
Abstract
Introduction: The cardiac sodium channel SCN5A regulates atrioventricular and ventricular depolarization as well as cardiac conduction. Patients with cardiac electrical abnormalities have an increased risk of sudden cardiac death (SCD) and cardio-embolic stroke. Optimal management of cardiac disease includes the understanding of association between the causative mutations and the clinical phenotype. A 12-lead electrocardiogram (ECG) is an easy and inexpensive tool for finding risk patients.
Materials and methods: A blood sample for DNA extraction was obtained in a Finnish family with 43 members; systematic 12-lead ECG analysis was performed in 13 of the family members carrying an SCN5A D1275N mutation. Conduction defects and supraventricular arrhythmias, including atrial fibrillation/flutter, atrioventricular nodal re-entry tachycardia (AVNRT) and junctional rhythm were searched for.
Results: Five (38%) mutation carriers had fascicular or bundle branch block, 10 had atrial arrhythmias; no ventricular arrhythmias were found. Notching of the R- and S waves – including initial QRS fragmentation – and prolonged S-wave upstroke were present in all the affected family members. Notably, four (31%) affected family members had a stroke before the age of 31 and two experienced premature death.
Conclusions: A 12-lead ECG can be used to predict arrhythmias in SCN5A D1275N mutation carriers.
The 12-lead ECG may reveal cardiac abnormalities even before clinical symptoms occur.
Specific ECG findings – initial QRS fragmentation, prolonged S-wave upstroke as well as supraventricular arrhythmias – were frequently encountered in all SCN5A D1257N mutation carriers.
ECG follow-up is recommended for all SCN5A D1275N mutation carriers.
Key messages
Introduction
Regulation of the expression and proper function of voltage-gated ion channels at the plasma membrane of excitable cells is essential in maintaining cellular excitability and electrical impulse propagation. In the heart, voltage-gated sodium (Na+) channels determine the amplitude and slope of the action potential upstroke, which are especially important in the control of impulse conduction velocity, and in the maintenance of appropriate waves of excitation through the working myocardium. Dysfunction of these channels can lead to life-threatening cardiac arrhythmias [Citation1,Citation2].
Some mutations associated with cardiac disease have been identified in the gene encoding cardiac sodium channel α-subunit SCN5A, which is responsible for the generation and propagation of action potentials in the atrium, ventricle and specialized conduction system [Citation3].
Genetic defects associated with cardiac electrical disorders are usually found in genes encoding for cardiac ion channels. Cardiac Na+ channel dysfunction caused by mutations in the SCN5A gene is associated with a number of relative uncommon arrhythmic diseases, including long QT syndrome type 3 (LQT3), Brugada syndrome, and short QT interval with Brugada-like electrocardiogram
(ECG) [Citation2,Citation4], progressive cardiac conduction disease (PCCD), sick sinus syndrome (SSS), familiar atrial fibrillation (FAF), and atrial standstill, which all potentially can lead to fatal arrhythmias at relatively young age. Various SCN5A mutations are known to present with mixed cardiac phenotypes including arrhythmias and dilated cardiomyopathy [Citation2,Citation5]. SCN5A mutations are also involved in overlap syndromes of cardiac sodium channelopathy. Remme et al. [Citation6,Citation7] provided an overview of the current knowledge on SCN5A mutations associated with sodium channel overlap syndromes. They also discussed the possible role of modifiers in determining disease expressivity in an individual patient [Citation6–12].
Cardiac conduction, as assessed by the PR interval and QRS duration, is an important electrophysiological trait and a determinant of arrhythmic risk [Citation13]. Atrial standstill is a rare cardiac disorder characterized by the absence of electrical and mechanical atrial activity. Atrial standstill can be persistent or transient, and either diffuse or partial in the atrial wall involving either one or both atria [Citation14,Citation15]. Atrial standstill is characterized by bradycardia, diminution or absence of the P wave, junctional (usually narrow complex) escape rhythm and failure of atrial excitation, either spontaneously or by pacemaker stimulation. Atrial standstill is associated with an increased risk of sudden death due to occurrence of very low escape rhythm and an increased risk of thromboembolic stroke due to the high incidence of atrial fibrillation/flutter [Citation3].
Familiar atrial standstill has been associated with the SCN5A D1275N mutation combined with rare polymorphisms in an atrial-specific connexin40 (Cx40) gene [Citation16–19]. In a large Finnish family, cardiac conduction defects and atrial arrhythmias were also associated with the same SCN5A D1275N mutation, although not linked to the polymorphism in the Cx40 gene [Citation20].
Although no sudden cardiac death was recorded in the family, at least four affected members suffered from cardio-embolic stroke by the age of 31 and two of them died of stroke [Citation20]. In this report, we present the ECG findings, including atrial arrhythmias in individuals carrying the SCN5A D1275N mutation.
Materials and methods
Subjects
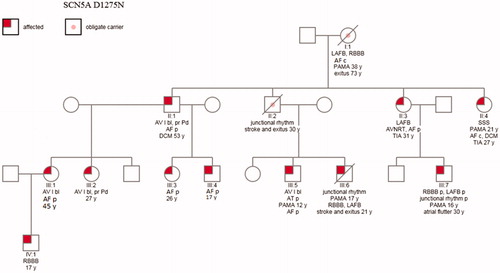
A Finnish family with 45 members was evaluated at the Heart Center, Tampere University Hospital. All participants gave their informed consent for study participation and the local Ethics Committee gave their approval for the study (R01128). A blood sample for DNA extraction was obtained from 43 family members. In this study, we report the ECG findings of eleven SCN5A D1275N mutation carriers and two deceased obligate carriers. The oldest family member had nine children and only the ECGs of the SCN5A D1275N mutation carriers, including the two obligate carriers were analyzed ().
Figure 1. Pedigree indicating the ECG findings. Four of nine children of the oldest obligate mutation carrier (patient I:1) had the SCN5A mutation, only these individuals significant to genetic analysis and their children are included. AF: atrial fibrillation or flutter; AT: atrial tachycardia; AV I: grade I atrioventricular block; AVNRT: atrioventricular nodal re-entry tachycardia; c: chronic; DCM: dilated cardiomyopathy; LAFB: left anterior fascicular block; p: paroxysmal; PAMA: implanted pacemaker; pr Pd: prolonged P duration; RBBB: right bundle branch block; SSS: sick sinus syndrome; TIA: transient ischemic attack; y: years.
Genetic analysis
The genotype analysis of the family members has been previously described in detail by Laitinen-Forsblom et al. [Citation20]. Based on candidate gene approach, a point mutation (guanine to adenine at position 3823 in exon 21) in the SCN5A gene was observed causing a substitution of aspartic acid with aspargine (D1275N).
ECG analysis
A 12-lead surface ECG was recorded at rest and the study participants were not allowed to smoke or drink coffee at least 6 h prior to the ECG recording. The ECG electrodes were positioned according to standardized protocol at a paper speed of 50 mm/s and 1 mV/cm standardization.
Heart rate, P-wave duration and amplitude, PR interval and deviation from the baseline, QRS duration, S-wave amplitude and up-stroke, QT, and corrected QT (QTc; Bazett′s formula) intervals were measured manually by one of the investigators (S. V.).
Normal P-wave duration was defined as 70–140 ms [Citation21]. The P wave was described as flat, if the amplitude was ≤0.05 mV in the limb leads.
We used 120–200 ms as normal values for the PR interval. The interval was measured in the lead with the largest and widest P wave and the longest QRS duration. The P and QRS axis was considered normal when it was between −30° and +90°. We defined PR-segment depression as ≥0.08 mV, and elevation as ≥0.05 mV [Citation21].
For QRS-complex fragmentation (fQRS), we used the definition by Das et al: additional R wave (R´ prime), notching in nadir of the S wave, notching of R wave, or the presence of more than one R prime (fragmentation). Typical bundle branch block (BBB) pattern (QRS ≥120 ms) – right or left – and incomplete RBBB were excluded from the definition of fQRS [Citation22,Citation23]. In left anterior fascicular block (LAFB), QRS duration may be normal or slightly prolonged [Citation21]. The upper limits of the S-wave amplitude in leads V1–V3 were 1.8, 2.6, and 2.1 mV [Citation20]. The QT-interval was measured according to established methodology [Citation21].
The diagnosis junctional escape rhythm required at least three successive complexes. Both the typical finding with a narrow QRS and possible cases with broad QRS due to pre-existing intraventricular conduction defect were included [Citation21].
Atrial standstill is characterized by bradycardia (HR< 50/bpm), the absence of P waves, and a junctional narrow or slightly wide QRS complex and regular escape rhythm [Citation16,Citation17,Citation21].
Results
Subjects
In genetic analysis, we found 11 SCN5A D1275N mutation carriers and two obligate carriers. The mutation in the SCN5A gene present in this family results in the substitution of asparagine for aspartic acid at aa 1275 (D1275N). One of the obligate carriers was the oldest family member (patient I:1; ) of this pedigree and another one (patient II:2) was the father of two mutation carriers (patients III:5 and III:6). Both of these obligate mutations’ carriers had passed away before this study was initiated. Four of nine children of the oldest obligate mutation carrier (patient I:1) had the SCN5A mutation. Only the mutation carriers of these children are presented in . Four mutation carriers (patients II:2, II:3, II:4, and III:6) of the family had encountered thromboembolic stroke by the age of 31 years, two of them (father II:2 and son III: 6) died at young age; the father at the age of 30 and the son even at the age of 21 ().
Electrocardiografic characteristics of the family
Five of the eleven (38%) mutation carriers had a VVIR-type of pacemaker implanted due to a bradycardic junctional rhythm (patients III:6 and III:7, ) or atrial fibrillation with symptomatic bradycardia (patients I:1, II:4, and III:5). The oldest family member (patient I:1) had atrial fibrillation with slow ventricular response at the age of 38 years and her ECG showed LAFB and RBBB before pacemaker implantation. She had anticoagulation therapy because of atrial fibrillation, but no history of stroke. Nine (69%) SCN5A D1275N mutation carriers had atrial fibrillation or flutter.
Transient atrial standstill was observed in one patient (III:7) (8%); he also had paroxysmal RBBB and LAFB. Two other patients – father and son – (II:2 and III:6) with junctional rhythm had no documented atrial tachyarrhythmias before premature death due to stroke. The son (III:6) also had RBBB and LAFB.
In all patients carrying the SCN5A D1275D mutation, we found a distinct initial pattern of QRS fragmentation with notching of the R wave (). This change was prominent in leads II, III, or aVF. The fragmentation seemed to be more prominent during junctional rhythm than during sinus rhythm (). All the SCN5A D1275N patients, who progressed from sinus rhythm to atrial standstill, showed an increase in the amplitude of the initial fragmentation of the QRS complex.
Figure 2. A typical 12-lead ECG in an asymptomatic patient with the SCN5A D1275N mutation. Wide arrow: low amplitude signal at the beginning QRS complex. Narrow arrow: notch in S wave. ![]()
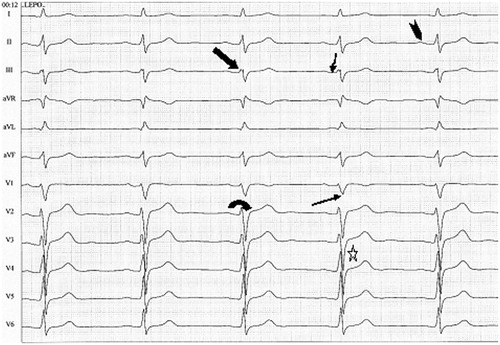
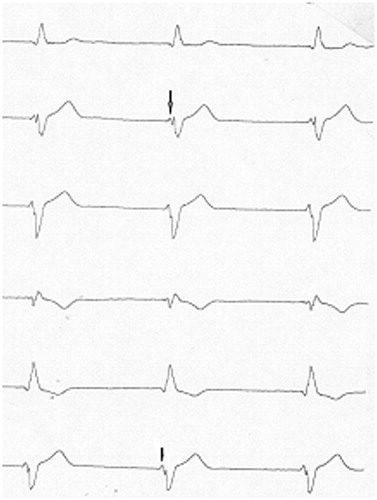
Figure 3. Typical large initial fragmentation of the QRS complex with junctional escape rhythm in an SCN5A gene D1275N mutation carrier. The arrow shows modification in the progressive growing initial fragmentation of QRS complex in inferior leads.
All 13 (100%) patients had fragmentation of the nadir of the S wave in at least one of the leads V1–V3 and also a prolonged S-wave upstroke.
The P wave was generally flat in all mutation carriers in conjunction with remarkable PR depression in three (23%) individuals (II:2, III:4, and III:7). The P wave could not be analyzed in three patients (II:2, III:5, and III:7) due to junctional rhythm. The QTc interval was normal in the 12-lead ECG in all mutation carriers ().
Table 1. Distribution of 12-leads ECG parameters of the SCN5A D1275N patients.
Discussion
We describe a large family with a mutation in the SCN5A gene (D1275N) with variable clinical phenotypes, including supraventricular arrhythmias, atrial standstill, and sudden death due to thromboembolic stroke. The 12-lead ECG proved to be abnormal in all affected family members. Atrial fibrillation or flutter and atrial standstill were probable aetiologic factors for thromboembolic stroke, and the deaths occurred at the age of 31 or younger. Typical changes of the 12-lead ECG predict conduction defects and atrial arrhythmias in the patients with the mutation SCN5A (D1275N). Many rare and common SCN5A variants are associated with cardiac conduction defects [Citation24] and atrial fibrillation with or without underlying heart disease [Citation25]. SCN5A mutations causing early-onset atrial fibrillation have demonstrated to been gain-of-function mutations [Citation26]. The mutation present in our family (SCN5A D1275N) causing conduction defects and atrial arrhythmias is, however, a loss-of-function mutation when analyzed in transfected cells [Citation27].
Cardiac conduction, as assessed by the PR interval and QRS duration, is an important electrophysiological trait and a determinant of arrhythmic risk [Citation13]. The ECG is characterized by different waves that represent specific phases of the cardiac activation sequence: the P wave reflects atrial depolarization, the QRS complex represents ventricular depolarization, and the T wave indicates ventricular repolarization [Citation28].
PR-segment changes
A wide, notched or low-amplitude Ta can predict an interatrial conduction disturbance with or without atrial enlargement. The Ta wave represents atrial repolarization and the interval from the beginning of the P wave to the end of Ta is the atrial equivalent of the ventricular QT interval [Citation29]. Theoretically, the analysis of atrial repolarization may reveal important information regarding arrhythmic propensity, as does QT-interval analysis in case of ventricular arrhythmias. PR depression may be considered as a marker of atrial repolarization abnormality with similar arrhythmogenic potential as the ST elevations for ventricular arrhythmias.
Conditions that significantly reduce Na+-current (i.e. Brugada syndrome mutations) may selectively shorten the duration of the epicardial action potential. This condition creates a temporal imbalance between endocardial and epicardial repolarization, and such electrical heterogeneity may underlie the ST-segment elevation and pro-arrhythmic manifestations of the Brugada syndrome [Citation30]. Three of our SCN5A gene mutation D1275N carriers in sinus rhythm had a remarkable PR depression in many leads just before junctional rhythm episodes. We propose that abnormal PR depression in many inferolateral leads may predict atrial conduction defects associated with P-wave prolongation.
Therefore, PR depression may be a precursor of atrial arrhythmias including conduction disorders, atrial fibrillation, and atrial standstill. Also, Makita et al. [Citation17] showed progressive atrial dysfunction leading to the atrial standstill in SCN5A mutation L212P.
P wave and PR interval
SCN5A mutations with loss-of-function properties have been identified in patients with cardiac conduction defects, SSS [Citation17], and atrial standstill [Citation16,Citation18]. Herfst et al. [Citation1] have shown that P-wave broadening and a tendency to PR-prolongation are manifested in the ECG of individuals with the SCN5A/5280delG mutation, presumably due to a reduction in Na+ current. PR prolongation was also associated with increased risk of atrial fibrillation [Citation31], need for pacemaker implantation, and excess mortality [Citation32]. We also found these changes in some of our SCN5A family member even though the mutation was different.
One of our patients, who underwent an electrophysiology study, proved to have fibrosis of the right atrial lateral wall. Loss of INa function could induce atrial fibrosis, which may cause atrial fibrillation in individuals with associated structural abnormalities including increased diastolic ventricular pressure in the setting of dilated cardiomyopathy with subsequent enlargement of the atria [Citation10]. Watanabe et al. studied the same SCN5A mutation as in our family and they observed slowed and disordered cardiac conduction and decreased contractile function in mice bearing an SCN5A mutation; mice with two D1275N alleles displayed worse phenotypes than those with one variant allele. In vitro electrophysiological studies identified reduced peak cardiac Na+ current as a key defect and this is consistent with the observed reduced conduction velocity [Citation33]. Two of our patients needed upgrading from a VVI-type to a physiologic pacemaker. However, after atrial lead implantation, they were more symptomatic of atrial arrhythmias, suggesting that atrial fibrosis could disturb pacemaker capture as was suggested by Chiang et al. [Citation34].
QRS fragmentation
Many investigators considered fragmented QRS (fQRS) as an independent predictor of cardiac events [Citation35–39]. When looking for an electrical link between the genotype and the phenotype in our patients, we noted a distinct ECG pattern with fragmentation of the R wave especially in leads II, III, and aVF. This fragmentation is a low-amplitude signal at the beginning of the QRS complex with similarities to the epsilon wave in arrhytmogenic right ventricular cardiomyopathy [Citation40], although in a different location. In the normal heart, an activated cell of the conduction system moves to positive potential because of the rapid inward surge of Na+-ions. Na+ provides the electrical energy for impulse conduction [Citation41]. Anomalous Na+ current handling in SCN5A D1275N gene mutation may affect the initial part of the QRS complex, resulting in the observed R-wave fragmentation. We have no definite explanation for the fact that this fragmented signal appeared to be larger during junctional rhythm. Interestingly, all our SCN5A D1275N patients, who progressed from sinus rhythm to atrial standstill, showed an increase in the amplitude of the initial fragmentation of the QRS complex. It could be speculated that progressive increase of this "spike" in the initial part of the QRS may signal disorders in atrial conduction and there by predict cardiac thromboembolic diseases.
All our gene mutation carriers also had fragmentation of the S wave and a prolonged upstroke in the right precordial leads in the 12-lead ECG. FQRS has been defined as a marker of arrhythmogenic right ventricular cardiomyopathy (ARVC) and the Brugada syndrome, both of which have been linked to Na+ channel dysfunction [Citation21]. Prolonged S-wave upstroke in leads V1 through V3 is a common ECG feature of arrhythmogenic right ventricular cardiomyopathy [Citation40]. Morita et al. [Citation39] showed an over-representation of patients with the SCN5A mutation in patients with fQRS (33% versus 5% in non-affected individuals). Our results are in line with these previous observations.
QRS duration
In the Cohorts for Heart and Aging Research in Genome Epidemiology (CHARGE) consortium QRS genome-wide association study meta-analysis, QRS duration was associated with future development of atrial fibrillation [Citation41]. Probst et al. [Citation42] described progressively increased QRS duration in patients with SCN5A-linked hereditary Lev–Lenegre disease affecting the His bundle and its branches. Our SCN5A D1275N family members had prolonged QRS duration; many of them had different atrial arrhythmias and some of them had BBB. It is possible that SCN5A D1275N mutation also causes degenerative intraventricular conduction defects. Types of intraventricular conduction defects (such as LBBB, LAHB, and RBBB) were present in the family with SCN5A mutation in the classical description of idiopathic Lev–Lenegre disease [Citation42]. Te Riele et al. speculated that Nav1.5 is a multifunctional protein with a variety of clinical phenotypes depending on genetic changes in Nav1.5. They showed using induced pluripotent stem cell-derived cardiomyocytes (hIPSC-CMs) from an ARVC patient with SCN5A variants that QRS duration was significantly prolonged [Citation43].
Progressively increased conduction defects predict atrial paralysis and atrial arrhythmias, which are possible reasons for cardio-embolic stroke without anticoagulation [Citation42,Citation44]. Prost et al. demonstrated that carriers of a SCN5A mutation need a clinical and ECG follow-up because of the risk associated with severe conduction defects [Citation45]. Mutations in the cardiac Na+ channel encoded by the gene SCN5A can result in many different phenotypes. The wide array of clinical phenotypes explains the variable outcomes of the same mutation even within one family.
Limitations
Our study did not include long-term follow-up. Therefore, we have no solid data on the consistency of the ECG findings. We were not able to perform reliable analysis of the P waves during stress test in the study population. Therefore, exercise test data were not included.
The findings presented represent one family with a specific gene mutation. These findings may not necessarily apply to other families with mutations in the same gene. More studies are needed to explore the complete ECG pattern of SCN5A gene mutations.
Conclusions
In a Finnish family with the SCN5A D1275N mutation, ECG changes were very frequent in the mutation carriers. In addition to P-wave and PR changes, typical abnormalities were fragmented R waves in the inferior leads II, III, and aVF as well as of the S wave in the leads V1–V3 accompanied by slow S-wave upstroke and prolonged QRS duration. Atrial electrical changes probably predispose to arrhythmias, including junctional rhythm, atrial standstill, and atrial fibrillation with increased cardio-embolic stroke risk at young age. Our findings confirm that complex and variable findings are present in the standard 12-lead ECG in individuals with a single SCN5A mutations and, therefore, ECG follow-up of asymptomatic individuals is recommended.
Disclosure statement
The authors report no conflicts of interest. The authors alone are responsible for the content and writing of this article.
References
- Herfst LJ, Potet F, Bezzina CR, et al. Na+ channel mutation leading to loss of function and non-progressive cardiac conduction defects. J Mol Cell Cardiol. 2003;35:549–557.
- Perez-Riera A, Raimundo RD, Watanabe RA, et al. Cardiac sodium channel, its mutations and their spectrum of arrhythmia phenotypes. J Hum Growth Dev. 2016;26:281–296.
- Castro A, Loricchio ML, Turreni F, et al. Role of electroanatomic mapping in assessing the extent of atrial standstill: diagnostic and therapeutic implications. J Cardiovasc Med (Hagerstown). 2009;10:787–791.
- Hong K, Hu J, Yu J, et al. Concomitant Brugada-like and short QT electrocardiogram linked to SCN5A mutation. Eur J Hum Genet. 2012;20:1189–1192.
- Zaklyazminskaya E, Dzemeshkevich S. The role of mutations in the SCN5A gene in cardiomyopathies. Biochim Biophys Acta. 2016;1863:1799–1805.
- Remme CA, Wilde AAM, Bezzina CR. Cardiac sodium channel overlap syndromes: different faces of SCN5A mutations. Trends Cardiovasc Med. 2008;18:78–87.
- Remme CA. Cardiac sodium channelopathy associated with SCN5A mutations: electrophysiological, molecular and genetic aspects. J Physiol. 2013;59:4099–4116.
- Remme CA, Bezzina CR. Sodium channel (dys)function and cardiac arrhythmias. Cardiovasc Ther. 2010;28:287–294.
- Amin AS, Asghari-Roodsari A, Tan HL. Cardiac sodium channelopathies. Pflugers Arch. 2010;460:223–237.
- Wilde AAM, Brugada R. Phenotypical manifestations of mutations in the genes encoding subunits of cardiac sodium channel. Circ Res. 2011;108:884–897.
- Wolf CM, Berul CI. Inherited conduction system abnormalities – one group of diseases, many genes. J Cardiovasc Electrophysiol. 2006;17:446–455.
- Tfelt-Hansen J, Winkel BG, Grunnet M, et al. Inherited cardiac diseases caused by mutations in the Nav1.5 sodium channel. J Cardiovasc Electrophysiol. 2010;21:107–115.
- Smith JG, Lowe JK, Kovvalim S, et al. Genome-wide association study of electrocardiographic conduction measures in an isolated founder population: Kosrae. Heart Rhythm. 2009;6:634–641.
- Fazelifar AF, Arya A, Haghjoo M, et al. Familial atrial standstill in association with dilated cardiomyopathy. Pacing Clin Electrophysiol. 2005;28:1005–1008.
- Jorat MV, Nikoo MH, Yousefi A. Persistent isolated right atrial standstill associated with left atrial tachycardia. Res Cardiovasc Med. 2014;3:1–3.
- Groenewegen WA, Firouzi M, Bezzina CR, et al. A cardiac sodium channel mutation cosegregates with a rare connexin40 genotype in familiar atrial standstill. Circ Res. 2003;92:14–22.
- Makita N, Sasaki K, Groenewegen WA, et al. Congenital atrial standstill associated with coinheritance of a novel SCN5A mutation and connexin 40 polymorphisms. Heart Rhythm. 2005;2:1128–1134.
- Makita N. Phenotypic overlap of cardiac sodium channelopathies: individual-specific or mutation-specific? Circ J. 2009;73:810–817.
- Olson TM, Michels VV, Ballew JD, et al. Sodium channel mutations and susceptibility to heart failure and atrial fibrillation. JAMA. 2005;293:447–454.
- Laitinen-Forsblom PJ, Mäkynen P, Mäkynen H, et al. SCN5A mutation associated with cardiac conduction defect and atrial arrhythmias. J Cardiovasc Electrophysiol. 2006;17:480–485.
- Surawicz B, Knilans TK. Chou´s electrocardiography in clinical practice: 6. Philadelphia: Saunders; 2008. p. 8–44;108–110; 355; 386–388.
- Das MK, Zipes DP. Fragmented QRS: a predictor of mortality and sudden cardiac death. Heart Rhythm. 2009;6:S8–S14.
- Basaran Y, Tigen K, Karaahmet T, et al. Fragmented QRS complexes are associated with cardiac fibrosis and significant intraventricular systolic dyssyncrony in nonischemic dilated cardiomyopathy patients with a narrow QRS interval. Echocardiography. 2011;28:62–68.
- Magnani JW, Brody JA, Prins BP, CHARGE Consortium; NHLBI Exome Sequencing Project (ESP); UK10K, et al. Sequencing of SCN5A identifies rare and common variants associated with cardiac conduction: cohorts for heart and aging research in genomic epidemiology (CHARGE) consortium. Circ Cardiovasc Genet. 2014;7:365–373.
- Darbar D, Kannankeril PJ, Donahue BS, et al. Cardiac sodium channel (SCN5A) variants associated with atrial fibrillation. Circulation. 2008;117:1927–1935.
- Olesen MS, Yuan L, Liang B, et al. High prevalence of long QT syndrome-associated SCN5A variants in patients with early-onset lone atrial fibrillation. Circ Cardiovasc Genet. 2012;5:450–459.
- Gui J, Wang T, Jones RPO, et al. Multiple loss-of-function mechanisms contribute to SCN5A-related familial sick sinus syndrome. PLoS One. 2010;5:e10985.
- Terrenoire C, Simhaee D, Kass RS. Role of sodium channels in propagation in heart muscle: how subtle genetic alternations result in major arrhythmic disorders. J Cardiovasc Electrophysiol. 2007;18:900–905.
- Childers R. Atrial repolarization: its impact on electrocardiography. J Electrocardiol. 2011;44:635–640.
- Balser JR. The cardiac sodium channel: gating function and molecular pharmacology. J Mol Cell Cardiol. 2001;33:599–613.
- Shulman E, Aagaard P, Kargoli F, et al. Validation of PR interval length as a criterion for development of atrial fibrillation in non-Hispanic whites, African Americans and Hispanics. J Electrocardiol. 2015;48:703–709.
- Cheng S, Keyes MJ, Larson MG, et al. Long-term outcomes in individuals with prolonged PR interval or first-degree atrioventricular block. JAMA. 2009;301:2571–2577.
- Watanabe H, Yang T, Stroud DM, et al. Striking in vivo phenotype of a disease-associated human SCN5A mutation producing minimal changes in vitro. Circulation. 2011;124:1001–1011.
- Chiang DY, Kim JJ, Valdes SO, et al. Loss-of-function SCN5A mutations associated with sinus node dysfunction, atrial arrhythmias, and poor pacemaker capture. Circ Arrhythm Electrophysiol. 2015;8:1105–1112.
- Sha J, Zhang S, Tang M, et al. Fragmented QRS is associated with all-cause mortality and ventricular arrhythmias in patient with idiopathic dilated cardiomyopathy. Ann Noninvasive Electrocardiol. 2011;16:270–275.
- Chatterjee S, Changawala N. Fragmented QRS Complex: a novel marker of cardiovascular disease. Clin Cardiol. 2010;33:68–71.
- Das MK, El Masry H. Fragmented QRS and other depolarization abnormalities as a predictor of mortality and sudden cardiac death. Curr Opin Cardiol. 2010;25:59–64.
- Das MK, Maskoun W, Shen C, et al. Fragmented QRS on twelve-lead electrocardiogram predicts arrhythmic events in patients with ischemic and nonischemic cardiomyopathy. Heart Rhythm. 2010;7:74–80.
- Morita H, Kusano KF, Miura D, et al. Fragmented QRS as a marker of conduction abnormality and a predictor of prognosis of Brugada syndrome. Circulation. 2008;118:1697–1704.
- Muhappan P, Calkins H. Arrhythmogenic right ventricular dysplasia. Prog Cardiovasc Dis. 2008;51:31–43.
- Ritchie MD, Denny JC, Zuvich RL, et al. Genome-and phenome-wide analyses of cardiac conduction identifies markers of arrhythmia risk. Circulation. 2013;127:1377–1385.
- Probst V, Kyndt F, Potet F, et al. Haploinsufficiency in combination with aging causes SCN5A-linked hereditary Lenègre disease. J Am Coll Cardiol. 2003;41:643–652.
- te Riele ASJM, Agullo-Pascual E, James CA, et al. Multilevel analyses of SCN5A mutations in arrhythmogenic right ventricular dysplasia/cardiomyopathy suggest non-canonical mechanisms for disease pathogenesis. Cardiovasc Res. 2017;113:102–111.
- Sajeev CG, Francis J, Sankar V, et al. Idiopathic dilatation of right atrium with atrial standstill presenting as stroke. Echocardiography. 2006;23:50–52.
- Probst V, Allouis M, Sacher F, et al. Progressive cardiac conduction defect is the prevailing phenotype in carriers of a Brugada syndrome SCN5A Mutation. J Cardiovasc Electrophysiol. 2006;17:270–275.