
Abstract
Calcific aortic valve disease (CAVD) represents a spectrum of disease spanning from milder degrees of calcification of valve leaflets, i.e., aortic sclerosis, to severe calcification i.e., aortic stenosis (AS) with hemodynamic instability. The prevalence of CAVD is increasing rapidly due to the aging of the population, being up to 2.8% among patients over 75 years of age. Even without significant aortic valve stenosis, aortic sclerosis is associated with a 50% increased risk of myocardial infarction and death from cardiovascular causes. To date, there is no pharmacological treatment available to reverse or hinder the progression of CAVD. So far, the cholesterol-lowering therapies (statins) and renin–angiotensin system (RAS) blocking drugs have been the major pharmacological agents investigated for treatment of CAVD. Especially angiotensin receptor blockers (ARB)s and angiotensin convertase enzyme inhibitors (ACEI)s, have been under active investigation in clinical trials, but have proven to be unsuccessful in slowing the progression of CAVD. Several studies have suggested that other vasoactive hormones, including endothelin and apelin systems are also associated with development of AS. In the present review, we discuss the role of vasoactive factors in the pathogenesis of CAVD as novel pharmacological targets for the treatment of aortic valve calcification.
Vasoactive factors are involved in the progression of calcific aortic valve disease.
Endothelin and renin–angiotensin systems seem to be most prominent targets for therapeutic interventions in the view of valvular pathogenesis.
Circulating vasoactive factors may provide targets for diagnostic tools of calcified aortic valve disease.
Key messages
Introduction
Calcific aortic valve disease (CAVD) represents a spectrum of disease spanning from mild aortic valve sclerosis to severe aortic stenosis (AS) with hemodynamic instability (Citation1). CAVD is the most common valvular heart disease in Europe and North America and its prevalence is increasing rapidly due to the aging of the population. The prevalence of aortic stenosis is only about 0.2% among adults over 50 years of age, but increases to 9.8% over 80 years of age, with an overall prevalence of 2.8% in adults older than 75 years of age (Citation2). Even without significant stenosis, aortic valve sclerosis is associated with a 50% increased risk of myocardial infarction and death from cardiovascular causes (Citation1–3). Once symptoms, including angina, syncope, or heart failure are present in AS, valve replacement is the only effective treatment (Citation1). The average survival without valve replacement is no more than 1.5–2 years after the onset of symptoms among patients with severe AS (Citation2,Citation4).
The pathogenesis of CAVD is initiated by endothelial injury, followed by lipid accumulation, infiltration of inflammatory cells, degradation or increased deposition of the extracellular matrix (ECM), neoangiogenesis, and extensive calcification combined with ossification (Citation5–7). These processes are also contributing factors of vascular atherosclerosis but despite pathophysiological similarities, only about 50% of patients with CAVD have clinically significant atherosclerosis (Citation8). To date there is no pharmacological treatment available to treat or even to slow down the disease progression of CAVD. Therefore, mechanisms of progression from an early inflammatory lesion to phenotype transformation of valve myofibroblasts and then to the end stage of severe valve calcification have been intensively characterized to reveal novel targets for managing the disease.
In the present review, we discuss the role of vasoactive peptides in CAVD. We summarize the current knowledge of renin-angiotensin system (RAS) counterparts, chymase, cathepsin G, bradykinin, and its receptors as well as endothelin (ET), natriuretic peptide and apelin-APJ system in the pathogenesis of aortic valve calcification. In addition, we discuss potential strategies to target the vasoactive peptides for the development of pharmacological treatment and diagnosis to CAVD.
The renin–angiotensin system (RAS)
During the last decades the focus of research has been changed from systemic and endocrine RAS to autocrine and paracrine effects of tissue RAS (Citation9). Tissue RAS functions by inducing fibroblast proliferation, expression of proinflammatory mediator ET-1 (Citation10) and ECM deposition, suggesting that paracrine/autocrine RAS may play a major role in the pathogenesis of aortic valve stenosis. In addition to the classical RAS (renin, Ang I, angiotensin converting enzyme [ACE], Ang II, and angiotensin II receptors type 1 and 2 [AT1 and AT2], newer components such as (pro)renin receptor, ACE2, its product angiotensin (1-7) [Ang-(1-7)] and its receptor Mas proto-oncogene have been recently discovered providing potential targets for pharmacological therapies. So far, RAS has been the major target for drugs in attempt to seek therapeutic strategies for patients with CAVD and several clinical trials have evaluated the effect of angiotensin receptor blockers (ARBs) and angiotensin convertase enzyme inhibitors (ACEI)s on the progression of CAVD.
Angiotensin II, AT1 and AT2 receptors
The functional effects of Ang II are mediated via two receptor isoforms AT1 and AT2. Although AT1 shares structural homology with AT2, they are functionally distinct and differentially distributed (Citation11,Citation12). The classical systemic effects of Ang II (vasoconstriction, cardiac hypertrophy, ECM formation, and increased release of aldosterone) are mediated via AT1 (Citation13). According to theory, the beneficial effects on cardiovascular system (antifibrotic effect, antihypertrophic effect, vasodilating effect and opposing the antinatriuretic effect) are mediated via AT2 receptor (Citation13). Several studies have described the existence of local RAS in aortic valves (Citation14–17). AT1 receptor mRNA levels are described to be both increased (Citation15) and unchanged (Citation16) in stenotic aortic valves. These results may be explained by the interindividual variability in the abundance of the AT1 receptor in the valvular tissue since in one study AT1 receptor was detected in only less than 20% of nonstenotic valves but in 75% of stenotic valves (Citation14). Even though a decrease in gene expression levels of AT2 receptor was seen in AS valves when compared with normal aortic valves, the AT2-receptors were not detected by immunohistochemistry in normal or calcified valves (Citation16). Ang II have been shown to promote aortic valve thickening independent of elevated blood pressure in apolipoprotein-E deficient mice (Citation18), and induce vascular calcification in vitro and in vivo through receptor activator of nuclear factor-κB ligand (RANKL) system activation (Citation19). In addition, RANKL activated RAS, especially ACE and AT1 receptor, providing RANKL as a novel therapeutic option to inhibit RAS and prevent vascular calcification. Previously, RANKL, a component of emerging regulatory pathway (nuclear factor-κB (RANK), RANKL, and osteoprotegerin (OPG)) for vascular calcification (Citation20), has been reported to contribute to vascular calcification by decreasing the calcification inhibitor, matrix Gla protein, in vascular smooth muscle cells, and elevating bone morphogenetic protein-2 (BMP-2) expression in endothelial cells (Citation19).
Three retrospective studies have been published according to which ARBs are useful in the treatment of AS (). Japanese Aortic Stenosis Study (JASS) showed that initiation of ARB treatment during the early stage of the CAVD may be effective in slowing the progression of AS (Citation21), and in a study with hypertensive patients, ARB (but not ACEI) treatment was associated with slower progression of CAVD (Citation22). In another retrospective study, the use of ARBs was associated with a lower remodelling score of stenotic aortic valves (Citation23). However, the treatment with candesartan for five months in patients with clinically significant AS did not have beneficial effects on mortality or left ventricular mass and function (Citation24) (). Additionally, treatment of cholesterol-fed rabbits with olmesartan has been associated with decreased macrophage infiltration and reductions in osteopontin and ACE in aortic valves (Citation25). Unfortunately, no hemodynamic valvular measurements of AS progression were performed in that study (Citation25). In addition, hemodynamic factors and effects of ARB fimasartan on LV remodeling is being investigated in ongoing study in patients with asymptomatic moderate to severe AS as well as effect of telmisartan and losartan in bicuspid aortic valve patients ().
Table 1. Retrospective cohort studies of angiotensin converting enzyme inhibitor (ACEI) and angiotensin receptor blocker (ARB) treatments in calcific aortic valve disease (CAVD).
Table 2. Clinical trials of renin–angiotensin system targeting treatments in calcific aortic valve disease (CAVD).
Angiotensin converting enzyme (ACE), ACE2 and Mas receptor
ACE is a metalloproteinase that hydrolyzes angiotensin I into Ang II. Additionally, there are two other enzymes, chymase and cathepsin G, capable of increasing Ang II amounts locally in stenotic aortic valves (Citation15,Citation30). ACE2 is thought to counteract actions of Ang II by hydrolysing Ang II to Ang-(1-7), which in turn has opposite effects to those of Ang II by mediating beneficial antifibrotic, antihypertrophic, antiproliferative and anti-inflammatory effects (Citation9,Citation13,Citation31).
ACE is present in aortic valvular lesions, where it colocalizes with its product, Ang II, and with retained plasma lipoproteins (Citation14). In calcified valves, both ACE mRNA levels and protein activity of ACE are increased (Citation15), whereas components of ACE2-Ang-(1-7) –Mas –axis were reported to be downregulated in stenotic aortic valves (Citation17). Due to decreased ACE2 mRNA levels in stenotic valves, the disequilibrium between ACE and ACE2 expressions may play a key role in fibrocalcific process in which Ang II generation by ACE, chymase and cathepsin G is increased (Citation14,Citation15,Citation17,Citation30). Interestingly, these enzymes can also inactivate the antifibrotic bradykinin (Citation50). Furthermore, Ang-(1-7) has antiangiogenic properties (Citation32) suggesting that decreased production of Ang-(1-7) may, in part, contribute to the angiogenesis in fibrocalcific process. All in all, ACE inhibition may result in antiatherosclerotic effects, reduced neointimal formation and improved endothelial function and plaque stabilization (Citation33), and ACEIs are on theoretical level likely to have beneficial effects among CAVD patients (Citation34).
There are several studies evaluating the effect of ACEIs in patients of AS (). Indeed, beneficial effects have been reported in a retrospective report concerning ACEI treatment and lowered aortic valve calcium (AVC) accumulation, as measured by electron beam computed tomography (Citation35). In three clinical trials, the beneficial effects of ACEIs can be explained by improved hemodynamics (Citation26–28) (). In addition, in a recent observational study of more than 2000 patients with AS, the use of RAS inhibitors was associated with a significantly lower risk of death and other serious cardiovascular events in AS, even in patients with severe valve obstruction (Citation36). In one prospective, double–blind, controlled trial the ramipril group showed a slower rate of progression of the AS (valve area 0.0 cm2 in the ramipril group vs. −0.2 cm2 in the placebo arm) but without statistical significance (Citation29). Furthermore, there is an ongoing study of effects of captopril or trandolapril on hemodynamic and functional parameters in patients with severe AS. As regards human studies, no prospective large trials of ACE have been published to date. High blood pressure can be considered as a major determinant of valve disease progression, maybe through mechanical stretching of valve leaflets and/or increased shear stress. In addition, increased vascular afterload (ventricular wall stress) induces left ventricular hypertrophy and dysfunction, and this together with chronic valvular and vascular pressure overload is a major cause of the morbidity and mortality of CAVD (Citation37). Thus, some beneficial effects of ACE inhibitors, and ARBs as well, on clinical outcomes in patients with AS, could be attributed to lowering of blood pressure.
There might also be differences between ACE inhibition and AT1 antagonism as pharmacologic approach for calcified aortic valve disease. In several studies ACE inhibitors have modulated various components of the atherosclerotic process by inhibiting Ang II formation and by reducing breakdown of anti-fibrotic bradykinin (Citation33). Ang II increases lipid peroxidation and oxyradical formation, stimulates the expression of proinflammatory genes, such as chemoattractant protein and leucocyte adhesion molecules, resulting in endothelial dysfunction (Citation14,Citation15,Citation17,Citation30). In addition, Ang II induces vascular smooth-muscle proliferation (Citation38). By favoring the balance between Ang II and bradykinin, ACEIs are likely to improve endothelial function and counteract initiation and progression of atherosclerosis (Citation33). However, quantitative differences do exist among ACEIs. Ramipril and perindopril are highly lipophilic and have strong enzyme-binding capabilities; such ACE inhibitors may probably provide greater penetration into the atherosclerotic plaque in valvular location as well. Interestingly, hemodynamic retardation of AS, concomitantly with reduction in calcification, macrophage infiltration, redox stress and improvement in endothelial function has been reported with ramipril treatment in a model of AS in New Zealand white rabbits (Citation39).
Both Mas and AT2-receptor mRNA levels are both significantly downregulated in stenotic aortic valves (Citation17). Since, Ang II-forming enzymes are upregulated and AT1-receptor protein levels are increased in stenotic valves (Citation14,Citation15), the downregulation of both Mas and AT2-receptor expression (Citation16,Citation17), via unopposed AT1-receptor activation, may augment the profibrotic and pro-inflammatory effects of RAS. Furthermore, Mas and AT2-receptor activation induce eNOS activation (Citation40), and mutant mice lacking the Mas exhibit impaired endothelial function, decreased NO production and lower endothelial NO synthase (eNOS) expression (Citation40). In humans, expression of Mas receptor is mainly restricted to endothelial cells (Citation41), so Mas, along with the AT2, may contribute to the downregulation of eNOS and endothelial dysfunction in calcific aortic valve disease as well. Moreover, the first selective AT2 agonist compound 21 (C21) has been shown to have anti-inflammatory effects (Citation13,Citation42) so it would be interesting to see whether AT2 agonism could prevent inflammatory response related to disease progression of AS.
Renin, prorenin and (pro)renin receptor
Renin is a proteolytic enzyme synthesized and secreted as an inactive proenzyme, prorenin. The only known substrate for renin is angiotensinogen, which is cleaved by renin to form angiotensin I (Citation43). A specific human renin receptor, (P)RR, binds both prorenin and renin and shows a dual function. First, binding of prorenin to (P)RR leads to its activation and to generation of Ang II locally (Citation43). Secondly, binding of renin or prorenin to (P)RR results in the activation signaling pathways associated to profibrotic and proliferative actions as well as endothelial cell function and neovascularization, independent of Ang II production (Citation43–45).
Both renin and prorenin gene expression have been reported to be downregulated in stenotic aortic valves compared to normal valves, whereas no change in (P)RR mRNA levels has been noted (Citation17). Instead, a reduced amount of (P)RR positivity was seen in the valvular endothelial cells of stenotic valves but more in neovessels, consistent with proangiogenic effect of (P)RR (Citation17). Prorenin receptor ((P)RR) is the first counterpart receptor of tissue RAS, therefore (P)RR antagonism as a strategy would lead lowered activity of whole RAS cascade downwards. One important aspect of (P)RR is that it appears to be a vital component of the wnt/b-catenin pathway (Citation46). With its component, low-density lipoprotein receptor-related protein 5 (Lrp5), this pathway seems to be upregulated in the valvular tissue of rabbit CAVD (Citation47), and is a known regulator of osteogenic differentiation (Citation48). These raise the intriguing possibility that inhibition of (P)RR may also disturb the wnt/Lrp5/b-catenin-mediated osteogenic differentiation of valvular interstitial cells.
Bradykinin and bradykinin receptors
Kinin peptides act via two types of kinin receptors: the bradykinin B1 receptor (B1R) and the bradykinin B2 receptor (B2R). The cardioprotective effect of stimulation of B2R trigger the production of nitric oxide (NO) and exerts anti-proliferative and anti-hypertrophic effects on myocytes and fibroblasts (Citation49). In stenotic valves, increased expression of B1R and B2R has been reported (Citation50). Furthermore, exposure to tumor necrosis factor-α (TNFα) increased gene expression of B1R and B2Rin valvular cell culture environment (Citation50). Ang II generation by ACE, chymase and cathepsin G is increased in AS valves (Citation14,Citation15,Citation17,Citation30). Interestingly, these enzymes can also inactivate the antifibrotic bradykinin (Citation31,Citation51). Conversely, bradykinin counteracts the negative action of Ang II and improves endothelial function by increasing expression and activity of the constitutive NO synthase (Citation50). Bradykinin also inhibits the expression of monocytes and adhesion molecules (Citation50), and has an antiproliferative effect (Citation49,Citation52). In addition, in inflammatory conditions, bradykinin receptors B1 and B2 can couple to activate NF-kB signaling through Akt and/or ERK1/2 pathways, which leads to inhibition of osteoblastic differentiation with subsequent activation of osteoclast formation (Citation53). Consequently, bradykinin receptor specific agonists and/or antagonists could be therapeutic agents also in development of CAVD. Additionally, neutral endopeptidase (NEP or neprilysin), another enzyme capable to degrading bradykinin, is upregulated in stenotic valves (Citation50). These findings suggest that downregulation of bradykinin contributes to the shift of balance between fibrotic and antifibrotic factors promoting fibrosis in calcific aortic valve stenosis (Citation17). Consequently, inhibiting ACE, chymase, cathepsin G, and NEP that all contribute in producing Ang II by inhibiting bradykinin might lead to success as pharmacological CAVD therapy.
Endothelin and endothelin receptors
Four structurally different endothelin (ET) isoforms have been described (i.e., ET-1, ET-2, ET-3, ET-4). Mature ET-1 is formed from pre-pro-ET-1 via an intermediate big ET-1 by a family of endothelin converting enzymes (ECEs) and other enzymes such as chymases, and endopeptidases (Citation54). The biological effects of ET-1 are mediated by ETA and ETB receptors, both of which are expressed in aortic valves. ETA receptor expression is increased in stenotic valves whereas no changes in ETB expression was reported (Citation55). In addition, increased plasma big ET-1 levels have been reported in patients with AS (Citation56). In stenotic aortic valves, ET-1 protein is increased without increase in ET-1 mRNA levels, likely reflecting ETB receptor mediated up-take of ET-1 from circulation (Citation55). However, the levels of ETB receptors were similar between control and stenotic valves in the recent study (Citation55). Another possibility would be the changes in the degradation of ET-1. This seems, however, unlikely due to the increase in NEP activity in stenotic aortic valves (Citation15). The pathophysiological significance of increased ET-1 peptide levels in aortic valve stenosis may be further enhanced by the concomitant upregulation of ETA receptor gene (Citation55). Since, continued stimulation of cells with agonists generally result in a down-regulation of receptors, one would expect either ETA or ETB or both receptors to be down-regulated due to higher ET-1 peptide levels in stenotic valves. So it is possible that the net effect of ET-1 can be considered to be determined by receptor localization and the balance between ETA and ETB receptors (Citation57). As increased ET-1 and ETA protein levels have been documented in stenotic aortic valves (Citation55), particularly ETA receptor antagonism could potentially reduce the actions of ET-1 and even slow down the development of CAVD.
Interestingly, ET-1 was found to regulate calcification in both in vivo and in cultured vascular smooth muscle cells (Citation58). Moreover, administration of bosentan attenuated vascular calcification induced by vitamin D3 plus administration of nicotine in rats, but whether ET-1 has a role in development in CAVD is not known (Citation58). Experimental studies have established a role for vitamin D metabolites in pathways that are integral to cardiovascular function and disease, including inflammation, thrombosis, and the renin–angiotensin system and low levels of vitamin D are associated with cardiovascular diseases (Citation59). Interestingly, VDR deficiency and diets low in vitamin D promote aortic valve and aortic vessel calcification in the VDR−/− and LDLR−/− mouse models (Citation60). The vascular calcification in this study was associated with an up-regulation of osteoblast transcription factors which could have triggered the differentiation of vascular cells into osteoblast-like cells (Citation60).
The effect of dualistic ETA/ETB inhibition by tezosentan on ET-1 uptake was recently studied in cultured human aortic valve tissue explants (Citation61). Promisingly, tezosentan reduced the uptake of ET-1 to valve tissue more effectively in sclerotic aortic valve cusp explants than in macroscopically normal sections (Citation61). However, it remains to be determined whether ET receptor antagonists might have beneficial effects in slowing down the progression of AS. Experimentally, ETA receptor blockade prevents endothelial dysfunction and structural vascular changes in atherosclerosis (Citation54). On the other hand, the vasodilating and blood pressure lowering properties of ET receptor antagonists might present a clinical problem in AS. In patients with heart failure, ETA receptor antagonists (e.g., tezosentan) have controversial effects, although neurohormones and natriuretic peptides decreased favorably by treatment (Citation62). In addition to ET-1, NO signaling pathway is involved in aortic valve calcification since both eNOS and iNOS are expressed in human aortic valves, and eNOS gene expression is downregulated in stenotic valves (Citation55). Thus, imbalance between ETA and ETB receptors is potentiated by downregulated eNOS gene expression, similarly to endothelial dysfunction in atherosclerosis (Citation57).
Apelin – APJ system
Apelin, an adipokine, and its receptor APJ are expressed in several tissues including the heart, vasculature and aortic valves (Citation16,Citation63). The proposed cardiovascular effects of the apelin-APJ system are opposite to the effects of the RAS. ACE2, breaking down Ang II to Ang (Citation1–7), acts on apelin as well, suggesting a dynamic interaction between apelin and Ang II pathways (Citation64). Interestingly, non-activated APJ suppresses AT1, whereas apelin-activated APJ acts conversely (Citation65).
In stenotic aortic valves, there are increased mRNA and protein levels of apelin which are potented by the up-regulation on APJ gene (Citation16). Therefore, the upregulated apelin-APJ axis may also act as a compensatory mechanism ameliorating the harmful effects of AT1 receptor activation in the pathogenesis of aortic valve stenosis. In endothelial cells, apelin is expressed primarily in cells at sites of active vascular growth (Citation66), and APJ receptor may play an important role in angiogenesis (Citation67). In vitro, apelin promotes chemotaxis in human endothelial cells (Citation68), a well-established phenomenon in the plaque formation in stenotic aortic valves (Citation69). In addition, apelin has been reported to stimulate proliferation and to suppress apoptosis of mouse osteoblastic cell line (Citation70), and importantly, apelin was shown to attenuate the osteoblastic differentiation of aortic valve interstitium cells (VICs) via the extracellular signal–regulated kinase (ERK) and phosphatidylinositol-3 kinase (PI3-K)/Akt pathway (Citation71). Consequently, APJ receptor antagonists might be beneficial in the treatment of aortic valve stenosis by suppressing chemotaxis, angiogenesis and osteoblast activity. Apelin can also block a number of Ang II-related pathological processes associated with atherosclerosis (Citation72) and inhibit fibrosis (Citation73). It could be negatively regulated by miR125b (Citation74), which are upregulated in stenotic valves (Citation75). So far, there are no drugs targeting the apelin/APJ system available. By development of suitable agonists/antagonists, it will be intriguing to evaluate the impact of drug treatment targeting apelin/APJ system on CAVD pathology.
Natriuretic peptides
Natriuretic peptide family consists of three members, namely ANP (atrial natriuretic peptide), BNP (B-type natriuretic peptide) and CNP (C-type natriuretic peptide), whose biological effects are mediated by specific cell surface guanylate cyclase (GC) -linked receptors. ANP, BNP and CNP are produced as pro-forms and they are converted into mature peptides by proteolytic processing of the respective precursor molecules. Corin has been identified as a proANP- and proBNP-converting enzyme (Citation76), whereas furin processes proCNP to its mature form (Citation77).
All natriuretic peptides, their receptors as well as their processing enzymes are expressed in aortic valves (Citation78). The CNP system, i.e., CNP, furin and target receptor GC-B, has been reported to be down-regulated in stenotic valves, when compared with non-calcified aortic valves while no changes in ANP and BNP were seen (Citation78). Degradation of CNP does not exist only via target receptor GC-B or clearance receptor natriuretic peptide receptor-C (NPR-C), but via proteolytic processing by NEP, which has been reported to be up regulated in AS both in mRNA and protein level (Citation50).
Interestingly, CNP has a role in the bone growth (Citation79,Citation80), and it seems to inhibit vascular calcification (Citation81), fibrosis (Citation82) and inflammation (Citation83), which suggests its downregulation may be a contributing factor in several underlying processes of CAVD. In one study, CNP attenuated calcification partly via a cGMP/protein kinase G pathway both in rat calcified aortas and cultured vascular smooth muscle cells affecting protein levels of osteopontin and bone morphogenetic protein (Citation81). In addition, CNP has been shown to suppress calcified aggregate formation in (VICs) in vitro, as well as inhibit differentiation of VICs to osteoclasts and myofibroblasts when cultured under osteogenic and myofibrogenic conditions, respectively (Citation84). Furthermore, CNP expression was stimulated by simvastatin in VICs grown in myofibrogenic conditions, whereas small interfering RNA knockdown of NPRP-C significantly reduced the antifibrotic effect of simvastatin, suggesting that statins may act in part via CNP/NPR-B autocrine/paracrine signaling in the aortic valve (Citation84).
The studies of mice with endothelial-specific deletion of CNP (ecCNP KO mouse) indicated that endothelium derived CNP could contribute to reversing the development of atherosclerotic lesions (Citation85) and intervention with endothelium derived CNP could ameliorate fibrosis by reducing the activity and release of several matrix metalloproteinases (MMPs), including MMP-9, (Citation86), whose mRNA expression and activation is increased in calcific valves (Citation87). However, if CNP agonism with valvular targeted delivery would be considered as pharmacological intervention, valvular target receptors should be available in large scale in order to mediate beneficial effects of CNP.
An intriguing pharmacological approach would also be to investigate potential inhibitors to NEP or other CNP-degrading enzymes, as is the case with LCZ696 (sacubitril/valsartan). Combined neprilysin inhibition with valsartan (sacubitril + valsartan) seems to be interesting dualistic strategy with solid theory of benefits of ARB as well as prolonging effect of natriuretic peptides. Sacubitril is a first-in-class angiotensin receptor neprilysin inhibitor, converted by esterases to LBQ657, which inhibits neprilysin, the enzyme responsible for the degradation of the natriuretic peptides and many other vasoactive peptides (Citation88). The neprilysin inhibition in combination with ARB therapy could theoretically improve prognosis in patients with CAVD due to its beneficial effects on congestive heart failure (Citation89).
Finally, BNP is the most investigated circulating biomarker for AS, and it has been shown to be related to severity and functional status of the disease (Citation90,Citation91). In a recent study, BNP clinical activation was detected as a predictor of long-term mortality in patients with moderate or severe AS (Citation92), whereas in a large prospective cohort NT-proBNP had a poor diagnostic value for severe symptomatic AS in elderly patients (Citation91). Consequently, BNP has not yet been shown to provide sufficient incremental value in risk stratification in managing patients with AS (Citation1,Citation93).
Conclusions
Several components of vasoactive peptide systems are expressed in aortic valves with significant up/downregulation in CAVD (). So far, local RAS is the most studied vasoactive system, and the expression of its components may contribute to ACE/AngII/AT1-receptor-mediated fibrosis, proliferation and inflammation in AS (). Clinical trials have shown beneficial effects in AS patients managed with ARB but not with ACEI (). Although vasoactive factors have been intensively studied at the molecular level, there is lack of pharmacological agents to manipulate these systems. Nevertheless, renin, (P)RR, and endothelin (ETA/ETB) receptor antagonists should be studied in more detail in prospective clinical studies in CAVD patients.
Figure 1. Vasoactive peptides in calcific aortic valve disease. A schematic illustration summarizing peptides involved in comprehensive shift in the balance of valvular RAS components towards activated ACE/AngII/AT1-receptor-mediated fibrosis, proliferation and inflammation in AS. Black arrows show reported upregulated (↑), downregulated (↓) or unchanged (↔) gene expression in stenotic aortic valves.
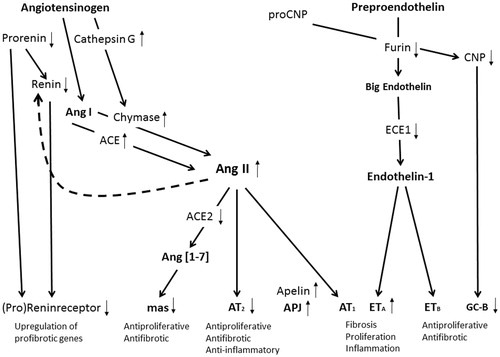
Table 3. Expression of vasoactive peptides in calcified aortic valves (vs normal) and their localization in calcified valves/normal valves.
Disclosure statement
T.P. is an employee of MSD Finland.
References
- Nishimura RA, Otto CM, Bonow RO, Carabello BA, Erwin JP, 3rd, Guyton RA, et al. 2014 AHA/ACC guideline for the management of patients with valvular heart disease: executive summary. A Report of the American College of Cardiology/American Heart Association Task Force on Practice Guidelines. J Am Coll Cardiol. 2014;63:2438–88.
- Otto CM, Prendergast B. Aortic-valve stenosis-from patients at risk to severe valve obstruction. N Engl J Med. 2014;371:744–56.
- Jander N, Minners J, Holme I, Gerdts E, Boman K, Brudi P, et al. Outcome of patients with low-gradient severe aortic stenosis and preserved ejection fraction. Circulation. 2011;123:887–95.
- Aronow WS. Valvular aortic stenosis in the elderly. Cardiol Rev. 2007;15:217–25.
- Otto CM, Kuusisto J, Reichenbach DD, Gown AM, O'Brien KD. Characterization of the early lesion of 'degenerative' valvular aortic stenosis. Histological and immunohistochemical studies. Circulation. 1994;90:844–53.
- Rosenhek R, Zilberszac R, Schemper M, Czerny M, Mundigler G, Graf S, et al. Natural history of very severe aortic stenosis. Circulation. 2010;121:151–6.
- Dweck MR, Boon NA, Newby DE. Calcific aortic stenosis: a disease of the valve and the myocardium. J Am Coll Cardiol. 2012;60:1854–63.
- Qian J, Chen Z, Ge J, Ma J, Chang S, Fan B, et al. Relationship between aortic valve calcification and the severity of coronary atherosclerotic disease. J Heart Valve Dis. 2010;194:466–70.
- Santos RA, Ferreira AJ, Verano-Braga T, Bader M. Angiotensin-converting enzyme 2, angiotensin-(1-7) and Mas: new players of the renin-angiotensin system. J Endocrinol. 2013;216:R1–17.
- Rossi GP, Sacchetto A, Cesari M, Pessina AC. Interactions between endothelin-1 and the renin-angiotensin-aldosterone system. Cardiovasc Res. 1999;43:300–7.
- Unger T, Chung O, Csikos T, Culman J, Gallinat S, Gohlke P, et al. Angiotensin receptors. J Hypertens Suppl. 1996;14:S95–S103.
- Kim S, Iwao H. Molecular and cellular mechanisms of angiotensin II-mediated cardiovascular and renal diseases. Pharmacol Rev. 2000;52:11–34.
- Steckelings UM, Paulis L, Namsolleck P, Unger T. AT2 receptor agonists: hypertension and beyond. Curr Opin Nephrol Hypertens. 2012;21:142–6.
- O'Brien KD, Shavelle DM, Caulfield MT, McDonald TO, Olin-Lewis K, Otto CM, et al. Association of angiotensin-converting enzyme with low-density lipoprotein in aortic valvular lesions and in human plasma. Circulation. 2002;106:2224–30.
- Helske S, Lindstedt KA, Laine M, Mayranpaa M, Werkkala K, Lommi J, et al. Induction of local angiotensin II-producing systems in stenotic aortic valves. J Am Coll Cardiol. 2004;44:1859–66.
- Peltonen T, Napankangas J, Vuolteenaho O, Ohtonen P, Soini Y, Juvonen T, et al. Apelin and its receptor APJ in human aortic valve stenosis. J Heart Valve Dis. 2009;18:644–52.
- Peltonen T, Napankangas J, Ohtonen P, Aro J, Peltonen J, Soini Y, et al. (Pro)renin receptors and angiotensin converting enzyme 2/angiotensin-(1-7)/Mas receptor axis in human aortic valve stenosis. Atherosclerosis. 2011;216:35–43.
- Fujisaka T, Hoshiga M, Hotchi J, Takeda Y, Jin D, Takai S, et al. Angiotensin II promotes aortic valve thickening independent of elevated blood pressure in apolipoprotein-E deficient mice. Atherosclerosis. 2013;226:82–7.
- Osako MK, Nakagami H, Shimamura M, Koriyama H, Nakagami F, Shimizu H, et al. Cross-talk of receptor activator of nuclear factor-κB ligand signaling with renin-angiotensin system in vascular calcification. Arterioscler Thromb Vasc Biol. 2013;33:1287–96.
- Ndip A, Wilkinson FL, Jude EB, Boulton AJ, Alexander MY. RANKL-OPG and RAGE modulation in vascular calcification and diabetes: novel targets for therapy. Diabetologia. 2014;57:2251–60.
- Yamamoto K, Yamamoto H, Yoshida K, Kisanuki A, Hirano Y, Ohte N, et al. Prognostic factors for progression of early- and late-stage calcific aortic valve disease in Japanese: the Japanese Aortic Stenosis Study (JASS) Retrospective Analysis. Hypertens Res. 2010;33:269–74.
- Capoulade R, Clavel MA, Mathieu P, Cote N, Dumesnil JG, Arsenault M, et al. Impact of hypertension and renin-angiotensin system inhibitors in aortic stenosis. Eur J Clin Invest. 2013;43:1262–72.
- Cote N, Couture C, Pibarot P, Despres JP, Mathieu P. Angiotensin receptor blockers are associated with a lower remodelling score of stenotic aortic valves. Eur J Clin Invest. 2011;41:1172–9.
- Helske-Suihko S, Laine M, Lommi J, Kaartinen M, Werkkala K, Kovanen PT, et al. Is blockade of the Renin-Angiotensin system able to reverse the structural and functional remodeling of the left ventricle in severe aortic stenosis? J Cardiovasc Pharmacol. 2015;65:233–40.
- Arishiro K, Hoshiga M, Negoro N, Jin D, Takai S, Miyazaki M, et al. Angiotensin receptor-1 blocker inhibits atherosclerotic changes and endothelial disruption of the aortic valve in hypercholesterolemic rabbits. J Am Coll Cardiol. 2007;49:1482–9.
- Chockalingam A, Venkatesan S, Subramaniam T, Jagannathan V, Elangovan S, Alagesan R, et al. Safety and efficacy of angiotensin-converting enzyme inhibitors in symptomatic severe aortic stenosis: symptomatic cardiac obstruction-pilot study of enalapril in aortic stenosis (SCOPE-AS). Am Heart J. 2004;147:E19.
- Jimenez-Candil J, Bermejo J, Yotti R, Cortina C, Moreno M, Cantalapiedra JL, et al. Effects of angiotensin converting enzyme inhibitors in hypertensive patients with aortic valve stenosis: a drug withdrawal study. Heart. 2005;91:1311–18.
- Dalsgaard M, Iversen K, Kjaergaard J, Grande P, Goetze JP, Clemmensen P, et al. Short-term hemodynamic effect of angiotensin-converting enzyme inhibition in patients with severe aortic stenosis: a placebo-controlled, randomized study. Am Heart J. 2014;167:226–34.
- Bull S, Loudon M, Francis JM, Joseph J, Gerry S, Karamitsos TD, et al. A prospective, double-blind, randomized controlled trial of the angiotensin-converting enzyme inhibitor ramipril in aortic stenosis (RIAS trial). Eur Heart J Cardiovasc Imaging. 2015;16:834–41.
- Helske S, Syvaranta S, Kupari M, Lappalainen J, Laine M, Lommi J, et al. Possible role for mast cell-derived cathepsin G in the adverse remodelling of stenotic aortic valves. Eur Heart J. 2006;27:1495–504.
- Paul M, Poyan Mehr A, Kreutz R. Physiology of local renin-angiotensin systems. Physiol Rev. 2006;86:747–803.
- Machado RD, Santos RA, Andrade SP. Mechanisms of angiotensin-(1-7)-induced inhibition of angiogenesis. Am J Physiol Regul Integr Comp Physiol. 2001;280:R994–R1000.
- Fox KM. European trial on reduction of cardiac events with perindopril in stable coronary artery disease investigators. Efficacy of perindopril in reduction of cardiovascular events among patients with stable coronary artery disease: randomised, double-blind, placebo-controlled, multicentre trial (the EUROPA study). Lancet. 2003;3629386:782–8.
- Elder DH, McAlpine-Scott V, Choy AM, Struthers AD, Lang CC. Aortic valvular heart disease: is there a place for angiotensin-converting-enzyme inhibitors? Expert Rev Cardiovasc Ther. 2013;11:107–14.
- O'Brien KD, Probstfield JL, Caulfield MT, Nasir K, Takasu J, Shavelle DM, et al. Angiotensin-converting enzyme inhibitors and change in aortic valve calcium. Arch Intern Med. 2005;165:858–62.
- Nadir MA, Wei L, Elder DH, Libianto R, Lim TK, Pauriah M, et al. Impact of renin-angiotensin system blockade therapy on outcome in aortic stenosis. J Am Coll Cardiol. 2011;58:570–6.
- Lindman BR, Bonow RO, Otto CM. Current management of calcific aortic stenosis. Circ Res. 2013;113:223–37.
- Stoll M, Steckelings UM, Paul M, Bottari SP, Metzger R, Unger T. The angiotensin AT2-receptor mediates inhibition of cell proliferation in coronary endothelial cells. J Clin Invest. 1995;95:651–7.
- Ngo DT, Stafford I, Sverdlov AL, Qi W, Wuttke RD, Zhang Y, et al. Ramipril retards development of aortic valve stenosis in a rabbit model: mechanistic considerations. Br J Pharmacol. 2011;162:722–32.
- Alenina N, Xu P, Rentzsch B, Patkin EL, Bader M. Genetically altered animal models for Mas and angiotensin-(1-7). Exp Physiol. 2008;93:528–37.
- Vaajanen A, Kalesnykas G, Vapaatalo H, Uusitalo H. The expression of mas-receptor of the renin-angiotensin system in the human eye. Graefes Arch Clin Exp Ophthalmol. 2015;253:1053–9.
- Wan Y, Wallinder C, Plouffe B, Beaudry H, Mahalingam AK, Wu X, et al. Design, synthesis, and biological evaluation of the first selective nonpeptide AT2 receptor agonist. J Med Chem. 2004;47:5995–6008.
- Nguyen G, Delarue F, Burckle C, Bouzhir L, Giller T, Sraer JD. Pivotal role of the renin/prorenin receptor in angiotensin II production and cellular responses to renin. J Clin Invest. 2002;109:1417–27.
- Satofuka S, Ichihara A, Nagai N, Noda K, Ozawa Y, Fukamizu A, et al. (Pro)renin receptor promotes choroidal neovascularization by activating its signal transduction and tissue renin-angiotensin system. Am J Pathol. 2008;173:1911–8.
- Uraoka M, Ikeda K, Nakagawa Y, Koide M, Akakabe Y, Nakano-Kurimoto R, et al. Prorenin induces ERK activation in endothelial cells to enhance neovascularization independently of the renin-angiotensin system. Biochem Biophys Res Commun. 2009;390:1202–7.
- Cruciat CM, Ohkawara B, Acebron SP, Karaulanov E, Reinhard C, Ingelfinger D, et al. Requirement of prorenin receptor and vacuolar H+-ATPase-mediated acidification for Wnt signaling. Science. 2010;327:459–63.
- Rajamannan NM, Subramaniam M, Caira F, Stock SR, Spelsberg TC. Atorvastatin inhibits hypercholesterolemia-induced calcification in the aortic valves via the Lrp5 receptor pathway. Circulation. 2005;112:I229–34. 9:
- Gong Y, Slee RB, Fukai N, Rawadi G, Roman-Roman S, Reginato AM, et al. LDL receptor-related protein 5 (LRP5) affects bone accrual and eye development. Cell. 2001;107:513–23.
- McAllister BS, Leeb-Lundberg F, Olson MS. Bradykinin inhibition of EGF- and PDGF-induced DNA synthesis in human fibroblasts. Am J Physiol. 1993;265:C477–84.
- Helske S, Laine M, Kupari M, Lommi J, Turto H, Nurmi L, et al. Increased expression of profibrotic neutral endopeptidase and bradykinin type 1 receptors in stenotic aortic valves. Eur Heart J. 2007;28:1894–903.
- Ferrario CM. New physiological concepts of the renin-angiotensin system from the investigation of precursors and products of angiotensin I metabolism. Hypertension. 2010;55:445–52.
- Morissette G, Houle S, Gera L, Stewart JM, Marceau F. Antagonist, partial agonist and antiproliferative actions of B-9870 (CU201) as a function of the expression and density of the bradykinin B1 and B2 receptors. Br J Pharmacol. 2007;150:369–79.
- Srivastava S, Sharma K, Kumar N, Roy P. Bradykinin regulates osteoblast differentiation by Akt/ERK/NFκB signaling axis. J Cell Physiol. 2014;229:2088–105.
- Barton M, Traupe T, Haudenschild CC. Endothelin, hypercholesterolemia and atherosclerosis. Coron Artery Dis. 2003;14:477–90.
- Peltonen T, Taskinen P, Napankangas J, Leskinen H, Ohtonen P, Soini Y, et al. Increase in tissue endothelin-1 and ETA receptor levels in human aortic valve stenosis. Eur Heart J. 2009;30:242–9.
- Bergler-Klein J, Klaar U, Heger M, Rosenhek R, Gabriel H, Binder T, et al. Big endothelin-1 is not a predictor in aortic stenosis, but is related to arterial blood pressure. Int J Cardiol. 2006;113:174–80.
- Bohm F, Pernow J. The importance of endothelin-1 for vascular dysfunction in cardiovascular disease. Cardiovasc Res. 2007;76:8–18.
- Wu SY, Zhang BH, Pan CS, Jiang HF, Pang YZ, Tang CS, et al. Endothelin-1 is a potent regulator in vivo in vascular calcification and in vitro in calcification of vascular smooth muscle cells. Peptides. 2003;24:1149–56.
- Norman PE, Powell JT. Vitamin D and cardiovascular disease. Circ Res. 2014;114:379–93.
- Schmidt N, Brandsch C, Kühne H, Thiele A, Hirche F, Stangl GI. Vitamin D receptor deficiency and low vitamin D diet stimulate aortic calcification and osteogenic key factor expression in mice. PLoS One. 2012;7:e35316.
- Leskelä HV, Vuolteenaho O, Koivula MK, Taskinen P, Ruskoaho H, Peltonen T, et al. Tezosentan inhibits uptake of proinflammatory endothelin-1 in stenotic aortic valves. J Heart Valve Dis. 2012;211:23–30.
- Anand I, McMurray J, Cohn JN, Konstam MA, Notter T, Quitzau K, et al. Long-term effects of darusentan on left-ventricular remodelling and clinical outcomes in the EndothelinA Receptor Antagonist Trial in Heart Failure (EARTH): randomised, double-blind, placebo-controlled trial. Lancet. 2004;364:347–54.
- Yu XH, Tang ZB, Liu LJ, Qian H, Tang SL, Zhang DW, et al. Apelin and its receptor APJ in cardiovascular diseases. Clin Chim Acta. 2014;428:1–8.
- Vickers C, Hales P, Kaushik V, Dick L, Gavin J, Tang J, et al. Hydrolysis of biological peptides by human angiotensin-converting enzyme-related carboxypeptidase. J Biol Chem. 2002;277:14838–43.
- Sun X, Iida S, Yoshikawa A, Senbonmatsu R, Imanaka K, Maruyama K, et al. Non-activated APJ suppresses the angiotensin II type 1 receptor, whereas apelin-activated APJ acts conversely. Hypertens Res. 2011;34:701–6.
- Saint-Geniez M, Masri B, Malecaze F, Knibiehler B, Audigier Y. Expression of the murine msr/apj receptor and its ligand apelin is upregulated during formation of the retinal vessels. Mech Dev. 2002;110:183–6.
- Goidescu CM, Vida-Simiti LA. The apelin-APJ system in the evolution of heart failure. Clujul Med. 2015;88:3–8.
- Cox CM, D'Agostino SL, Miller MK, Heimark RL, Krieg PA. Apelin, the ligand for the endothelial G-protein-coupled receptor, APJ, is a potent angiogenic factor required for normal vascular development of the frog embryo. Dev Biol. 2006;296:177–89.
- Helske S, Oksjoki R, Lindstedt KA, Lommi J, Turto H, Werkkala K, et al. Complement system is activated in stenotic aortic valves. Atherosclerosis. 2008;196:190–200.
- Eyries M, Siegfried G, Ciumas M, Montagne K, Agrapart M, Lebrin F, et al. Hypoxia-induced apelin expression regulates endothelial cell proliferation and regenerative angiogenesis. Circ Res. 2008;103:432–40.
- Yuan ZS, Zhou YZ, Liao XB, Luo JW, Shen KJ, Hu YR, et al. Apelin attenuates the osteoblastic differentiation of aortic valve interstitial cells via the ERK and PI3-K/Akt pathways. Amino Acids. 2015;47:2475–82.
- Chun HJ, Ali ZA, Kojima Y, Kundu RK, Sheikh AY, Agrawal R, et al. Apelin signaling antagonizes Ang II effects in mouse models of atherosclerosis. J Clin Invest. 2008;118:3343–54.
- Pchejetski D, Foussal C, Alfarano C, Lairez O, Calise D, Guilbeau-Frugier C, et al. Apelin prevents cardiac fibroblast activation and collagen production through inhibition of sphingosine kinase 1. Eur Heart J. 2012;33:2360–9.
- Nagpal V, Rai R, Place AT, Murphy SB, Verma SK, Ghosh AK, et al. MiR-125b is critical for fibroblast-to-myofibroblast transition and cardiac fibrosis. Circulation. 2016;1333:291–301.
- Ohukainen P, Syvaranta S, Napankangas J, Rajamaki K, Taskinen P, Peltonen T, et al. MicroRNA-125b and chemokine CCL4 expression are associated with calcific aortic valve disease. Ann Med. 2015;47:423–9.
- Yan W, Wu F, Morser J, Wu Q. Corin, a transmembrane cardiac serine protease, acts as a pro-atrial natriuretic peptide-converting enzyme. Proc Natl Acad Sci USA. 2000;97:8525–9.
- Wu C, Wu F, Pan J, Morser J, Wu Q. Furin-mediated processing of Pro-C-type natriuretic peptide. J Biol Chem. 2003;278:25847–52.
- Peltonen TO, Taskinen P, Soini Y, Rysa J, Ronkainen J, Ohtonen P, et al. Distinct downregulation of C-type natriuretic peptide system in human aortic valve stenosis. Circulation. 2007;116:1283–9.
- Chusho H, Tamura N, Ogawa Y, Yasoda A, Suda M, Miyazawa T, et al. Dwarfism and early death in mice lacking C-type natriuretic peptide. Proc Natl Acad Sci USA. 2001;98:4016–21.
- Komatsu Y, Chusho H, Tamura N, Yasoda A, Miyazawa T, Suda M, et al. Significance of C-type natriuretic peptide (CNP) in endochondral ossification: analysis of CNP knockout mice. J Bone Miner Metab. 2002;20:331–6.
- Chen JJ, Zhang J, Cai Y, Zhou YB, Wen GB, Tang CS, et al. C-type natriuretic peptide inhibiting vascular calcification might involve decreasing bone morphogenic protein 2 and osteopontin levels. Mol Cell Biochem. 2014;392:65–76.
- Kimura T, Nojiri T, Hino J, Hosoda H, Miura K, Shintani Y, et al. C-type natriuretic peptide ameliorates pulmonary fibrosis by acting on lung fibroblasts in mice. Respir Res. 2016;17:19
- Qian JY, Haruno A, Asada Y, Nishida T, Saito Y, Matsuda T, et al. Local expression of C-type natriuretic peptide suppresses inflammation, eliminates shear stress-induced thrombosis, and prevents neointima formation through enhanced nitric oxide production in rabbit injured carotid arteries. Circ Res. 2002;91:1063–9.
- Yip CY, Blaser MC, Mirzaei Z, Zhong X, Simmons CA. Inhibition of pathological differentiation of valvular interstitial cells by C-type natriuretic peptide. Arterioscler Thromb Vasc Biol. 2011;31:1881–9.
- Moyes AJ, Khambata RS, Villar I, Bubb KJ, Baliga RS, Lumsden NG, et al. Endothelial C-type natriuretic peptide maintains vascular homeostasis. J Clin Invest. 2014;124:4039–51.
- Krejci P, Masri B, Fontaine V, Mekikian PB, Weis M, Prats H, et al. Interaction of fibroblast growth factor and C-natriuretic peptide signaling in regulation of chondrocyte proliferation and extracellular matrix homeostasis. J Cell Sci. 2005;118:5089–100.
- Satta J, Oiva J, Salo T, Eriksen H, Ohtonen P, Biancari F, et al. Evidence for an altered balance between matrix metalloproteinase-9 and its inhibitors in calcific aortic stenosis. Ann Thorac Surg. 2003;76:681–8.
- Hubers SA, Brown NJ. Combined angiotensin receptor antagonism and neprilysin inhibition. Circulation. 2016;13311:1115–24.
- McMurray JJ, Packer M, Desai AS, Gong J, Lefkowitz MP, Rizkala AR, et al. Angiotensin-neprilysin inhibition versus enalapril in heart failure. N Engl J Med. 2014;371:993–1004.
- Steadman CD, Ray S, Ng LL, McCann GP. Natriuretic peptides in common valvular heart disease. J Am Coll Cardiol. 2010;55:2034–48.
- Cimadevilla C, Cueff C, Hekimian G, Dehoux M, Lepage L, Iung B, et al. Prognostic value of B-type natriuretic peptide in elderly patients with aortic valve stenosis: the COFRASA-GENERAC study. Heart. 2013;99:461–7.
- Clavel MA, Malouf J, Michelena HI, Suri RM, Jaffe AS, Mahoney DW, et al. B-type natriuretic peptide clinical activation in aortic stenosis: impact on long-term survival. J Am Coll Cardiol. 2014;63:2016–25.
- Vahanian A, Alfieri O, Andreotti F Antunes MJ, et al. Joint task force on the management of valvular heart disease of the European Society of Cardiology (ESC), European Association for Cardio-Thoracic Surgery (EACTS). Guidelines on the management of valvular heart disease (version 2012). Eur Heart J. 2012;3319:2451–96.
- Peltonen T, Ohtonen P, Näpänkangas J, Ohukainen P, Ruskoaho H, Taskinen P. Statin treatment and gene expression of anti-atherogenic factor C-type natriuretic peptide system in stenotic aortic valves. J Heart Valve Dis. 2011;205:545–51.