study design, the type of diabetes mellitus, and type of antidiabetic treatment. One of the studies showed that IDE activity of erythrocytes was increased in T2DM patients taking sulphonylureas, in a subgroup with well-controlled diabetes, and in patients with secondary failure to oral therapy, but it was unmodified in type 1 diabetic patients in good control (Citation60). In another study, an increase of IDE activity in plasma and erythrocytes was demonstrated both in insulin-dependent and in non-insulin-dependent diabetic patients (Citation61). In rodents, a decrease of hepatic insulin degradation was observed in diabetic rats, which was restored to near normal levels following insulin treatment (Citation62,Citation63). A decreased IDE activity in adipocytes of pre-diabetic and diabetic subjects was demonstrated (Citation64). Amazingly, extremely few reports to date concerning the functional regulation of the IDE in liver or liver cells have been published (Citation62,Citation65,Citation66).
In our study in human hepatoma cells, we observed an increase of IDE activity after insulin treatment under normal glucose concentration (Citation9) which obviously represents a negative physiological feedback mechanism of the regulation of insulin action. Notably, in high glucose conditions, this effect was abolished (Citation9). Thus, our findings suggested that hyperglycemia may provoke the disturbance of IDE activity in T2DM. Moreover, we found a decrease of IDE mRNA expression in the liver of diabetic subjects (Citation67). Further, decreased hepatic insulin degradation is an early marker of a disturbed insulin metabolism in obesity and T2DM (Citation68,Citation69), and prodiabetic genetic polymorphisms of the IDE gene are associated with reduced insulin clearance (Citation56). Thus, despite some inconsistency, most data suggest a decrease of IDE function in subjects with T2DM.
Modulators of IDE activity as attractive drugs for T2DM treatment
I. Arthur Mirsky, who discovered IDE, reasoned that inhibitors of IDE would offer an ideal antidiabetic therapy, as they would slow the degradation of insulin. In the last decade, a range of novel potent IDE inhibitors were developed (Citation32,Citation47,Citation49,Citation50).
Particularly, Maianti et al. (Citation47) proposed the use of an IDE inhibitor, 6bK, as a new therapeutic strategy to treat T2DM. This conclusion was based on a decrease of postprandial glucose concentrations after oral glucose administration in lean and obese mice following acute 6bK treatment (Citation47). Whether the long-term inhibition of IDE activity is also efficient as an antidiabetic strategy in humans still remains to be established, and several opposed findings might be critical and need to be taken into account ().
Figure 1. Possible side effects of pharmacological IDE inhibition. Pharmacological inhibition of IDE activity can increase levels of its targets – insulin, glucagon, amylin, and beta-amyloid (Aβ). Increased insulin acutely improves glucose tolerance; however, a long-term hyperinsulinemia can deteriorate insulin resistance. Increased activation of insulin-mediated proliferation pathway can facilitate cancerogenesis. Increased postprandial glucagon can induce gluconeogenesis in the liver which will worsen hyperglycemia. Increased levels of amylin in the pancreas and Aβ monomers in the brain can lead to the formation of toxic oligomers following by the function impairments and death of beta-cell and neurons.
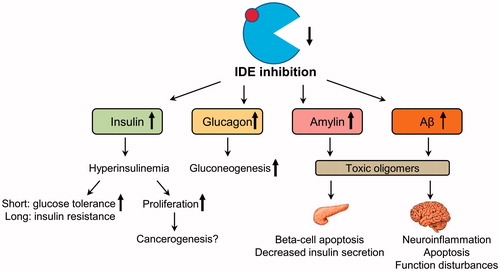
Firstly, as mentioned above, most data suggest a decrease of IDE activity in subjects with T2DM (Citation9,Citation62–64,Citation67). It can be speculated that a further decrease of IDE activity in subjects with T2DM by treatment with IDE inhibitors will not have a beneficial impact, but additional studies on IDE regulation in diabetic humans are definitely needed. Furthermore, chronical hyperinsulinemia induced by IDE inhibition can increase insulin resistance and impair insulin secretion (Citation69) (). Indeed, rodents with genetic defects of IDE activity have elevated insulin levels upon fasting, as predicted, but they also develop glucose intolerance (Citation57,Citation70). Interestingly, effects of the whole body IDE deletion in mice are age-dependent (Citation71). Whereas two-month-old IDE-KO mice exhibited improved glycemic control and lower body mass, six-month-old IDE-KO mice showed a T2DM phenotype characterized by increased body weight and pronounced glucose intolerance which obviously represents a compensatory response to chronic hyperinsulinemia (Citation71).
Secondly, due to the fact that IDE degrades many metabolically active substrates including glucagon, amylin, and Aβ, numerous side effects of IDE inhibition can be expected. Indeed, after the 6bK treatment, insulin, glucagon, and amylin levels were increased in the intraperitoneally glucose tolerance test; although no significant changes of basal hormone levels were found (Citation47). After the oral glucose administration in healthy subjects, higher insulin and lower glucagon responses are usually observed; therefore, the IDE inhibition will expectedly result primarily in an increase in insulin signaling and lower blood glucose levels (Citation47). However, the glucagon level is increased in T2DM subjects in fasting and postprandial state (Citation72). This imbalance in the insulin-to-glucagon ratio in diabetic humans may result in slower degradation of glucagon, and intensify glucose intolerance via an increase of postprandial gluconeogenesis. Further, prolonged IDE inhibition may lead to amyloid deposition and cytotoxicity in pancreatic beta-cells (Citation46) (). Paradoxically, Durham et al. found no action of another (dual-exosite) IDE inhibitor NTE-1 on insulin and glucagon degradation in vivo, although it resulted in elevated plasma amylin levels and improved glucose excursion in oral glucose tolerance test (Citation50). Authors hypothesized that these contradictory findings may be explained by differences in biochemical mechanisms of action or biodistribution of 6bK and NTE-1, but further experimentation is needed to clarify this.
Moreover, the systemic IDE inhibition may lead to an increase of cerebral Aβ that accumulates abnormally in AD. To avoid this, IDE inhibitors used for T2DM treatment should not cross the blood–brain barrier as was shown for 6bK (Citation47) (). Additionally, IDE controls the dynamics of cellular processes such as proteasomal degradation and autophagic flux (Citation45,Citation73) as well as lipogenesis and glucose uptake in adipocytes (Citation71,Citation74), which may also be affected by chronic IDE inhibition (as well as activation).
Thirdly, the intracellular hyperinsulinemia induced by IDE inhibition can lead to up-regulation of cell proliferation. In skeletal myoblasts, the up-regulation of proliferation upon RNAi knockdown of IDE was shown (Citation75). T2DM is known to be an independent risk factor for hepatocellular carcinoma (Citation76) possibly via chronic hyperinsulinemia. We recently found that RNAi IDE knockdown induces dysregulation of p53 pathway and an increase of proliferation markers in HepG2 hepatoma cells, although the proliferation rate was lower in IDE knockdown cells than in controls (Citation67). Analysis of microarray datasets from liver samples of diabetic subjects revealed a decrease of IDE expression accompanied by downregulation of the p53-dependent genes FAS and CCNG2, but not by upregulation of proliferation markers (Citation67). Detailed mechanisms of this phenomenon should be a subject of future studies.
Taken together, human data suggest that IDE inhibition could intensify instead of improving glucose disturbances in T2DM, and may facilitate a proliferative process in different tissues. Alternatively, enhanced degradation of insulin by an IDE activator, accompanied by insulin sensitization in peripheral tissues, may be a perspective strategy for T2DM treatment and needs further investigation.
IDE and Alzheimer’s disease
IDE role in the pathogenesis of AD
Alzheimer’s disease is a neurodegenerative process characterized by Aβ agglomeration, neuroinflammation and cognitive dysfunction (Citation77). AD is caused by the deposition of Aβ plaques and tau-neurofibrillary tangles in the brain that disturb the neuronal organization and function. Because of many common pathological mechanisms of T2DM and late-onset AD, including insulin resistance, neuroinflammation, oxidative stress, the presence of advanced glycation end products etc., AD is often proposed as a form of diabetes and termed “Type 3 diabetes” (Citation78–80). T2DM, hyperinsulinemia, and systemic insulin resistance are known to increase the risk of developing AD and vice versa (Citation78,Citation79,Citation81). Clinical trials showed beneficial effects of T2DM treatment with peroxisome proliferator-activated receptor gamma agonists, thiazolidinediones and intranasal insulin in AD patients (Citation82).
Because IDE is required for both insulin and Aβ degradation in neurons and microglia, it represents one of the possible links between T2DM and AD pathogeneses (Citation79,Citation81). In the brain, IDE is secreted from microglial cells and neurons and degrades Aβ extracellularly and on the cell surface (Citation4,Citation83). Similarly to T2DM risk, some studies reported an association of polymorphisms within IDE locus with AD risk and plasma Aβ levels, although this effect is modified by the presence or absence of an apolipoprotein E epsilon 4 allele (Citation84–87). Moreover, animal models of IDE inhibition demonstrated both features of AD and T2DM. Goto–Kakizaki rats and IDE knockout mice showed an increase of cerebral endogenous Aβ and beta-amyloid precursor protein intracellular domain (Citation58,Citation70).
Most human studies found decreased Aβ-degrading IDE activity and expression in the brain of AD subjects and subjects with high AD risk (Citation88–90), although some authors observed no changes or even the increase of IDE activity (Citation91,Citation92). Similarly, discordant results were received concerning the IDE regulation in the elderly (Citation93–95). Interestingly, Kim et al. found the reduction in IDE activity but not in IDE expression in chromosome 10-linked late onset AD, suggesting the possibility of systemic functional defects in IDE activity (Citation8). Thus, AD may be caused in some cases by the failure of Aβ degradation by IDE or by IDE deficiency.
The role of IDE in a common pathological mechanism of T2DM and late-onset AD is shown in . T2DM is characterized by hyperinsulinemia and systemic insulin resistance; however we know relatively little about cerebral insulin level and its signaling in diabetic humans (Citation96). The widely-used hypothesis postulates that high insulin concentrations decrease the IDE-mediated Aβ degradation via competitive inhibition (Citation81). Nevertheless, insulin levels in the brain (pM) are much lower than the insulin Km (∼100 nM) and therefore unlikely to competitively inhibit Aβ degradation (Citation96). Therefore, the statement about competitive inhibition of IDE needs to be qualified. However, insulin is known to increase Aβ via other mechanisms, such as increased secretion (Citation97).
Figure 2. IDE as a pathological link between type 2 diabetes and Alzheimer’s disease. Type 2 diabetes is characterized by hyperinsulinemia and systemic insulin resistance. Insulin is hypothesized to decrease the IDE-mediated beta-amyloid (Aβ) degradation via the competitive inhibition, but this statement needs to be qualified. Insulin also increases Aβ production via other mechanisms, such as increased secretion. The central insulin resistance leads to the reduction of insulin signaling in the brain, which induces the hyperphosphorylation of tau protein and formation of toxic Aβ oligomers by the multiple mechanisms. Particularly, insulin resistance lowers the IDE expression and in this way decreases IDE-mediated Aβ degradation and additionally increases Aβ oligomer levels, which can in turn aggravate insulin resistance in the brain. All these molecular events finally lead to the formation of Aβ plaques and neurofibrillary tangles disturbing the neuronal organization and function.
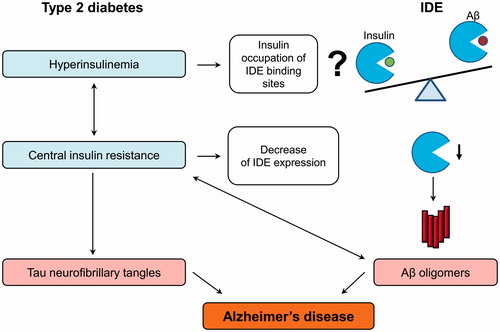
Insulin receptors are widely expressed in the brain where insulin is involved in the regulation of cognitive functions, feeding, energy balance and glucose uptake in some types of neurons (Citation78,Citation79). The AD brain demonstrates deficits in insulin signaling resulting from the combined effects of insulin resistance and deficiency (Citation78). Central insulin resistance is manifested by reduced levels of insulin receptor binding and decreased responsiveness to insulin stimulation due to reduced expression of insulin signaling molecules (Citation78,Citation80,Citation98,Citation99). Brain insulin resistance and its consequences can account for most of the metabolic, structural, and functional abnormalities in AD, including expression and metabolism of Aβ and tau-protein (Citation78) (). Increases in the activities of GSK-3β, JNK/p38, caspases, oxidative and ER stress, and other molecular mechanisms lead to the hyperphosphorylation of tau-protein and increase of toxic Aβ oligomer level followed by the formation of neurofibrillary tangles and Aβ plaques, respectively (Citation78–80). Because insulin increases IDE protein level through the phosphatidylinositol-3 kinase pathway (Citation100), central insulin resistance can lower the IDE expression which additionally decreases IDE-mediated Aβ degradation and increases Aβ oligomer levels (). Furthermore, dysregulated lipid metabolism in the AD brain may contribute to a decrease of IDE-dependent Aβ degradation (Citation25,Citation33). Alternatively, the decrease of IDE activity in AD may be caused by genetic factors (Citation8). An increase of Aβ oligomers can, in turn, aggravate insulin resistance in the brain which results in a deleterious positive feedback cycle (Citation101). The worsening of insulin resistance leads to further production of toxic Aβ oligomers.
Use of IDE activators for AD treatment
Upregulation of proteases that degrade Aβ represents a logical therapeutic approach for AD treatment. Indeed, transgenic IDE overexpression in neurons decreased brain Aβ levels, prevented amyloid plaque formation, and reduced episodes of early lethality in amyloid precursor protein (APP) transgenic mice (Citation102). Leissring et al. also discovered two compounds activating degradation of Aβ by 700% and 400% (Citation48). Another group investigated cell-permeable compounds stimulated rat IDE activity in a cell-based assay (Citation103), but no IDE activators were yet tested in vivo. Expectedly, such IDE activators will increase Aβ degradation, prevent aggregation and, ideally, prevent the neuronal loss that leads to AD symptoms.
Again, multiple side effects of the systemic IDE activation should be expected, including impaired glucose tolerance because of increased insulin degradation and risk of cardiovascular events caused by increased natriuretic peptide degradation. Moreover, upregulated IDE-mediated glucagon degradation in fasting state can lead to hypoglycemic episodes. Therefore, the development of substrate-specific IDE activators or brain-specific delivery of IDE activators would be of great therapeutic interest. However, IDE infusion or systems of gene delivery to the brain such as lentiviral vector, adeno-associated viral vector, siRNA, and recombinant gene therapy-based delivery successfully tested in animals (Citation77) are less practical in humans at present. Alternatively, other, more specific Aβ-degrading enzymes such as neprilysin, endothelin-converting enzyme, angiotensin-converting enzyme, and matrix metalloproteinase-9 are intensively discussed as therapeutic targets in AD (Citation104).
Conclusions
IDE represents a pathophysiological link between T2DM and AD. Modern technologies of drug design allow the development of potent and selective modulators of IDE activity which represent potential drugs for therapies of both diseases. However, because of possible side effects, the sustained treatment with systemic IDE modulators should be carefully tested in animal studies. Development of substrate-selective IDE modulators will allow avoiding adverse effects of IDE modulators associated with multiplicity of IDE targets.
Acknowledgements
We thank Ms. June Inderthal (Department of Clinical Nutrition, German Institute of Human Nutrition, Germany) for editing the manuscript.
Disclosure statement
The authors declare no conflict of interest.
Additional information
Funding
Related Research Data
References
- Duckworth WC, Bennett RG, Hamel FG. Insulin degradation: progress and potential. Endocr Rev. 1998;19:608–24.
- Mirsky IA, Broh-Kahn RH. The inactivation of insulin by tissue extracts; the distribution and properties of insulin inactivating extracts. Arch Biochem. 1949;20:1–9.
- Affholter JA, Fried VA, Roth RA. Human insulin-degrading enzyme shares structural and functional homologies with E. coli protease III. Science. 1988;242:1415–18.
- Vekrellis K, Ye Z, Qiu WQ, Walsh D, Hartley D, Chesneau V, et al. Neurons regulate extracellular levels of amyloid beta-protein via proteolysis by insulin-degrading enzyme. J Neurosci. 2000;20:1657–65.
- Farris W, Leissring MA, Hemming ML, Chang AY, Selkoe DJ. Alternative splicing of human insulin-degrading enzyme yields a novel isoform with a decreased ability to degrade insulin and amyloid beta-protein. Biochemistry. 2005;44:6513–25.
- Leissring MA, Farris W, Wu X, Christodoulou DC, Haigis MC, Guarente L, et al. Alternative translation initiation generates a novel isoform of insulin-degrading enzyme targeted to mitochondria. Biochem J. 2004;383:439–46.
- Rubenstein AH, Pottenger LA, Mako M, Getz GS, Steiner DF. The metabolism of proinsulin and insulin by the liver. J Clin Invest. 1972;51:912–21.
- Kim M, Hersh LB, Leissring MA, Ingelsson M, Matsui T, Farris W, et al. Decreased catalytic activity of the insulin-degrading enzyme in chromosome 10-linked Alzheimer disease families. J Biol Chem. 2007;282:7825–32.
- Pivovarova O, Gogebakan O, Pfeiffer AF, Rudovich N. Glucose inhibits the insulin-induced activation of the insulin-degrading enzyme in HepG2 cells. Diabetologia. 2009;52:1656–64.
- Shen Y, Joachimiak A, Rosner MR, Tang WJ. Structures of human insulin-degrading enzyme reveal a new substrate recognition mechanism. Nature. 2006;443:870–4.
- Kupfer SR, Wilson EM, French FS. Androgen and glucocorticoid receptors interact with insulin degrading enzyme. J Biol Chem. 1994;269:20622–8.
- Hamel FG, Bennett RG, Upward JL, Duckworth WC. Insulin inhibits peroxisomal fatty acid oxidation in isolated rat hepatocytes. Endocrinology. 2001;142:2702–6.
- Semple JW, Lang Y, Speck ER, Delovitch TL. Processing and presentation of insulin. III. Insulin degrading enzyme: a neutral metalloendoproteinase that is non-homologous to classical endoproteinases mediates the processing of insulin epitopes for helper T cells. Int Immunol. 1992;4:1161–7.
- Kayalar C, Wong WT. Metalloendoprotease inhibitors which block the differentiation of L6 myoblasts inhibit insulin degradation by the endogenous insulin-degrading enzyme. J Biol Chem. 1989;264:8928–34.
- Bennett RG, Fawcett J, Kruer MC, Duckworth WC, Hamel FG. Insulin inhibition of the proteasome is dependent on degradation of insulin by insulin-degrading enzyme. J Endocrinol. 2003;177:399–405.
- Hamel FG, Bennett RG, Duckworth WC. Regulation of multicatalytic enzyme activity by insulin and the insulin-degrading enzyme. Endocrinology. 1998;139:4061–6.
- Codogno P, Meijer AJ. Autophagy and signaling: their role in cell survival and cell death. Cell Death Differ. 2005;12:1509–18.
- Jung T, Catalgol B, Grune T. The proteasomal system. Mol Aspects Med. 2009;30:191–296.
- Fawcett J, Permana PA, Levy JL, Duckworth WC. Regulation of protein degradation by insulin-degrading enzyme: analysis by small interfering RNA-mediated gene silencing. Arch Biochem Biophys. 2007;468:128–33.
- Bennett RG, Hamel FG, Duckworth WC. Identification and isolation of a cytosolic proteolytic complex containing insulin degrading enzyme and the multicatalytic proteinase. Biochem Biophys Res Commun. 1994;202:1047–53.
- Duckworth WC, Heinemann MA, Kitabchi AE. Purification of insulin-specific protease by affinity chromatography. Proc Natl Acad Sci USA. 1972;69:3698–702.
- Hong L, Huang HC, Jiang ZF. Relationship between amyloid-beta and the ubiquitin-proteasome system in Alzheimer's disease. Neurol Res. 2014;36:276–82.
- Balasubramanyam M, Sampathkumar R, Mohan V. Is insulin signaling molecules misguided in diabetes for ubiquitin-proteasome mediated degradation? Mol Cell Biochem. 2005;275:117–25.
- Song ES, Juliano MA, Juliano L, Hersh LB. Substrate activation of insulin-degrading enzyme (insulysin). A potential target for drug development. J Biol Chem. 2003;278:49789–94.
- Hamel FG, Upward JL, Bennett RG. In vitro inhibition of insulin-degrading enzyme by long-chain fatty acids and their coenzyme A thioesters. Endocrinology. 2003;144:2404–8.
- Camberos MC, Perez AA, Udrisar DP, Wanderley MI, Cresto JC. ATP inhibits insulin-degrading enzyme activity. Exp Biol Med (Maywood). 2001;226:334–41.
- George S, Petit GH, Gouras GK, Brundin P, Olsson R. Nonsteroidal selective androgen receptor modulators and selective estrogen receptor beta agonists moderate cognitive deficits and amyloid-beta levels in a mouse model of Alzheimer's disease. ACS Chem Neurosci. 2013;4:1537–48.
- Grasso G, Satriano C, Milardi D. A neglected modulator of insulin-degrading enzyme activity and conformation: the pH. Biophys Chem. 2015;203–204:33–40.
- Ahuja N, Schwer B, Carobbio S, Waltregny D, North BJ, Castronovo V, et al. Regulation of insulin secretion by SIRT4, a mitochondrial ADP-ribosyltransferase. J Biol Chem. 2007;282:33583–92.
- Saric T, Muller D, Seitz HJ, Pavelic K. Non-covalent interaction of ubiquitin with insulin-degrading enzyme. Mol Cell Endocrinol. 2003;204:11–20.
- Mirsky IA, Perisutti G. Effect of insulinase-inhibitor on hypoglycemic action of insulin. Science. 1955;122:559–60.
- Leissring MA, Malito E, Hedouin S, Reinstatler L, Sahara T, Abdul-Hay SO, et al. Designed inhibitors of insulin-degrading enzyme regulate the catabolism and activity of insulin. PLoS One. 2010;5:e10504.
- Du J, Zhang L, Liu S, Wang Z. Palmitic acid and docosahexaenoic acid opposingly regulate the expression of insulin-degrading enzyme in neurons. Pharmazie. 2010;65:231–2.
- Dobarro M, Gerenu G, Ramirez MJ. Propranolol reduces cognitive deficits, amyloid and tau pathology in Alzheimer's transgenic mice. Int J Neuropsychopharmacol. 2013;16:2245–57.
- Shin MK, Kim HG, Baek SH, Jung WR, Park DI, Park JS, et al. Neuropep-1 ameliorates learning and memory deficits in an Alzheimer's disease mouse model, increases brain-derived neurotrophic factor expression in the brain, and causes reduction of amyloid beta plaques. Neurobiol Aging. 2014;35:990–1001.
- Ciaccio C, Tundo GR, Grasso G, Spoto G, Marasco D, Ruvo M, et al. Somatostatin: a novel substrate and a modulator of insulin-degrading enzyme activity. J Mol Biol. 2009;385:1556–67.
- Ohyagi Y. A drug targeting intracellular amyloid-beta and oxidative stress: apomorphine. Rinsho Shinkeigaku. 2011;51:884–7.
- Marwarha G, Ghribi O. Leptin signaling and Alzheimer's disease. Am J Neurodegener Dis. 2012;1:245–65.
- Adessi C, Enderle T, Grueninger F, Roth D. AG FH-LR. Activator for insulin degrading enzyme. World Intellectual Property Organization. Patent WO2006/066847; 2006.
- Charton J, Gauriot M, Guo Q, Hennuyer N, Marechal X, Dumont J, et al. Imidazole-derived 2-[N-carbamoylmethyl-alkylamino]acetic acids, substrate-dependent modulators of insulin-degrading enzyme in amyloid-beta hydrolysis. Eur J Med Chem. 2014;79:184–93.
- Kalinin S, Richardson JC, Feinstein DL. A PPARdelta agonist reduces amyloid burden and brain inflammation in a transgenic mouse model of Alzheimer's disease. Curr Alzheimer Res. 2009;6:431–7.
- Wang P, Su C, Li R, Wang H, Ren Y, Sun H, et al. Mechanisms and effects of curcumin on spatial learning and memory improvement in APPswe/PS1dE9 mice. J Neurosci Res. 2014;92:218–31.
- Kong Y, Ruan L, Qian L, Liu X, Le Y. Norepinephrine promotes microglia to uptake and degrade amyloid beta peptide through upregulation of mouse formyl peptide receptor 2 and induction of insulin-degrading enzyme. J Neurosci. 2010;30:11848–57.
- Liu Y, Gao M, Ma L, Zhang L, Pan N. Sevoflurane alters the expression of receptors and enzymes involved in Abeta clearance in rats. Acta Anaesthesiol Scand. 2013;57:903–10.
- Hamel FG, Fawcett J, Tsui BT, Bennett RG, Duckworth WC. Effect of nelfinavir on insulin metabolism, proteasome activity and protein degradation in HepG2 cells. Diabetes Obes Metab. 2006;8:661–8.
- Bennett RG, Hamel FG, Duckworth WC. An insulin-degrading enzyme inhibitor decreases amylin degradation, increases amylin-induced cytotoxicity, and increases amyloid formation in insulinoma cell cultures. Diabetes. 2003;52:2315–20.
- Maianti JP, McFedries A, Foda ZH, Kleiner RE, Du XQ, Leissring MA, et al. Anti-diabetic activity of insulin-degrading enzyme inhibitors mediated by multiple hormones. Nature. 2014;511:94–8.
- Cabrol C, Huzarska MA, Dinolfo C, Rodriguez MC, Reinstatler L, Ni J, et al. Small-molecule activators of insulin-degrading enzyme discovered through high-throughput compound screening. PLoS One. 2009;4:e5274.
- Cakir B, Dagliyan O, Dagyildiz E, Baris I, Kavakli IH, Kizilel S, et al. Structure based discovery of small molecules to regulate the activity of human insulin degrading enzyme. PLoS One. 2012;7:e31787.
- Durham TB, Toth JL, Klimkowski VJ, Cao JX, Siesky AM, Alexander-Chacko J, et al. Dual exosite-binding inhibitors of insulin-degrading enzyme challenge its role as the primary mediator of insulin clearance in vivo. J Biol Chem. 2015;290:20044–59.
- Noinaj N, Bhasin SK, Song ES, Scoggin KE, Juliano MA, Juliano L, et al. Identification of the allosteric regulatory site of insulysin. PLoS One. 2011;6:e20864.
- Abdul-Hay SO, Lane AL, Caulfield TR, Claussin C, Bertrand J, Masson A, et al. Optimization of peptide hydroxamate inhibitors of insulin-degrading enzyme reveals marked substrate-selectivity. J Med Chem. 2013;56:2246–55.
- Sladek R, Rocheleau G, Rung J, Dina C, Shen L, Serre D, et al. A genome-wide association study identifies novel risk loci for type 2 diabetes. Nature. 2007;445:881–5.
- Gu HF, Efendic S, Nordman S, Ostenson CG, Brismar K, Brookes AJ, et al. Quantitative trait loci near the insulin-degrading enzyme (IDE) gene contribute to variation in plasma insulin levels. Diabetes. 2004;53:2137–42.
- Karamohamed S, Demissie S, Volcjak J, Liu C, Heard-Costa N, Liu J, et al. Polymorphisms in the insulin-degrading enzyme gene are associated with type 2 diabetes in men from the NHLBI Framingham Heart Study. Diabetes. 2003;52:1562–7.
- Rudovich N, Pivovarova O, Fisher E, Fischer-Rosinsky A, Spranger J, Mohlig M, et al. Polymorphisms within insulin-degrading enzyme (IDE) gene determine insulin metabolism and risk of type 2 diabetes. J Mol Med (Berl). 2009;87:1145–51.
- Fakhrai-Rad H, Nikoshkov A, Kamel A, Fernstrom M, Zierath JR, Norgren S, et al. Insulin-degrading enzyme identified as a candidate diabetes susceptibility gene in GK rats. Hum Mol Genet. 2000;9:2149–58.
- Farris W, Mansourian S, Chang Y, Lindsley L, Eckman EA, Frosch MP, et al. Insulin-degrading enzyme regulates the levels of insulin, amyloid beta-protein, and the beta-amyloid precursor protein intracellular domain in vivo. Proc Natl Acad Sci USA. 2003;100:4162–7.
- Seta KA, Roth RA. Overexpression of insulin degrading enzyme: cellular localization and effects on insulin signaling. Biochem Biophys Res Commun. 1997;231:167–71.
- Standl E, Kolb HJ. Insulin degrading enzyme activity and insulin binding of erythrocytes in normal subjects and type 2 (non-insulin-dependent) diabetic patients. Diabetologia. 1984;27:17–22.
- Snehalatha C, Timothy H, Mohan V, Ramachandran A, Viswanathan M. Immunoreactive insulin and insulin degrading enzymes in erythrocytes. A preliminary report. J Assoc Phys India. 1990;38:558–61.
- Nikolaev SL, Strelkova MA, Komov VP. Insulin degradation in hepatocytes and erythrocytes of rats in normal condition and in experimental diabetes. Vopr Med Khim. 2001;47:329–37.
- Hern EP, Shroyer LA, Varandani PT. Insulin-degrading neutral cysteine proteinase activity of adipose tissue and liver of nondiabetic, streptozotocin-diabetic, and insulin-treated diabetic rats. Arch Biochem Biophys. 1987;254:35–42.
- Fawcett J, Sang H, Permana PA, Levy JL, Duckworth WC. Insulin metabolism in human adipocytes from subcutaneous and visceral depots. Biochem Biophys Res Commun. 2010;402:762–6.
- Jurcovicova J, Nemeth S, Vigas M. Effect of insulin and glucose on the activity of insulin-degrading enzymes in rat liver. Endocrinol Exp. 1977;11:209–13.
- Li CZ, Zhang SH, Shu CD, Ren W. Relationship between insulin-degrading enzyme activity and insulin sensitivity in cell model of insulin-resistance. Di Yi Jun Yi Da Xue Xue Bao. 2002;22:151–4.
- Pivovarova O, von Loeffelholz C, Ilkavets I, Sticht C, Zhuk S, Murahovschi V, et al. Modulation of insulin degrading enzyme activity and liver cell proliferation. Cell Cycle. 2015;14:2293–300.
- Rudovich NN, Rochlitz HJ, Pfeiffer AF. Reduced hepatic insulin extraction in response to gastric inhibitory polypeptide compensates for reduced insulin secretion in normal-weight and normal glucose tolerant first-degree relatives of type 2 diabetic patients. Diabetes. 2004;53:2359–65.
- Pivovarova O, Bernigau W, Bobbert T, Isken F, Mohlig M, Spranger J, et al. Hepatic insulin clearance is closely related to metabolic syndrome components. Diabetes Care. 2013;36:3779–85.
- Farris W, Mansourian S, Leissring MA, Eckman EA, Bertram L, Eckman CB, et al. Partial loss-of-function mutations in insulin-degrading enzyme that induce diabetes also impair degradation of amyloid beta-protein. Am J Pathol. 2004;164:1425–34.
- Abdul-Hay SO, Kang D, McBride M, Li L, Zhao J, Leissring MA. Deletion of insulin-degrading enzyme elicits antipodal, age-dependent effects on glucose and insulin tolerance. PLoS One. 2011;6:e20818.
- Godoy-Matos AF. The role of glucagon on type 2 diabetes at a glance. Diabetol Metab Syndr. 2014;6:91.
- Steneberg P, Bernardo L, Edfalk S, Lundberg L, Backlund F, Ostenson CG, et al. The type 2 diabetes-associated gene ide is required for insulin secretion and suppression of α-synuclein levels in β-cells. Diabetes. 2013;62:2004–14.
- Fawcett J, Tsui BT, Duckworth WC. Nelfinavir alters both insulin and lipid metabolism in 3T3-L1 adipocytes. ADA, 63th Scientific Sessions, 2003.
- Epting CL, King FW, Pedersen A, Zaman J, Ritner C, Bernstein HS. Stem cell antigen-1 localizes to lipid microdomains and associates with insulin degrading enzyme in skeletal myoblasts. J Cell Physiol. 2008;217:250–60.
- Blonski W, Kotlyar DS, Forde KA. Non-viral causes of hepatocellular carcinoma. World J Gastroenterol. 2010;16:3603–15.
- Jha NK, Jha SK, Kumar D, Kejriwal N, Sharma R, Ambasta RK, et al. Impact of insulin degrading enzyme and neprilysin in Alzheimer's disease biology: characterization of putative cognates for therapeutic applications. J Alzheimers Dis. 2015;48:891–917.
- de la Monte SM, Tong M. Brain metabolic dysfunction at the core of Alzheimer's disease. Biochem Pharmacol. 2014;88:548–59.
- Mittal K, Katare DP. Shared links between type 2 diabetes mellitus and Alzheimer's disease: a review. Diabetes Metab Syndr. 2016. [Epub ahead of print]. doi: 10.1016/j.dsx.2016.01.021.
- Sims-Robinson C, Kim B, Rosko A, Feldman EL. How does diabetes accelerate Alzheimer disease pathology? Nat Rev Neurol. 2010;6:551–9.
- Qiu WQ, Folstein MF. Insulin, insulin-degrading enzyme and amyloid-beta peptide in Alzheimer's disease: review and hypothesis. Neurobiol Aging. 2006;27:190–8.
- Akter K, Lanza EA, Martin SA, Myronyuk N, Rua M, Raffa RB. Diabetes mellitus and Alzheimer's disease: shared pathology and treatment? Br J Clin Pharmacol. 2011;71:365–76.
- Qiu WQ, Walsh DM, Ye Z, Vekrellis K, Zhang J, Podlisny MB, et al. Insulin-degrading enzyme regulates extracellular levels of amyloid beta-protein by degradation. J Biol Chem. 1998;273:32730–8.
- Cheng H, Wang L, Shi T, Shang Y, Jiang L. Association of insulin degrading enzyme gene polymorphisms with Alzheimer's disease: a meta-analysis. Int J Neurosci. 2015;125:328–35.
- Edland SD, Wavrant-De Vriese F, Compton D, Smith GE, Ivnik R, Boeve BF, et al. Insulin degrading enzyme (IDE) genetic variants and risk of Alzheimer's disease: evidence of effect modification by apolipoprotein E (APOE). Neurosci Lett. 2003;345:21–4.
- Carrasquillo MM, Belbin O, Zou F, Allen M, Ertekin-Taner N, Ansari M, et al. Concordant association of insulin degrading enzyme gene (IDE) variants with IDE mRNA, Abeta, and Alzheimer's disease. PLoS One. 2010;5:e8764.
- Zhang Y, Wang B, Wan H, Zhou Q, Li T. Meta-analysis of the insulin degrading enzyme polymorphisms and susceptibility to Alzheimer's disease. Neurosci Lett. 2013;541:132–7.
- Zhao Z, Xiang Z, Haroutunian V, Buxbaum JD, Stetka B, Pasinetti GM. Insulin degrading enzyme activity selectively decreases in the hippocampal formation of cases at high risk to develop Alzheimer's disease. Neurobiol Aging. 2007;28:824–30.
- Perez A, Morelli L, Cresto JC, Castano EM. Degradation of soluble amyloid beta-peptides 1-40, 1-42, and the Dutch variant 1-40Q by insulin degrading enzyme from Alzheimer disease and control brains. Neurochem Res. 2000;25:247–55.
- Del Campo M, Stargardt A, Veerhuis R, Reits E, Teunissen CE. Accumulation of BRI2-BRICHOS ectodomain correlates with a decreased clearance of Aβ by insulin degrading enzyme (IDE) in Alzheimer's disease. Neurosci Lett. 2015;589:47–51.
- Wang S, Wang R, Chen L, Bennett DA, Dickson DW, Wang DS. Expression and functional profiling of neprilysin, insulin-degrading enzyme, and endothelin-converting enzyme in prospectively studied elderly and Alzheimer's brain. J Neurochem. 2010;115:47–57.
- Miners JS, Baig S, Tayler H, Kehoe PG, Love S. Neprilysin and insulin-degrading enzyme levels are increased in Alzheimer disease in relation to disease severity. J Neuropathol Exp Neurol. 2009;68:902–14.
- Runyan K, Duckworth WC, Kitabchi AE, Huff G. The effect of age on insulin-degrading activity in rat tissue. Diabetes. 1979;28:324–5.
- Caccamo A, Oddo S, Sugarman MC, Akbari Y, LaFerla FM. Age- and region-dependent alterations in Abeta-degrading enzymes: implications for Abeta-induced disorders. Neurobiol Aging. 2005;26:645–54.
- Miners JS, van Helmond Z, Kehoe PG, Love S. Changes with age in the activities of beta-secretase and the Abeta-degrading enzymes neprilysin, insulin-degrading enzyme and angiotensin-converting enzyme. Brain Pathol. 2010;20:794–802.
- Heni M, Kullmann S, Preissl H, Fritsche A, Haring HU. Impaired insulin action in the human brain: causes and metabolic consequences. Nat Rev Endocrinol. 2015;11:701–11.
- Gasparini L, Gouras GK, Wang R, Gross RS, Beal MF, Greengard P, et al. Stimulation of beta-amyloid precursor protein trafficking by insulin reduces intraneuronal beta-amyloid and requires mitogen-activated protein kinase signaling. J Neurosci. 2001;21:2561–70.
- Steen E, Terry BM, Rivera EJ, Cannon JL, Neely TR, Tavares R, et al. Impaired insulin and insulin-like growth factor expression and signaling mechanisms in Alzheimer's disease – is this type 3 diabetes? J Alzheimers Dis. 2005;7:63–80.
- Bosco D, Fava A, Plastino M, Montalcini T, Pujia A. Possible implications of insulin resistance and glucose metabolism in Alzheimer's disease pathogenesis. J Cell Mol Med. 2011;15:1807–21.
- Zhao L, Teter B, Morihara T, Lim GP, Ambegaokar SS, Ubeda OJ, et al. Insulin-degrading enzyme as a downstream target of insulin receptor signaling cascade: implications for Alzheimer's disease intervention. J Neurosci. 2004;24:11120–6.
- Li X, Song D, Leng SX. Link between type 2 diabetes and Alzheimer's disease: from epidemiology to mechanism and treatment. Clin Interv Aging. 2015;10:549–60.
- Leissring MA, Farris W, Chang AY, Walsh DM, Wu X, Sun X, et al. Enhanced proteolysis of beta-amyloid in APP transgenic mice prevents plaque formation, secondary pathology, and premature death. Neuron. 2003;40:1087–93.
- Kukday SS, Manandhar SP, Ludley MC, Burriss ME, Alper BJ, Schmidt WK. Cell-permeable, small-molecule activators of the insulin-degrading enzyme. J Biomol Screen. 2012;17:1348–61.
- Nalivaeva NN, Fisk LR, Belyaev ND, Turner AJ. Amyloid-degrading enzymes as therapeutic targets in Alzheimer's disease. Curr Alzheimer Res. 2008;5:212–24.