
Abstract
According to the current paradigm, chronic vascular inflammation plays a central role in the pathogenesis of atherosclerosis. The plaque progression is typically completed with rupture and subsequent acute cardiovascular complications. Previously, the role of adventitial vasa vasorum in atherogenesis was underestimated. However, investigators then revealed that vasa vasorum neovascularization can be observed when no clinical manifestation of atherosclerosis is present. Vasa vasorum is involved in various proatherogenic processes such as intimal accumulation of inflammatory leukocytes, intimal thickening, necrotic core formation, intraplaque haemorrhage, lesion rupture and atherothrombosis. Due to the destabilizing action of the intraplaque microenvironment, lesional vasa vasorum neovessels experience serious defects and abnormalities during development that leads to their immaturity, fragility and leakage. Indeed, intraplaque neovessels are a main cause of intraplaque haemorrhage. Visualization techniques showed that presence of neovascularization/haemorrhage can serve as a good indicator of lesion instability and higher risk of rupture. Vasa vasorum density is a strong predictor of acute cardiovascular events such as sudden death, myocardial infarction and stroke. At present, arterial vasa vasorum neovascularization is under intensive investigation along with development of therapeutic tools focused on the control of formation of vasa vasorum neovessels in order to prevent plaque haemorrhage/rupture and thromboembolism.
Neovascularization plays an important role in atherosclerosis, being involved in unstable plaque formation.
Presence of neovascularization and haemorrhage indicates plaque instability and risk of rupture.
Various imaging techniques are available to study neovascularization.
KEY MESSAGE
Introduction
Atherosclerosis is a chronic inflammatory disorder. Late atherosclerotic complications mainly include myocardial infarction (MI) and stroke that are triggered by plaque disruption. Analyses of autopsy samples provided a conclusion that major attributes of unstable lesions are high lipid content, thin fibrous cap highly infiltrated by proinflammatory leukocytes, and intensive intraplaque neovascularization [Citation1]. Recent findings indicate that intralesional neovascularization plays a critical role in plaque destabilization.
Plaque neovessels arise from the vasa vasorum, a network of small blood vessels, which supply the vascular wall and neighbouring tissues with oxygen and nutrients and remove the products of cellular metabolism [Citation2]. Barger et al. first hypothesized that vasa vasorum can be implicated in atherogenic process [Citation3]. Pathomorphological studies of human autopsy material and animal models suggested for higher density of vasa vasorum in unstable lesions [Citation4,Citation5]. However, over 80% of neovessels in human coronary atherosclerotic lesions are weakly integrated, which often leads to intraplaque leakage [Citation6]. In experimental models, advanced vasa vasorum growth correlates with endothelial dysfunction and intimal thickening. Blockers of angiogenesis can improve this condition [Citation7]. Vascular inflammation plays a primary role in the induction of plaque haemorrhages and the recruitment of inflammatory cells to the adventitial neovessels [Citation8]. Leaky intraplaque neovessels represent a direct gateway for entrance of inflammatory immune cells into the lesion [Citation9].
Due to the significance of formation of lesional neovessels as an indicator of future development of intraplaque haemorrhage and plaque disruption, new imaging techniques were constructed to observe the neovascularization in the early atherogenic steps. Accordingly, therapies that target vasa vasorum are under development to represent a new strategy in cardiovascular treatment.
Vasa vasorum structural organization and function
In humans, vasa vasorum becomes visible during the first gestation week [Citation10]. The vasa vasorum lies mainly in the arterial adventitia and outer tunica media. In cattle and swine aortas, three vasa vasorum types were found: vasa vasorum externae (VVE), vasa vasorum VV internae (VVI) and venous vasa vasorum (VVV). While VVE originates from the main branches, VVI arises from the main aortic lumen. The VVV supports blood efflux from the arterial wall to the partner veins. The absolute majority (over 96%) of vasa vasorum neovessels belongs to VVE, with only a small portion coming from the lumen [Citation11].
Current imaging technique such as high-resolution micro-computed tomography (micro-CT) resolves a more delicate structural organization of the vasa vasorum. Vasa vasorum arises from the main arterial vessel and runs along the vascular wall to form the first-order vasa vasorum, which then branches to form a second-order vasa vasorum [Citation12]. In normal pig heart, the density of first-order vessels was 1.5-fold greater than that of the second-order vessels. By contrast, in diseased hearts, the density of second-order vessels was two-fold higher than that of first-order vessels [Citation4]. Moreover, the vasa vasorum branching in affected arteries is significantly more chaotic compared with dichotomically organized tree structure of the vasa vasorum in normal vasculature [Citation13].
Atherosclerotic vasa vasorum neovessels are immature and fragile, with weakly developed inter-endothelial junctions. The permeability of neovessels is greater in the areas close to the plaque [Citation14]. Changes of the endothelium include abnormal morphology of endothelial cells (ECs), presence of intercellular gaps, detached basement membrane, and impaired contacts with pericytes [Citation6]. The incomplete coverage of neovessels with pericytes disturbs vessel integrity and can be one of the causes of haemorrhage [Citation15]. Vascular smooth muscle cells (VSMCs) are poorly attached to the intraplaque vessels, which cases defects in vasodilatation/vasoconstriction and other abnormalities [Citation16].
Vasa vasorum provides the oxygen and nutrient supply to the outer one-third of the vessel media thickness. Oxygen levels are highest in the outer vascular wall layers and gradually decrease towards the lumen [Citation17]. The lowest oxygen level measured in the arterial wall corresponds to 10 mm/Hg at the distance of 300 μm from the lumen [Citation18]. Noteworthy, vasa vasorum is involved in the lipid transfer, and can therefore play a role in the lesional lipid core development [Citation19].
The proximal vasa vasorum vessels have the endothelial layer and adventitial VSMCs surrounded by connective tissue, which points to the possibility that such vessels might be capable of regulating their own tone [Citation20]. Studies on dog and pig tissues demonstrated that animal aorta was able to contract/relax in response to treatment with endothelin-1 (ET-1) and other vasoactive agents [Citation21,Citation22].
Under normal conditions, vasa vasorum is involved in the repair of vascular damage by providing an increased supply of oxygen and nutrients [Citation12]. However, in atherosclerotic conditions, the vasa vasorum function is compromised by hypoxic inflammatory microenvironment and influx of inflammatory cells. Since the neovessels serve as a gateway for inflammatory leukocytes, red blood cells and lipids, the plaque-associated vasa vasorum plays a complex role in atherosclerosis progression.
Stimulation of intraplaque neovascularization
Hypoxia/ischaemia
Vasa vasorum neovascularization plays an important role in the plaque development and progression. Oxygen deprivation in the plaque environment is a potent inducer of neovascularization (). In atherosclerosis, progressive intimal thickening associated with luminal narrowing is an essential event leading to hypoxia [Citation23]. Further plaque growth leads to the enlargement of hypoxic area. Vasa vasorum vessels are particularly sensitive to hypoxia due to their location in arterial ends [Citation24]. Blood supply itself cannot permeate deep enough from the adventitia to the media due to the pressure in the arterial wall. Aging and hypertension stimulate elevation of arterial tone that intervenes with the vasa vasorum-mediated blood supply of the inner layers and decreases oxygen content in vasa vasorum capillaries [Citation2]. Smoking causes contraction of arteries in the periphery, which limits peripheral blood supply and blood influx to the vasa vasorum.
Figure 1. Hypoxia/ischaemia induces vasa vasorum neovascularization in atherosclerotic plaques. Intimal thickening, reduced oxygen supply and oxygen consumption due to the active metabolism of macrophages represent factors that lead to the induction of the intraplaque hypoxia (left panel). Indeed, hypoxia stimulates hypoxia-inducible factor 1 (HIF1) that directs transcription of proangiogenic factors such as vascular endothelial growth factor A (VEGF-A) and E26 transformation-specific-1 (Ets-1). Ets-1 is a transcription factor that activates VEGF expression. VEGF reciprocally stimulates Ets-1 activity. Indeed, induction of VEGF and Ets-1 activates angiogenesis and formation of neovessels. However, due to the deregulated angiogenic control, influence of proinflammatory plaque microenvironment and lipid core, neovessels have the immature phenotype associated with fragility, morphological defects, increased permeability and leakage. The leaky neovessels is the main cause of intraplaque haemorrhage. Intraplaque neovascularization is a strong indicator of future plaque destabilization and rupture.
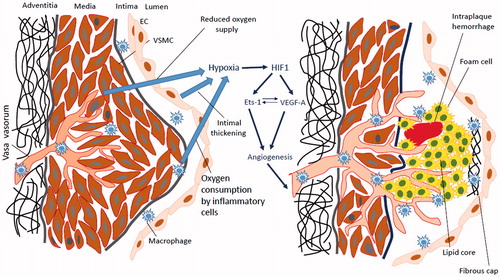
In advanced plaques, the active metabolism observed in lipid-laden macrophages and foam cells may contribute to the intraplaque oxygen deficiency and hypoxia [Citation25]. Hypoxia correlates with the plaque metabolic status, reflected by the presence of the angiogenic process or cells characterized by high metabolic activity, such as macrophages [Citation26]. Indeed, these results suggest that influence of the lesional metabolic activity on hypoxia induction has a more profound impact than intimal thickness. Altogether, intimal thickness, reduced blood influx to the vasa vasorum, and higher oxygen consumption are the primary contributors to the hypoxia development.
In the adaptive response to hypoxia, vasa vasorum tend to grow across the vascular wall towards the lumen to provide oxygen supply to the inner layers [Citation27]. In animal models of coronary balloon injury, damaged arteries cannot support the myocardium with sufficient oxygen levels, but vasa vasorum neovessels are able to compensate for this deficiency [Citation28]. In atherosclerotic pig model, low density of the vasa vasorum inversely correlated with hypoxia (i.e. reduced oxygen supply) and higher oxidative stress (i.e. increased superoxide formation) [Citation29].
Studies of signalling mechanisms in hypoxia-induced vasculogenesis revealed the key role of hypoxia-induced factor (HIF) and HIF-dependent downstream factors (). Apart from the central role in angiogenesis, HIF influences numerous atherogenic pathways, including cell proliferation, foam cell formation, intraplaque haemorrhage and plaque rupture [Citation30].
HIF-1 is a heterodimeric transcription factor composed of two subunits (HIF-1α and HIF-1β), which drives expression of hypoxia-inducible genes, including vascular endothelial growth factor (VEGF), heme oxygenase-1 (HO1) and E26 transformation-specific-1 (Ets-1) [Citation31]. Among HIF-1-induced genes, VEGF and Ets-1, transcription factors, are the most critical regulators of hypoxia-dependent angiogenesis.
Proangiogenic growth factors belonging to the VEGF family exhibit powerful mitogenic and promigratory properties focused on ECs and control neovascularization in physiological and pathological conditions [Citation32]. VEGF-A, the central member of the VEGF family, is crucially involved in neovessels formation through binding to the VEGF receptor type-2 (VEGFR2) [Citation33]. VEGF-A is induced in response to hypoxic conditions. In the aorta of hypertensive rats, expression of HIF-1α and VEGF-A changes simultaneously and is associated with the vasa vasorum development. Similar link between HIF-1α and VEGF-A was observed in lesions [Citation34].
VEGF-C and VEGF-D bind to both VEGFR2 and VEGFR3 [Citation35,Citation36]. Apart from lymphatic vessels, VEGFR3 is also expressed in the adult aorta and other fenestrated blood vessels [Citation37]. In atherosclerotic lesions, expression of VEGFR3 was observed in adventitial ECs and VSMCs, but not in the luminal endothelium [Citation38,Citation39]. Constitutive VEGF-D expression was observed in healthy vessels, fatty streaks and lesions. Since VEGFR3 expression is mainly restricted to the adventitial ECs, VEGF-D/VEGFR3 axis was suggested to play a role in neovascularization [Citation38]. However, overexpression of the VEGFR3 transgene in hypercholesterolemic mice did not influence the vasa vasorum density in atherosclerotic plaques, but greatly inhibited growth of lymphatic vessels and led to higher cholesterol and triglyceride levels and atherosclerosis progression [Citation40]. Indeed, in atherosclerosis, VEGFR3 is more relevant to alterations of lipid profile and lymphoid system rather than neovascularization.
Ets-1 drives expression of matrix metalloproteinases (MMPs), which are involved in tissue remodelling and support EC migration in angiogenesis [Citation41]. The Ets-1 promoter contains a hypoxia-responsive element (HRE) recognized by HIF-1, which directs Ets-1 transcription in response to hypoxia [Citation42]. Ets-1 stimulates angiogenesis by activating expression of VEGF and hepatocyte growth factor (HGF) and generating a positive feedback loop that leads to the up-regulation of Ets-1 [Citation43]. HIF-1, VEGF and Ets-1 play a prominent role in intraplaque neovascularization, which is reflected by a markedly increased expression of these factors in deep plaque regions characterized by active angiogenesis. Increased levels of VEGF were found in instable human lesions obtained at carotid endarterectomy. VEGF and Ets-1 levels were higher in plaques with haemorrhage [Citation44]. It remains to be investigated whether VEGF and Ets-1 play independent roles in angiogenesis or have overlapping activities. Apart from HIF-1-dependent induction, endothelial Ets-1 expression can be also initiated by other factors, such as epidermal growth factor (EGF), basic fibroblast growth factor (bFGF) and acidic fibroblast growth factor (FGF) [Citation45,Citation46]. This indicates that numerous mechanisms can be involved in hypoxia-induced neovascularization.
Stimulation with lipids
Atherosclerotic lesions contain many types of lipids, including oxidized low density lipoproteins (oxLDL), cholesterol (as a part of LDL, high density lipoprotein (HDL) and triglycerides), 7-ketocholesterol (7KC), soluble phospholipids and eicosanoids [Citation47,Citation48]. Plaque lipid deposits originate from dead foam cells, serum lipoproteins and cell membranes. Circulating LDL is a substantial contributor to lesion lipid content [Citation49]. Along with plaque progression, foam cells are subjected to apoptosis accompanied by liberation of non-esterified cholesterol and formation of a lipid-rich necrotic core [Citation50]. Red blood cell remnants and components of erythrocyte membranes with deposits of glycophorin A, an erythrocyte-specific membrane protein, were also found in unstable lesions, at the sites of intraplaque haemorrhage [Citation51].
Lesional neocapillaries serve as a route for lipid entry into the plaque, especially in the areas where the neovessels permeate to the necrotic core [Citation52]. Medial VSMC activation/proliferation may be caused not only by hypoxia, but also by lipids delivered through the vasa vasorum or accumulated in the necrotic core, that can serve as activators of smooth muscle peroxisome proliferator-activated receptors γ (PPARγ) [Citation48]. Conditioned media taken from early atheromatous plaques contain higher levels of 14-prostaglandin J2, lipid 15-deoxy-δ-12, and their intermediates that act as PPARγ stimulators in neighbouring VSMCs [Citation53]. PPARγ activation leads to the production of VEGF-A by VSMCs and stimulation of neovascularization. Cholesterol can influence angiogenesis via the lipid raft-mediated effects and endothelial VEGFR2-dependent signalling. Down-regulation of lipid raft formation leads to decreased membrane VEGFR2 levels and therefore to suppression of the proangiogenic VEGFR2 signalling [Citation54].
A number of VEGF-independent mechanisms of angiogenesis induction have been described. For example, polyunsaturated fatty acids (PUFA) are sensitive to oxidation, and their oxidized adducts can be sensed by Toll-like receptor 2 (TLR2). TLR2 then transfers signal to myeloid differentiation primary response gene 88 (MyD88) followed by the activation of Rac1 (i.e. Ras-related C3 botulinum toxin substrate 1) and NF-κB signalling. Finally, this leads to cell migration and formation of neovessels [Citation55,Citation56].
In atherogenesis, perivascular adipose tissue (PVAT) also contributes to vascular inflammation. PVAT has direct contacts with the upper adventitia at sites that are not surrounded by the connective tissue and participates in production of signalling mediators, such as cytokines and adipokines, releasing them primarily to the adventitia or through the vasa vasorum vessels that penetrate the PVAT [Citation57,Citation58]. The ability of mature adipocytes to secrete vasoactive substances was shown by Barandier et al., who observed activation of aortic VSMC proliferation under exposure to conditioned medium from mature 3T3-L1 adipocytes, but not from premature or non-differentiated adipocytes [Citation59]. Adipokines, such as leptin and visfatin were shown to activate VSMC proliferation and mobility [Citation60,Citation61]. Conditioned medium from human perivascular adipocytes has a potent angiogenic capacity and supports formation of vessel-like structures by ECs. In late atherogenesis, vasa vasorum neovessels can mediate transfer of inflammatory molecules from PVAT to the vascular wall [Citation62]. Heterogeneity of adipocytes in various tissues and even within PVAT necessitates a more detailed study of adipocytes originating from different locations.
Vascular inflammation
The classical paradigm of the induction of vascular inflammation in atherogenesis involves an “inside-out” hypothesis, which embraces the disease initiation from recruitment of monocytes and oxidation of lipids. However, a growing body of evidence suggests for a new “outside-in” hypothesis, in which vascular inflammation arises from the adventitia and enlarges inwards towards the intima. The hypothesis can be supported by observations about accumulation of the adventitia by exogenous cell types (monocytes, macrophages, lymphocytes, etc.), phenotypical conversion of resident (adventitial) fibroblasts to migratory myofibroblasts, and enhanced formation of the vasa vasorum neovessels [Citation8]. Neovascularization occurs preferentially at the intimal sites of chronic accumulation of inflammatory immune cells. Activated inflammatory cells secrete various cytokines, growth factors and angiogenic factors that stimulate formation of vasa vasorum neovessels [Citation63]. Some studies report a link between atherogenic inflammation and metabolism-related hypoxia, neovascularization, intraplaque haemorrhage and lesion breakage [Citation64]. However, the exact pathways mediating vascular inflammation-initiated lesional vasculogenesis remain to be uncovered.
Stimulators of immature phenotype of intraplaque neovessels
Imbalance between angiopoietin 1 and angiopoietin 2 and VEGF overproduction
Several factors contributing to the fragility and immaturity of newly formed vasa vasorum capillaries in atherosclerotic plaques have been considered. One of them is the enrichment of the lesion site with various proteases, including MMPs, elastase, plasmin, cathepsins, plasminogen activators and coagulation factors [Citation65]. Proteases participate in pathogenic vascular remodelling, plaque rupture and lesional angiogenesis. Intraplaque levels of MMP1, MMP2, MMP3 and MMP9 were found to be significantly elevated [Citation66]. Accordingly, expression of the receptor for urokinase-type plasminogen activator (UPAR) was found to be significantly up-regulated in macrophages and carotid plaques from subjects with carotid stenosis [Citation67].
Compared with non-haemorrhagic plaques, intraplaque levels of angiogenic factors, such as VEGF, angiopoietin-1 and placental growth factor (PIGF) were decreased in carotid lesions with haemorrhage, but nevertheless remained higher than in normal individuals. VEGF-dependent induction of angiogenesis results in formation of immature vessels. Angiopoietin 1 is required for final vascular maturation and stability [Citation68]. Levels of soluble Tie-2, which serves as a receptor for angiopoietin-1 and angiopoietin-2 (a natural antagonist for angiopoietin-1), were greater in haemorrhagic lesions, while angiopoietin-2 concentrations did not significantly differ between non-haemorrhagic and haemorrhagic plaques [Citation69]. Post et al. reported a higher content of angiopoietin-2 than angiopoietin-1 in lesions with higher rates of neovascularization [Citation70]. Therefore, in haemorrhagic lesions, the balance between angiogenic and anti-angiogenic factors, which is crucial for maintaining the integrity and functional maturity of developing vessels, can be seriously disturbed. Poor content and activity of proangiogenic factors in plaque microvessels of haemorrhagic lesions can be explained by the increased activity of proteases, such as elastase and plasmin that inactivate these factors by proteolytic cleavage [Citation71]. Apart from vessel maturation and maintaining of the barrier function, angiopoietin 1 also positively regulates stable interactions between ECs and adjacent VSMCs and pericytes [Citation72]. By contrast, angiopoietin 2 possesses destabilizing effects on vessel architecture and integrity [Citation73].
Increased expression of VEGFR2, which reflects the active VEGF-dependent signalling and intensive neovascularization, was observed in rupture-prone shoulder regions of carotid plaques in diabetic patients [Citation74]. Papalambros et al. showed strong activation of VEGF in human endarterectomized atherosclerotic carotid plaques [Citation75]. Yang et al. reported that VEGF overexpression leads to the formation of immature and leaky microvessels and intracranial haemorrhage in murine neonates, which is associated with increased neonatal mortality [Citation76]. Therefore, increased expression of VEGF in unstable plaques can also contribute to the immature phenotype of intraplaque neovessels [Citation77].
Role of proteases, cholesterol crystals and iron
As mentioned above, weak vascular integrity increases the risk of vessel leakage and haemorrhage. Along with leukocytes, red blood cells can frequently extravasate from leaky intraplaque microvessels to the necrotic core, where they undergo a rapid lysis releasing large amounts of haemoglobin. Through oxidation, extracellular haemoglobin transforms to ferrihaemoglobin, with release of heme that contains a redox reactive iron [Citation78]. The iron participates in various redox reactions including oxidative modification of LDL [Citation79]. Increase in extracellular content of iron and heme induces a compensatory up-regulation of ferritin and HO1 needed to utilize iron and neutralize heme and prevent or minimize iron-mediated vessel damage [Citation80,Citation81]. Atheroprotective intraplaque macrophages with a specific iron/heme-induced phenotype are involved in utilization of haemoglobin–haptoglobin complexes and clearance of erythrocyte remnants by phagocytosis [Citation8].
The necrotic core is enriched by free cholesterol, high concentrations of which can induce formation of cytotoxic cholesterol crystals. Growing cholesterol crystals can protrude cell membranes and lesional neocapillaries causing cell damage, vascular leakage, and expansion of a lipid core [Citation82]. New sites of vascular damage form new haemorrhages accompanied with extravasation of erythrocytes that have membranes especially rich in cholesterol. This cholesterol rapidly crystallizes after the cell lysis forming new crystals [Citation83]. This aggravates intraplaque inflammation, which can influence neovascularization. For instance, Papalambros et al. observed especially active formation of plaque neovessels around cholesterol crystals, which may reflect a stimulatory effect on neovascularization [Citation75]. Cholesterol crystals are also characterized by a high cytotoxicity, damaging vascular cells and cell membranes [Citation84].
Proteases that are activated in the plaque microenvironment, especially mast cell tryptase and chymase, can be involved in neovascularization due to their angiogenic and matrix-degrading activity [Citation85]. Mast cells were shown to accumulate in the shoulder plaque region, a site of intensive neovascularization and preferential area of lesion rupture [Citation86]. Protease-rich intraplaque microenvironment promotes neovascularization and plaque destabilization [Citation87], and also disturbs the balance between angiogenic/antiangiogenic factors by proteolytic degradation. Indeed, this can indirectly influence generation of immature and leaky neovessels. Protease-induced vascular injury due to the proteolytic damage of vessels may exist, but is unlikely to play a central role in induction of haemorrhage and plaque rupture.
Imaging of intraplaque vasa vasorum
The development of advanced imaging systems is necessary in order to monitor plaque progression, detect unstable lesions as early as possible, and prevent plaque disruption and acute atherothrombotic events like MI and stroke. Angiography showed that non-obstructive lesions account for ∼75% acute coronary occlusion cases [Citation88]. Currently, traditional detection and measure of plaque-induced stenosis may be insufficient for prediction of future plaque evolution and rupture. More detailed investigation of plaque regions, including the necrotic core, fibrous cap, presence of neovessels, and inflammation features is required. Visual detection and analysis of neovessels were recently suggested as an early surrogate indicator of the presence of an initial plaque [Citation89]. The safety and non-invasiveness of imaging techniques provide their great clinical value.
Anatomical imaging
In anatomical imaging, main efforts were done not only for plaque detection but also for identification of stable and unstable lesions. The micro-CT was the first visualization approach, which provided evidence for a role of neovascularization in atherogenesis. In the coronary artery of pigs with hypercholesterolemia, implication of this technique allows detecting the zones with intensive and dense vasa vasorum neovascularization [Citation90]. Compared with normal and calcified arterial segments, a two-fold denser neovascularization was observed with help of micro-CT in non-calcified lesions [Citation5]. However, since the volume of subject imaging is limited, micro-CT is mainly used for analysis of autopsy specimens or scan of small animals like mice.
Multi-slice CT is applicable to humans and provides speedy whole-body scanning [Citation91]. Researchers aimed to improve CT resolution for visualization of vasa vasorum. [131I]-labelled nanoparticles were used as a contrast agent to increase imaging resolution [Citation92]. CT angiography is another approach that provides high spatiotemporal resolution to get comprehensive anatomical depiction of affected arteries. CT angiographic examinations of patients with moderate (50–70%) carotid stenosis found plaques from patients with neurological symptoms that had elevated neovascularization. Indeed, this observation suggested that patients with increased neovascularization were at increased risk of stroke [Citation93]. However, the American Heart Association and American College of Cardiology did not recommend CT angiography for general screening of asymptomatic patients due to the risk of radiation exposure [Citation94].
Intravascular ultrasound (IVUS) is a commonly used approach to obtain high-resolution luminal images and perform exact plaque measurements in vivo. Initially, IVUS tools were designed to evaluate blood flow in the lumen of large arteries and are not well adapted to investigate vasa vasorum structure [Citation95]. IVUS enhancement with a contrast agent resulted in the possibility to visualize vasa vasorum with high resolution [Citation96].
Other IVUS modifications such as contrast-harmonic and contrast-subharmonic IVUS are also useful for vasa vasorum imaging [Citation97,Citation98]. The subharmonic imaging was shown to support obtaining images of better quality compared to the harmonic IVUS in terms of the contrast-to-noise and contrast-to-tissue ratio improvement [Citation97]. However, contrast-enhanced IVUS has a serious limitation because this technique cannot provide quantification of neovessels.
Contrast-enhanced ultrasonography (CEU) is a currently developed visualization technique that was able to observe a high-resolution imaging of the carotid vasa vasorum network in a preliminary feasibility study [Citation99]. In a follow-up study of atherosclerotic progression in familial hypercholesterolemia pigs, constitutive signal enhancement reflected an increase in the density of the vasa vasorum network and intima-media thickness [Citation100]. Implication of CEU demonstrated a significant correlation between high dense vasa vasorum and the location of vulnerable plaques [Citation101]. The capacity of CEU to visualize the vasa vasorum network in the adventitia and detect lesional neovascularization makes this imaging approach suitable for longitudinal plaque progression monitoring, early identification of rupture-susceptible lesions, and analysis of efficiency of antiatherogenic therapy. CEU-generated data were shown to correlate well with histologically measured vasa vasorum density quantified by counting the number of stained microvessels and their total cross-sectional area [Citation102].
In CEU, normalized maximal-video intensity enhancement (MVE) (represents vasa vasorum density) positively correlated with lesion volume. As demonstrated on a rabbit model, treatment with atorvastatin led to reduction in MVE enhancement (and therefore, decrease in plaque size) compared with non-treated animals that experienced increase in MVE enhancement [Citation103]. In humans, CEU implication showed feasibility in various clinical applications such as identification of patients at high risk of coronary artery disease (CAD) [Citation104] and prediction of acute coronary events in patients with stable CAD [Citation105,Citation106].
In optical coherence tomography (OCT), the near-infrared light is used to obtain cross-sectional intravascular images [Citation107]. The major advantage of OCT is high resolution (10–20 µm) that is compatible with the micro-CT resolution and 10-fold higher than that of IVUS [Citation108]. The main limitation of the vasa vasorum identification with OCT help is the extensive movement artefacts that arise from arterial pulsations and other physiological motions. These restraints were minimized in a recently developed intensity kurtosis OCT [Citation109]. This technique can distinguish vasa vasorum from the surrounding tissues in the presence of extensive noises and dynamic motions of the arterial wall.
Contrast-enhanced IVUS and other modalities such as CT angiography and contrast-enhanced MRI are useful imaging technologies to evaluate the vasa vasorum development in the human carotid artery in vivo [Citation110–112]. However, OCT is the only visualization technique that provides an option to precisely detect the vasa vasorum architecture in coronary artery plaques [Citation113]. The resolution of IVUS is not high enough to visualize a fine microstructure and longitudinal running of vasa vasorum neovessels in the adventitia. In addition, IVUS can provide proven evidence for vasa vasorum detection in stable but not in unstable plaques/plaque ruptures [Citation112] and has a limited resolution in the proper identification of plaque phenotype [Citation114]. For example, Vavuranakis et al. [Citation115] tried to use contrast-enhanced IVUS for assessment of the vasa vasorum density in the coronary plaques of patients with acute MI but can observe only an enhancement in the grey-scale intensity of the intima-media and adventitia that corresponds to the plexus of vasa vasorum neovessels. In contrast, OCT is able to evaluate the delicate vasa vasorum branching in coronary lesions with a high resolution [Citation116–118].
Optical frequency domain imaging (OFDI) is a new generation OCT that provides the possibility to obtain fast three-dimensional pullback imaging during administration of a non-occlusive dye of an optically clear media. The feasibility of this approach for visualization of vasa vasorum was recently validated in animals (pigs) and humans. OFDI displayed adventitial vasa vasorum with high resolution that was compatible with histological observations and was much better than that of other OCT generations [Citation117,Citation119]. Vasa vasorum count determined with three-dimensional OCT well correlated with vasa vasorum amount detected by micro-CT [Citation120]. OCT imaging was recommended by the International Working Group as a standard reference in clinical practice [Citation121].
With help of the OFDI approach, Taruya et al. [Citation118] reported interesting results about the evolution of the vasa vasorum intraplaque neovascularization at different stages of plaque progression. Analyses of cross-sectional images of human coronary microvasculatures revealed a positive correlation between vasa vasorum volume and lesional volume. Fibrous plaques have more vasa vasorum microvessels than fibrocalcific lesions [Citation118]. This correlation was previously shown in animal atherosclerotic models [Citation122,Citation123]. Compared with the fibrocalcific plaque, the fibrous plaque represents a dynamic phase in lesional progression and hence required more intensive supply with oxygen and nutrients, which can be provided by advanced density of vasa vasorum microcapillary network [Citation124]. Second, Taruya et al. [Citation118] observed association between the volume of vasa vasorum neovessels and plaque rupture. Mechanistically, intraplaque vasa vasorum acts as a gate for entrance of various inflammatory cells to the plaque [Citation125]. These cells can easily extravasate through immature plaque neovessels, infiltrate and then destruct the fibrous cap that leads to plaque rupture [Citation126]. In addition, increased intraplaque neovascularization destabilizes lesion itself [Citation127].
Briefly, coronary imaging studies showed that, in addition to the vasa vasorum first-order vessels running from the coronary artery lumen longitudinally along the adventitial layer, new second-order vasa vasorum vessels with internal longitudinal running from the adventitia or coronary artery branches appear at the stage of fibroatheroma [Citation118]. These vessels actively proliferate to form an extensive branching network [Citation113] and have an aberrant immature leaking morphology, a source for recurrent haemorrhages, which contribute to the necrotic core formation and increase plaque instability [Citation9].
With help of the OFDI technique, the involvement of excessive vasa vasorum formation to the development of coronary artery vasospasm was demonstrated [Citation128,Citation129]. The coronary vasospasm contributes to the pathogenesis of ischemic heart disease. The spasm is induced by the coronary artery overcontraction in a Rho-kinase-dependent manner and serves as a signal for enhanced vasa vasorum formation (to provide additional oxygen and nutritional supply) that plays an important role in further propagation of vascular inflammation [Citation130]. These observations therefore indicate that atherogenic process is conducted not only in the “outside-in” direction, i.e. from the arterial lumen, but also in the “inside-out” course, i.e. from the adventitia, with an active involvement of the enhanced vasa vasorum network.
Molecular imaging
Molecular imaging systems were implemented for vasa vasorum investigations when researchers and clinicians discovered that biological properties of lesions can influence the prognosis of atherosclerosis. Indeed, investigators switched from the anatomical evaluation to mixed anatomical and molecular imaging. Molecular imaging is focused on in vivo measurement and characterization of biological mechanisms at different levels such as molecular, cellular, whole-organ and entire-body levels [Citation131].
To investigate angiogenesis, various radiolabeled agents were used such as [111In]-αvβ3 integrin, [64Cu]-DOTA-VEGF and others were used in single-photon emission computed tomography (SPECT) and nuclear positron emission tomography (PET) [Citation132,Citation133]. However, the resolution of PET/SPECT is not enough for detailed investigation of vasa vasorum neovascularization. Autoradiography is another nuclear approach. Matter et al. labelled antibody specific to the extradomain D of fibronectin with [125I] and injected to apolipoprotein E-deficient mice fed a fat-rich diet [Citation134]. The authors observed stable antibody uptake and incorporation to the atherosclerotic plaques that was significantly higher than the same antibody uptake by control animals. Kuge et al., using a monoclonal antibody [99mTc]-MT1-MMP mAb, detected the highest accumulation of active MMP2 and MMP13 in atheromatous lesions, which indicates the possible involvement of these proteases in matrix remodelling and formation of neovessels [Citation135].
Magnetic resonance imaging (MRI) has advanced properties of high-resolution characterization of a target without radiation use. Developing αvβ3 integrin-targeted gadolinium chelates led to a great burst in MRI applying. With help of paramagnetic gadolinium-based nanoparticles, enhanced angiogenesis and irregular neovessel distribution were found in hypercholesterolemic rabbits fed a lipid-rich diet [Citation136]. Dynamic contrast enhanced (DCE) MRI, a new MRI generation, which was previously used to visualize neovessel formation in tumours, was then applied to observe vasa vasorum neovascularization in atherosclerotic lesions and quantify neovessel volume and density [Citation137]. DCE MRI and traditional MRI were used in multiple clinical applications for analysing plaque composition, measuring lesion size, assessment of efficiency of cardiovascular therapy, monitoring plaque regression and progression, early detection of unstable lesions and plaque rupture [Citation138]. A good correlation was observed between quantitative MRI and histological measurement of a total neovessel area [Citation139] and vasa vasorum extent [Citation110]. The transfer constant (Ktrans) of gadolinium enhancement reflects the transfer of the contrast agent to the extravascular space (i.e. adventitia). Ktrans depends on vasa vasorum density and therefore correlates with neovascularization [Citation140]. Indeed, an evolution of plaque haemorrhage and progress in neovessel formation could be observed with DCE MRI with gadolinium chelates. Indeed, it is possible to access the risk of lesion disruption and thrombus formation. The intraplaque haemorrhage detected by MRI is a strong predictor of acute cardiovascular events (MI, stroke, peripheral vascular disease, coronary stenting and angioplasty), especially in patients with vulnerable lesions [Citation141,Citation142].
Implication of conjugated microbubbles that bind to thrombus-specific ligands or neovessels introduced the term “molecular imaging” in CEU scanning. VEGFR-bound microbubbles were used for a detection of neovascularization. Using dual ET-1/VEGFsp receptor-targeted microbubbles, higher expression of VEGF and ET-1 was found in carotid plaques and vasa vasorum neovessel expansion was observed in rats with carotid atherosclerosis [Citation143]. CEU with microbubbles targeted to P-selectin and vascular cell adhesion molecule 1 (VCAM-1), key molecules in leukocyte adhesion, was used to detect an inflammatory lesion phenotype [Citation144]. Despite a significant progress achieved, prospective clinical studies are required to validate the use of CEU in patients at high risk of repeating large artery strokes. Especially, this concerns the identification of lesional neovascularization, a well-established marker in preclinical and observational studies.
Proangiogenic treatment
The fragility of neovessels is frequently leads to their breakage and haemorrhage. Indeed, the anti-angiogenic treatment should be applied to inhibit intraplaque neovascularization. It is difficult to estimate effects of inhibitors of angiogenesis because these agents exploit specific mechanisms of action that are dependent on cell type and local microenvironment. However, angiogenic inhibitors can be roughly divided into three groups: angiostatics (i.e. agents that directly inhibit angiogenesis), inhibitors that act on angiogenic factors such as VEGF, and others that employ incompletely distinguished pathways.
Angiostatics such as endostatin or angiostatin influence VSMCs and ECs but do not act on native angiogenic factors. Angiostatin is a proteolytic product of plasminogen that reduces the angiogenic capacity of atherosclerotic vessels and lesional macrophage numbers [Citation122]. Activated macrophages release VEGF and attract more inflammatory cells that stimulate angiogenesis [Citation33]. In addition, macrophages transmit proangiogenic effects of oxLDL through induction of the proangiogenic transcriptional factor HIF-1α [Citation145]. Angiostatin interrupts this mechanism and inhibits vasculogenesis in plaques. Endostatin is a proteolytic fragment produced by cleavage of collagen XVIII [Citation146], which affects angiogenesis via multiple mechanisms that to date remain to be clearly characterized. Generally, this molecule inhibits proliferation and trafficking of ECs and VSMCs but does not impair intracellular signalling [Citation147]. In ApoE-deficient mice, administration of endostatin significantly down-regulated the formation of intimal neovessels and decreased plaque progression by 85% [Citation148].
In atherosclerosis-associated angiogenesis, inhibitors of angiogenic factors were frequently used to target the VEGF/VEGFR1 axis. Bevacizumab is a monoclonal antibody against VEGF. Stefanadis et al. evaluated anti-angiogenic properties of bevacizumab-releasing stents implanted to rabbits fed an atherosclerotic diet and showed significant decrease in neovessel density without any effects on reendothelization and inflammation [Citation149]. However, the systemic VEGF inhibition had proatherogenic effects by increasing plaque size by 33%, and stimulating mitochondrial reactive oxygen species production by ECs that uncouples endothelial NO synthase (eNOS) [Citation150]. Similarly, anti-VEGF therapy with bevacizumab of cancer patients (especially those who had atherosclerotic disease) showed serious side effects associated with endothelial dysfunction, development of hypertension, and even thromboembolism [Citation151].
Blocking VEGFR1 with an antibody had no visible anti-atherogenic effects [Citation152]. Genetic deletion of PlGF, which binds to VEGFR1, resulted in suppression of early atherogenesis. By contrast, overexpression of PIGF in the adventitia favoured vasa vasorum extension and intimal thickening [Citation153]. PIGF-specific antibodies were found to suppress angiogenesis only in affected vessels and did not disturb normal vessels, an observation that provides a serious advantage in clinical practice [Citation154].
ET-1 is a vasoactive factor that constricts vessels including vasa vasorum [Citation20]. This factor contributes to the neovascularization as a mitogenic agent that stimulates VSMC growth and may be involved in intimal hyperplasia in atherosclerosis [Citation155]. Chronic inhibition of the ET-1 receptor with antagonists prevented increase in VEGF expression and vasa vasorum density of coronary arteries in hypercholesterolemic pigs [Citation156], suggesting that ET-1 receptor blockade may be of potential interest for anti-angiogenic therapy.
Thalidomide belongs to the pharmaceutical agents that possess immunoregulatory, anti-inflammatory and anti-angiogenic properties. Usually, this drug is used in therapy of some cancers and inflammatory disease. In atherosclerotic swine, thalidomide caused almost two-fold decrease in vasa vasorum density. In ApoE/LDL receptor (LDLR)-deficient mice, the anti-angiogenic action of thalidomide led to the inhibition of vasa vasorum neovascularization and reduced plaque growth. In atherosclerosis, the anti-angiogenic activity of thalidomide was associated with down-regulation of VEGF expression [Citation157]. Overall, thalidomide shows efficient anti-atherogenic and anti-angiogenic activities but there are some reports notifying about side effects of this drug such as venous thromboembolism associated with up-regulation of procoagulant factors and endothelial damage [Citation158,Citation159].
Winter et al. showed efficacy of αvβ3 integrin-targeted paramagnetic nanoparticles loaded with fumagillin (potent anti-angiogenic agent) in inhibiting vasa vasorum neovessels formation in atherosclerotic rabbits [Citation160]. In combination in atorvastatin, the inhibitory effect of fumagillin-containing nanoparticles can be even more prolonged.
The anti-angiogenic properties of fumagillin can be attributed to its suppression of methionine aminopeptidase-2 that stimulates proliferation of many cell types including ECs and VSMCs [Citation161]. rPAI-123, a short isoform of plasminogen activator inhibitor-1 (PAI-1), is an important regulator of vasa vasorum density and regression through activation of plasmin, a protease that degrades extracellular matrix proteins. Without a support from these proteins, ECs can undergo increased apoptosis resulting in vasa vasorum regression [Citation162]. Indeed, rPAI-123-mediated reduction of vasa vasorum neovessels density has anti-atherogenic effect by preventing intraplaque haemorrhage and lesion destabilization [Citation163]. Since rPAI-123 shows a potent atheroprotective activity, it is necessary to stimulate its expression in the plaque or develop rPAI-123 peptides that mimic the angiogenic activity of this factor.
Overall, the anti-angiogenic treatment is beneficial to minimize intraplaque generation of neovessels and reduce plaque instability and rupture. However, as illustrated by the case of bevacizumab, not all anti-angiogenic medications can be safe for this group of patients.
Antiangiogenic treatment
The ability of some anti-angiogenic drugs to induce thrombosis suggests for a more complicated role of formation of neovessels in atherosclerotic plaques. The development of obstruction in atherosclerotic arteries may stimulate increase in vasa vasorum density and area to serve as a side way for blood flow to get round the occluded vascular segment [Citation164]. Due to the high cellular content, VSMC-enriched plaques are stable, while problematic lesions with low cellular content and enlarged lipid-rich necrotic core are sensitive to rupture [Citation165]. In the latter case, vasa vasorum is an adaptive vascular response in order to restore supply with oxygen and nutrients, which becomes malfunctioned due to the atherosclerotic proinflammatory and prooxidant microenvironment. However, increased blood flow through vasa vasorum network could increase lesion cellularity and contribute to plaque stabilization. This may serve as a rationale for using proangiogenic treatment in atherogenesis. At higher doses, statins such as cerivastatin block vasa vasorum neovascularization while they exhibit proangiogenic activity at low doses [Citation166]. However, statins exhibit anti-atherogenic activity, inhibiting the plaque development at low and high doses. This may indicate that proangiogenic effects are not strictly associated with proatherogenic effects, and suppression of vasa vasorum neovessels formation is not indispensable therapeutic approach for plaque stabilization. It may appear more beneficial to support the maturity and normal phenotype of neovessels in order to prevent the development of immature and leaky lesional capillaries.
During the recent years, studies in the field of application of proangiogenic treatment against atherosclerosis were initiated. For example, nerve growth factor (NGF) exhibits angiogenic properties but its levels are reduced in atherosclerotic vessels [Citation167]. Compared with VEGF, NGF supports development of large and mature vessels and increases maturation of VEGF-induced vessels by two-fold [Citation168]. Therefore, restoration and enhancement of NGF expression in atherosclerotic vessels may be a promising goal of future cardiovascular therapy.
Another angiogenic growth factor that maintains vasa vasorum network stability in LDLR/ApoB-100-deficient mice is bFGF. This factor was demonstrated to possess atheroprotective activity, since it improved endothelial function and decreased lesional macrophage accumulation and VCAM-1 expression in hypercholesterolemic rabbits [Citation169,Citation170]. However, increased endothelial bFGF-dependent signalling (i.e. overproduction of FGFR2, a receptor for bFGF) impairs EC function via activation of p21Cip, a negative cell cycle regulator, that may stimulate endothelial senescence and display proatherogenic effects [Citation171]. Indeed, bFGF can act in a different manner depending on the pathogenic mechanism and microenvironment. In treatment of ischemic disease, bFGF displays proangiogenic properties, which can be switched to harmful effects in another pathogenic process (i.e. atherosclerotic disease) [Citation172]. Accordingly, it is important to take into consideration the atherogenic stage in order to choose which (proangiogenic or anti-angiogenic) therapy would be beneficial.
Conclusions
Enhanced development of vasa vasorum neovessels in atherogenesis is a compensatory mechanism aimed to restore the supply of oxygen and nutrients in affected arteries. However, inflammatory conditions presented in the plaque impair neovascularization by inducing imbalance between pro- and anti-angiogenic factors and affecting vascular cell function that leads to the formation of immature and leaky neovessels. The leakage of the vasa vasorum, aggravation of inflammation, and intraplaque necrosis contribute to lesional haemorrhage.
Vasa vasorum evaluation is considered as a useful marker of early identification of rupture-prone lesions. So far, a number of imaging modalities were used to visualize vasa vasorum neovascularization and find vulnerable lesions. Anatomical imaging approaches such as IU and OCT allow obtaining of high-resolution images but cannot provide information about biological processes. Indeed, combination of anatomical and molecular imaging techniques, such as DCE MRI and CEU, represents an optimal strategy to visualize and quantify vasa vasorum along with targeting biomolecules that influence neovascularization. However, certain problems in vasa vasorum visualization, such as toxicity of contrast and enhancing agents and insufficient imaging resolution still persist.
At present, various anti-angiogenic therapies are using or under development in order to block vasa vasorum neovascularization in atherosclerotic plaques. However, some anti-angiogenic agents including bevacizumab, an anti-VEGF monoclonal antibody and thalidomide were shown to exhibit serious side effects such as induction of thromboembolic events. A proper therapeutic strategy should aim for prevention of intraplaque haemorrhage and lesion instability. It is necessary to take into account the atherosclerotic stage and plaque microenvironment to make a choice whether implementation of anti-angiogenic or proangiogenic therapy will provide an advantage. The use of proangiogenic agents is warrant for stimulation of formation of normal, mature and non-leaky vasa vasorum neovessels with concomitant improvement of a local intraplaque microenvironment through inhibition of inflammation, stimulation of anti-apoptosis, or lowering the intraplaque lipid levels.
Disclosure statement
The authors declare no conflict of interest.
Additional information
Funding
Related Research Data
References
- Staub D, Patel MB, Tibrewala A, et al. Vasa vasorum and plaque neovascularization on contrast-enhanced carotid ultrasound imaging correlates with cardiovascular disease and past cardiovascular events. Stroke J Cereb Circ. 2010;41:41–47.
- Ritman EL, Lerman A. The dynamic vasa vasorum. Cardiovasc Res. 2007;75:649–658.
- Barger AC, Beeuwkes R III, Lainey LL, et al. Hypothesis: vasa vasorum and neovascularization of human coronary arteries. A possible role in the pathophysiology of atherosclerosis. N Engl J Med. 1984;310:175–177.
- Kwon HM, Sangiorgi G, Ritman EL, et al. Enhanced coronary vasa vasorum neovascularization in experimental hypercholesterolemia. J Clin Invest. 1998;101:1551–1556.
- Gössl M, Versari D, Hildebrandt HA, et al. Segmental heterogeneity of vasa vasorum neovascularization in human coronary atherosclerosis. JACC Cardiovasc Imaging. 2010;3:32–40.
- Sluimer JC, Kolodgie FD, Bijnens AP, et al. Thin-walled microvessels in human coronary atherosclerotic plaques show incomplete endothelial junctions relevance of compromised structural integrity for intraplaque microvascular leakage. J Am Coll Cardiol. 2009;53:1517–1527.
- Herrmann J, Lerman LO, Rodriguez-Porcel M, et al. Coronary vasa vasorum neovascularization precedes epicardial endothelial dysfunction in experimental hypercholesterolemia. Cardiovasc Res. 2001;51:762–766.
- Maiellaro K, Taylor WR. The role of the adventitia in vascular inflammation. Cardiovasc Res. 2007;75:640–648.
- Chistiakov DA, Orekhov AN, Bobryshev YV. Contribution of neovascularization and intraplaque haemorrhage to atherosclerotic plaque progression and instability. Acta Physiol. 2015;213:539–553.
- Clarke JA. An X-ray microscopic study of the postnatal development of the vasa vasorum of normal human coronary arteries. Acta Anat (Basel). 1966;64:506–516.
- Bitar R, Moody AR, Leung G, et al. In vivo 3D high-spatial-resolution MR imaging of intraplaque hemorrhage. Radiology. 2008;249:259–267.
- Xu J, Lu X, Shi GP. Vasa vasorum in atherosclerosis and clinical significance. Int J Mol Sci. 2015;16:11574–11608.
- Gössl M, Rosol M, Malyar NM, et al. Functional anatomy and hemodynamic characteristics of vasa vasorum in the walls of porcine coronary arteries. Anat Rec. 2003;272:526–537.
- Rademakers T, Douma K, Hackeng TM, et al. Plaque-associated vasa vasorum in aged apolipoprotein E-deficient mice exhibit proatherogenic functional features in vivo. Arterioscler Thromb Vasc Biol. 2013;33:249–256.
- Mulligan-Kehoe M. The vasa vasorum in diseased and nondiseased arteries. Am J Physiol Heart Circ Physiol. 2010;298:H295–H305.
- Dunmore BJ, McCarthy MJ, Naylor AR, et al. Carotid plaque instability and ischemic symptoms are linked to immaturity of microvessels within plaques. J Vasc Surg. 2007;45:155–159.
- Werber AH, Heistad DD. Diffusional support of arteries. Am J Physiol. 1985;248:H901–H906.
- Crawford DW, Back LH, Cole MA. In vivo oxygen transport in the normal rabbit femoral arterial wall. J Clin Invest. 1980;65:1498–1508.
- Bratzler RL, Chisolm GM, Colton CK, et al. The distribution of labeled low-density lipoproteins across the rabbit thoracic aorta in vivo. Atherosclerosis. 1977;28:289–307.
- Scotland RS, Vallance PJ, Ahluwalia A. Endogenous factors involved in regulation of tone of arterial vasa vasorum: implications for conduit vessel physiology. Cardiovasc Res. 2000;46:403–411.
- Ohhira A, Ohhashi T. Effects of aortic pressure and vasoactive agents on the vascular resistance of the vasa vasorum in canine isolated thoracic aorta. J Physiol (Lond). 1992;453:233–245.
- Scotland R, Vallance P, Ahluwalia A. Endothelin alters the reactivity of vasa vasorum: mechanisms and implications for conduit vessel physiology and pathophysiology. Br J Pharmacol. 1999;128:1229–1234.
- Nakashima Y, Chen YX, Kinukawa N, et al. Distributions of diffuse intimal thickening in human arteries: preferential expression in atherosclerosis-prone arteries from an early age. Virchows Arch. 2002;441:279–288.
- Järvilehto M, Tuohimaa P. Vasa vasorum hypoxia: initiation of atherosclerosis. Med Hypotheses. 2009;73:40–41.
- Björnheden T, Bondjers G. Oxygen consumption in aortic tissue from rabbits with diet-induced atherosclerosis. Arteriosclerosis. 1987;7:238–247.
- Sluimer JC, Gasc JM, van Wanroij JL, et al. Hypoxia, hypoxia-inducible transcription factor, and macrophages in human atherosclerotic plaques are correlated with intraplaque angiogenesis. J Am Coll Cardiol. 2008;51:1258–1265.
- Zemplenyi T, Crawford DW, Cole MA. Adaptation to arterial wall hypoxia demonstrated in vivo with oxygen microcathodes. Atherosclerosis. 1989;76:173–179.
- Kwon HM, Sangiorgi G, Ritman EL, et al. Adventitial vasa vasorum in balloon-injured coronary arteries: visualization and quantitation by a microscopic three-dimensional computed tomography technique. J Am Coll Cardiol. 1998;32:2072–2079.
- Gössl M, Versari D, Lerman LO, et al. Low vasa vasorum densities correlate with inflammation and subintimal thickening: potential role in location-determination of atherogenesis. Atherosclerosis. 2009;206:362–368.
- Lim CS, Kiriakidis S, Sandison A, et al. Hypoxia-inducible factor pathway and diseases of the vascular wall. J Vasc Surg. 2013;58:219–230.
- Loboda A, Jozkowicz A, Dulak J. HIF-1 and HIF-2 transcription factors-similar but not identical. Mol Cells. 2010;29:435–442.
- Holmes DI, Zachary I. The vascular endothelial growth factor (VEGF) family: angiogenic factors in health and disease. Genome Biol. 2005;6:209.
- Shibuya M. VEGF-VEGFR system as a target for suppressing inflammation and other diseases. EMIDDT. 2015;15:135–144.
- Kuwahara F, Kai H, Tokuda K, et al. Hypoxia-inducible factor-1alpha/vascular endothelial growth factor pathway for adventitial vasa vasorum formation in hypertensive rat aorta. Hypertension. 2002;39:46–50.
- Joukov V, Pajusola K, Kaipainen A, et al. A novel vascular endothelial growth factor, VEGF-C, is a ligand for the Flt4 (VEGFR-3) and KDR (VEGFR-2) receptor tyrosine kinases. EMBO J. 1996;15:290–298.
- Achen MG, Jeltsch M, Kukk E, et al. Vascular endothelial growth factor D (VEGF-D) is a ligand for the tyrosine kinases VEGF receptor 2 (Flk1) and VEGF receptor 3 (Flt4). Proc Natl Acad Sci USA. 1998;95:548–553.
- Partanen TA, Arola J, Saaristo A, et al. VEGF-C and VEGF-D expression in neuroendocrine cells and their receptor, VEGFR-3, in fenestrated blood vessels in human tissues. FASEB J. 2000;14:2087–2096.
- Rutanen J, Leppänen P, Tuomisto TT, et al. Vascular endothelial growth factor-D expression in human atherosclerotic lesions. Cardiovasc Res. 2003;59:971–979.
- Belgore F, Blann A, Neil D, et al. Localisation of members of the vascular endothelial growth factor (VEGF) family and their receptors in human atherosclerotic arteries. J Clin Pathol. 2004;57:266–272.
- Vuorio T, Nurmi H, Moulton K, et al. Lymphatic vessel insufficiency in hypercholesterolemic mice alters lipoprotein levels and promotes atherogenesis. Arterioscler Thromb Vasc Biol. 2014;34:1162–1170.
- Iwasaka C, Tanaka K, Abe M, et al. Ets-1 regulates angiogenesis by inducing the expression of urokinase-type plasminogen activator and matrix metalloproteinase-1 and the migration of vascular endothelial cells. J Cell Physiol. 1996;169:522–531.
- Oikawa M, Abe M, Kurosawa H, et al. Hypoxia induces transcription factor ETS-1 via the activity of hypoxia-inducible factor-1. Biochem Biophys Res Commun. 2001;289:39–43.
- Tomita N, Morishita R, Taniyama Y, et al. Angiogenic property of hepatocyte growth factor is dependent on upregulation of essential transcription factor for angiogenesis, ets-1. Circulation. 2003;107:1411–1417.
- Higashida T, Kanno H, Nakano M, et al. Expression of hypoxia-inducible angiogenic proteins (hypoxia-inducible factor-1alpha, vascular endothelial growth factor, and E26 transformation-specific-1) and plaque hemorrhage in human carotid atherosclerosis. J Neurosurg. 2008;109:83–91.
- Kitange G, Shibata S, Tokunaga Y, et al. Ets-1 transcription factor-mediated urokinase-type plasminogen activator expression and invasion in glioma cells stimulated by serum and basic fibroblast growth factors. Lab Invest. 1999;79:407–416.
- Paumelle R, Tulasne D, Kherrouche Z, et al. Hepatocyte growth factor/scatter factor activates the ETS1 transcription factor by a RAS-RAF-MEK-ERK signaling pathway. Oncogene. 2002;21:2309–2319.
- Choy PC, Siow YL, Mymin D. Lipids and atherosclerosis. Biochem Cell Biol. 2004;82:212–224.
- Ho-Tin-Noé B, Le Dall J, Gomez D, et al. Early atheroma-derived agonists of peroxisome proliferator-activated receptor-γ trigger intramedial angiogenesis in a smooth muscle cell-dependent manner. Circ Res. 2011;109:1003–1014.
- Sahebkar A, Watts GF. New LDL-cholesterol lowering therapies: pharmacology, clinical trials, and relevance to acute coronary syndromes. Clin Ther. 2013;35:1082–1098.
- Bentzon JF, Otsuka F, Virmani R, et al. Mechanisms of plaque formation and rupture. Circ Res. 2014;114:1852–1866.
- Kolodgie FD, Gold HK, Burke AP, et al. Intraplaque hemorrhage and progression of coronary atheroma. N Engl J Med. 2003;349:2316–2325.
- Moulton KS. Angiogenesis in atherosclerosis: gathering evidence beyond speculation. Curr Opin Lipidol. 2006;17:548–555.
- Ricote M, Li AC, Willson TM, et al. The peroxisome proliferator-activated receptor-gamma is a negative regulator of macrophage activation. Nature. 1998;391:79–82.
- Fang L, Liu C, Miller YI. Zebrafish models of dyslipidemia: relevance to atherosclerosis and angiogenesis. Transl Res. 2014;163:99–108.
- West XZ, Malinin NL, Merkulova AA, et al. Oxidative stress induces angiogenesis by activating TLR2 with novel endogenous ligands. Nature. 2010;467:972–976.
- Salomon RG, Hong L, Hollyfield JG. Discovery of carboxyethylpyrroles (CEPs): critical insights into AMD, autism, cancer, and wound healing from basic research on the chemistry of oxidized phospholipids. Chem Res Toxicol. 2011;24:1803–1816.
- Chatterjee TK, Stoll LL, Denning GM, et al. Proinflammatory phenotype of perivascular adipocytes: influence of high-fat feeding. Circ Res. 2009;104:541–549.
- Britton KA, Fox CS. Perivascular adipose tissue and vascular disease. Clin Lipidol. 2011;6:79–91.
- Barandier C, Montani JP, Yang Z. Mature adipocytes and perivascular adipose tissue stimulate vascular smooth muscle cell proliferation: effects of aging and obesity. Am J Physiol Heart Circ Physiol. 2005;289:H1807–H1813.
- Wang P, Xu TY, Guan YF, et al. Perivascular adipose tissue-derived visfatin is a vascular smooth muscle cell growth factor: role of nicotinamide mononucleotide. Cardiovasc Res. 2009;81:370–380.
- Noblet JN, Goodwill AG, Sassoon DJ, et al. Leptin augments coronary vasoconstriction and smooth muscle proliferation via a Rho-kinase-dependent pathway. Basic Res Cardiol. 2016;111:25.
- Rajsheker S, Manka D, Blomkalns AL, et al. Crosstalk between perivascular adipose tissue and blood vessels. Curr Opin Pharmacol. 2010;10:191–196.
- Yamashita A, Shoji K, Tsuruda T, et al. Medial and adventitial macrophages are associated with expansive atherosclerotic remodeling in rabbit femoral artery. Histol Histopathol. 2008;23:127–136.
- Sluimer JC, Daemen MJ. Novel concepts in atherogenesis: angiogenesis and hypoxia in atherosclerosis. J Pathol. 2009;218:7–29.
- Garcia-Touchard A, Henry TD, Sangiorgi G, et al. Extracellular proteases in atherosclerosis and restenosis. Arterioscler Thromb Vasc Biol. 2005;25:1119–1127.
- Liu XQ, Mao Y, Wang B, et al. Specific matrix metalloproteinases play different roles in intraplaque angiogenesis and plaque instability in rabbits. PLoS One. 2014;9:e107851.
- Svensson PA, Olson FJ, Hägg DA, et al. Urokinase-type plasminogen activator receptor is associated with macrophages and plaque rupture in symptomatic carotid atherosclerosis. Int J Mol Med. 2008;22:459–464.
- Ahmed A, Fujisawa T. Multiple roles of angiopoietins in atherogenesis. Curr Opin Lipidol. 2011;22:380–385.
- Le Dall J, Ho-Tin-Noé B, Louedec L, et al. Immaturity of microvessels in haemorrhagic plaques is associated with proteolytic degradation of angiogenic factors. Cardiovasc Res. 2010;85:184–193.
- Post S, Peeters W, Busser E, et al. Balance between angiopoietin-1 and angiopoietin-2 is in favor of angiopoietin-2 in atherosclerotic plaques with high microvessel density. J Vasc Res. 2008;45:244–250.
- Leclercq A, Houard X, Philippe M, et al. Involvement of intraplaque hemorrhage in atherothrombosis evolution via neutrophil protease enrichment. J Leukoc Biol. 2007;82:1420–1429.
- Nishishita T, Lin PC. Angiopoietin 1, PDGF-B, and TGF-beta gene regulation in endothelial cell and smooth muscle cell interaction. J Cell Biochem. 2004;91:584–593.
- Maisonpierre PC, Suri C, Jones PF, et al. Angiopoietin-2, a natural antagonist for Tie2 that disrupts in vivo angiogenesis. Science. 1997;277:55–60.
- Olson FJ, Strömberg S, Hjelmgren O, et al. Increased vascularization of shoulder regions of carotid atherosclerotic plaques from patients with diabetes. J Vasc Surg. 2011;54:1324–1331.e5.
- Papalambros E, Sigala F, Georgopoulos S, et al. Vascular endothelial growth factor and matrix metalloproteinase 9 expression in human carotid atherosclerotic plaques: relationship with plaque destabilization via neovascularization. Cerebrovasc Dis. 2004;18:160–165.
- Yang D, Baumann JM, Sun YY, et al. Overexpression of vascular endothelial growth factor in the germinal matrix induces neurovascular proteases and intraventricular hemorrhage. Sci Transl Med. 2013;5:193ra90.
- Pelisek J, Well G, Reeps C, et al. Neovascularization and angiogenic factors in advanced human carotid artery stenosis. Circ J. 2012;76:1274–1282.
- Nagy E, Eaton JW, Jeney V, et al. Red cells, hemoglobin, heme, iron, and atherogenesis. Arterioscler Thromb Vasc Biol. 2010;30:1347–1353.
- Potor L, Bányai E, Becs G, et al. Atherogenesis may involve the prooxidant and proinflammatory effects of ferryl hemoglobin. Oxid Med Cell Longev. 2013;2013:676425.
- Juckett MB, Balla J, Balla G, et al. Ferritin protects endothelial cells from oxidized low density lipoprotein in vitro. Am J Pathol. 1995;147:782–789.
- Lee FY, Lee TS, Pan CC, et al. Colocalization of iron and ceroid in human atherosclerotic lesions. Atherosclerosis. 1998;138:281–288.
- Abela GS, Aziz K, Vedre A, et al. Effect of cholesterol crystals on plaques and intima in arteries of patients with acute coronary and cerebrovascular syndromes. Am J Cardiol. 2009;103:959–968.
- Michel JB, Delbosc S, Ho-Tin-Noé B, et al. From intraplaque haemorrhages to plaque vulnerability: biological consequences of intraplaque haemorrhages. J Cardiovasc Med (Hagerstown). 2012;13:628–634.
- Gadeela N, Rubinstein J, Tamhane U, et al. The impact of circulating cholesterol crystals on vasomotor function: implications for no-reflow phenomenon. JACC Cardiovasc Interv. 2011;4:521–529.
- Kaartinen M, Penttilä A, Kovanen PT. Mast cells accompany microvessels in human coronary atheromas: implications for intimal neovascularization and hemorrhage. Atherosclerosis. 1996;123:123–131.
- Kaartinen M, Penttilä A, Kovanen PT. Accumulation of activated mast cells in the shoulder region of human coronary atheroma, the predilection site of atheromatous rupture. Circulation. 1994;90:1669–1678.
- Lijnen HR. Extracellular proteolysis in the development and progression of atherosclerosis. Biochem Soc Trans. 2002;30:163–167.
- Ambrose JA, Tannenbaum MA, Alexopoulos D, et al. Angiographic progression of coronary artery disease and the development of myocardial infarction. J Am Coll Cardiol. 1988;12:56–62.
- Staub D, Schinkel AF, Coll B, et al. Contrast-enhanced ultrasound imaging of the vasa vasorum: from early atherosclerosis to the identification of unstable plaques. JACC Cardiovasc Imaging. 2010;3:761–771.
- Gössl M, Versari D, Mannheim D, et al. Increased spatial vasa vasorum density in the proximal LAD in hypercholesterolemia-implications for vulnerable plaque-development. Atherosclerosis. 2007;192:246–252.
- Moritz R, Eaker DR, Langheinrich AC, et al. Quantification of vasa vasorum density in multi-slice computed tomographic coronary angiograms: role of computed tomographic image voxel size. J Comput Assist Tomogr. 2010;34:273–278.
- Hyafil F, Cornily JC, Feig JE, et al. Noninvasive detection of macrophages using a nanoparticulate contrast agent for computed tomography. Nat Med. 2007;13:636–641.
- Romero JM, Pizzolato R, Atkinson W, et al. Vasa vasorum enhancement on computerized tomographic angiography correlates with symptomatic patients with 50% to 70% carotid artery stenosis. Stroke. 2013;44:3344–3349.
- Sadeghi MM, Glover DK, Lanza GM, et al. Imaging atherosclerosis and vulnerable plaque. J Nucl Med. 2010;51 Suppl 1:51S–65S.
- Li W, van der Steen AF, Lancée CT, et al. Blood flow imaging and volume flow quantitation with intravascular ultrasound. Ultrasound Med Biol. 1998;24:203–214.
- Papaioannou TG, Vavuranakis M, Androulakis A, et al. In-vivo imaging of carotid plaque neoangiogenesis with contrast-enhanced harmonic ultrasound. Int J Cardiol. 2009;134:e110–e112.
- Goertz DE, Frijlink ME, Tempel D, et al. Contrast harmonic intravascular ultrasound: a feasibility study for vasa vasorum imaging. Invest Radiol. 2006;41:631–638.
- Goertz DE, Frijlink ME, Tempel D, et al. Subharmonic contrast intravascular ultrasound for vasa vasorum imaging. Ultrasound Med Biol. 2007;33:1859–1872.
- Magnoni M, Coli S, Marrocco-Trischitta MM, et al. Contrast-enhanced ultrasound imaging of periadventitial vasa vasorum in human carotid arteries. Eur J Echocardiogr. 2009;10:260–264.
- Schinkel AF, Krueger CG, Tellez A, et al. Contrast-enhanced ultrasound for imaging vasa vasorum: comparison with histopathology in a swine model of atherosclerosis. Eur J Echocardiogr. 2010;11:659–664.
- Coli S, Magnoni M, Sangiorgi G, et al. Contrast-enhanced ultrasound imaging of intraplaque neovascularization in carotid arteries: correlation with histology and plaque echogenicity. J Am Coll Cardiol. 2008;52:223–230.
- Moguillansky D, Leng X, Carson A, et al. Quantification of plaque neovascularization using contrast ultrasound: a histologic validation. Eur Heart J. 2011;32:646–653.
- Tian J, Hu S, Sun Y, et al. Vasa vasorum and plaque progression, and responses to atorvastatin in a rabbit model of atherosclerosis: contrast-enhanced ultrasound imaging and intravascular ultrasound study. Heart. 2013;99:48–54.
- Deyama J, Nakamura T, Takishima I, et al. Contrast-enhanced ultrasound imaging of carotid plaque neovascularization is useful for identifying high-risk patients with coronary artery disease. Circ J. 2013;77:1499–1507.
- Zhu Y, Deng YB, Liu YN, et al. Use of carotid plaque neovascularization at contrast-enhanced US to predict coronary events in patients with coronary artery disease. Radiology. 2013;268:54–60.
- Nakamura J, Nakamura T, Deyama J, et al. Assessment of carotid plaque neovascularization using quantitative analysis of contrast-enhanced ultrasound imaging is useful for risk stratification in patients with coronary artery disease. Int J Cardiol. 2015;195:113–119.
- Kubo T, Tanaka A, Ino Y, et al. Assessment of coronary atherosclerosis using optical coherence tomography. JAT. 2014;21:895–903.
- Kume T, Akasaka T, Kawamoto T, et al. Assessment of coronary intima—media thickness by optical coherence tomography: comparison with intravascular ultrasound. Circ J. 2005;69:903–907.
- Cheng KH, Sun C, Vuong B, et al. Endovascular optical coherence tomography intensity kurtosis: visualization of vasa vasorum in porcine carotid artery. Biomed Opt Express. 2012;3:388–399.
- Kerwin WS, Oikawa M, Yuan C, et al. MR imaging of adventitial vasa vasorum in carotid atherosclerosis. Magn Reson Med. 2008;59:507–514.
- Staub D, Patel MB, Tibrewala A, et al. Vasa vasorum and plaque neovascularization on contrast-enhanced carotid ultrasound imaging correlates with cardiovascular disease and past cardiovascular events. Stroke. 2010;41:41–47.
- Vavuranakis M, Sigala F, Vrachatis DA, et al. Quantitative analysis of carotid plaque vasa vasorum by CEUS and correlation with histology after endarterectomy. Vasa. 2013;42:184–195.
- Tanaka K, Sata M. Visualization of the human coronary vasa vasorum in vivo. Circ J. 2015;79:1211–1212.
- Alfonso F, De la Torre Hernández JM. Vasa vasorum and coronary artery disease progression: optical coherence tomography findings. Eur Heart J Cardiovasc Imaging. 2016;17:280–282.
- Vavuranakis M, Kakadiaris IA, O'Malley SM, et al. A new method for assessment of plaque vulnerability based on vasa vasorum imaging, by using contrast-enhanced intravascular ultrasound and differential image analysis. Int J Cardiol. 2008;130:23–29.
- Vorpahl M, Nakano M, Virmani R. Small black holes in optical frequency domain imaging matches intravascular neoangiogenesis formation in histology. Eur Heart J. 2010;31:1889.
- Nishimiya K, Matsumoto Y, Takahashi J, et al. In vivo visualization of adventitial vasa vasorum of the human coronary artery on optical frequency domain imaging. Validation study. Circ J. 2014;78:2516–2518.
- Taruya A, Tanaka A, Nishiguchi T, et al. Vasa vasorum restructuring in human atherosclerotic plaque vulnerability: a clinical optical coherence tomography study. J Am Coll Cardiol. 2015;65:2469–2477.
- Nishimiya K, Matsumoto Y, Uzuka H, et al. Accuracy of optical frequency domain imaging for evaluation of coronary adventitial vasa vasorum formation after stent implantation in pigs and humans – a validation study. Circ J. 2015;79:1323–1331.
- Aoki T, Rodriguez-Porcel M, Matsuo Y, et al. Evaluation of coronary adventitial vasa vasorum using 3D optical coherence tomography-animal and human studies. Atherosclerosis. 2015;239:203–208.
- Tearney GJ, Regar E, Akasaka T, et al. Consensus standards for acquisition, measurement, and reporting of intravascular optical coherence tomography studies: a report from the International Working Group for Intravascular Optical Coherence Tomography Standardization and Validation. J Am Coll Cardiol. 2012;59:1058–1072.
- Moulton KS, Vakili K, Zurakowski D, et al. Inhibition of plaque neovascularization reduces macrophage accumulation and progression of advanced atherosclerosis. Proc Natl Acad Sci USA. 2003;100:4736–4741.
- Langheinrich AC, Michniewicz A, Sedding DG, et al. Correlation of vasa vasorum neovascularization and plaque progression in aortas of apolipoprotein E(−/−)/low-density lipoprotein(−/−) double knockout mice. Arterioscler Thromb Vasc Biol. 2006;26:347–352.
- Heistad DD, Marcus ML, Larsen GE, et al. Role of vasa vasorum in nourishment of the aortic wall. Am J Physiol. 1981;240:H781–H787.
- Wolinsky H, Glagov S. Nature of species differences in the medial distribution of aortic vasa vasorum in mammals. Circ Res. 1967;20:409–421.
- van der Wal AC, Becker AE, van der Loos CM, et al. Site of intimal rupture or erosion of thrombosed coronary atherosclerotic plaques is characterized by an inflammatory process irrespective of the dominant plaque morphology. Circulation. 1994;89:36–44.
- Uemura S, Ishigami K, Soeda T, et al. Thin-cap fibroatheroma and microchannel findings in optical coherence tomography correlate with subsequent progression of coronary atheromatous plaques. Eur Heart J. 2012;33:78–85.
- Nishimiya K, Matsumoto Y, Takahashi J, et al. Enhanced adventitial vasa vasorum formation in patients with vasospastic angina: assessment with OFDI. J Am Coll Cardiol. 2016;67:598–600.
- Nishimiya K, Matsumoto Y, Uzuka H, et al. Focal vasa vasorum formation in patients with focal coronary vasospasm: an optical frequency domain imaging study. Circ J. 2016;80:2252–2254.
- Nishimiya K, Matsumoto Y, Shindo T, et al. Association of adventitial vasa vasorum and inflammation with coronary hyperconstriction after drug-eluting stent implantation in pigs in vivo. Circ J. 2015;79:1787–1798.
- Dobrucki LW, Sinusas AJ. PET and SPECT in cardiovascular molecular imaging. Nat Rev Cardiol. 2010;7:38–47.
- Meoli DF, Sadeghi MM, Krassilnikova S, et al. Noninvasive imaging of myocardial angiogenesis following experimental myocardial infarction. J Clin Invest. 2004;113:1684–1691.
- Rodriguez-Porcel M, Cai W, Gheysens O, et al. Imaging of VEGF receptor in a rat myocardial infarction model using PET. J Nucl Med. 2008;49:667–673.
- Matter CM, Schuler PK, Alessi P, et al. Molecular imaging of atherosclerotic plaques using a human antibody against the extra-domain B of fibronectin. Circ Res. 2004;95:1225–1233.
- Kuge Y, Takai N, Ogawa Y, et al. Imaging with radiolabelled anti-membrane type 1 matrix metalloproteinase (MT1-MMP) antibody: potentials for characterizing atherosclerotic plaques. Eur J Nucl Med Mol Imaging. 2010;37:2093–2104.
- Winter PM, Morawski AM, Caruthers SD, et al. Molecular imaging of angiogenesis in early-stage atherosclerosis with alpha(v)beta3-integrin-targeted nanoparticles. Circulation. 2003;108:2270–2274.
- Calcagno C, Mani V, Ramachandran S, et al. Dynamic contrast enhanced (DCE) magnetic resonance imaging (MRI) of atherosclerotic plaque angiogenesis. Angiogenesis. 2010;13:87–99.
- Corti R. Noninvasive imaging of atherosclerotic vessels by MRI for clinical assessment of the effectiveness of therapy. Pharmacol Ther. 2006;110:57–70.
- Kerwin W, Hooker A, Spilker M, et al. Quantitative magnetic resonance imaging analysis of neovasculature volume in carotid atherosclerotic plaque. Circulation. 2003;107:8516.
- Sun J, Song Y, Chen H, et al. Adventitial perfusion and intraplaque hemorrhage: a dynamic contrast-enhanced MRI study in the carotid artery. Stroke. 2013;44:1031–1036.
- Saam T, Hetterich H, Hoffmann V, et al. Meta-analysis and systematic review of the predictive value of carotid plaque hemorrhage on cerebrovascular events by magnetic resonance imaging. J Am Coll Cardiol. 2013;62:1081–1091.
- Singh N, Moody AR, Rochon-Terry G, et al. Identifying a high risk cardiovascular phenotype by carotid MRI-depicted intraplaque hemorrhage. Int J Cardiovasc Imaging. 2013;29:1477–1483.
- Decano JL, Moran AM, Ruiz-Opazo N, et al. Molecular imaging of vasa vasorum neovascularization via DEspR-targeted contrast-enhanced ultrasound micro-imaging in transgenic atherosclerosis rat model. Mol Imaging Biol. 2011;13:1096–1106.
- Alonso A, Artemis D, Hennerici MG. Molecular imaging of carotid plaque vulnerability. Cerebrovasc Dis. 2015;39:5–12.
- Hutter R, Speidl WS, Valdiviezo C, et al. Macrophages transmit potent proangiogenic effects of oxLDL in vitro and in vivo involving HIF-1α activation: a novel aspect of angiogenesis in atherosclerosis. J Cardiovasc Trans Res. 2013;6:558–569.
- Marneros AG, Olsen BR. Physiological role of collagen XVIII and endostatin. FASEB J. 2005;19:716–728.
- Eriksson K, Magnusson P, Dixelius J, et al. Angiostatin and endostatin inhibit endothelial cell migration in response to FGF and VEGF without interfering with specific intracellular signal transduction pathways. FEBS Lett. 2003;536:19–24.
- Moulton KS, Heller E, Konerding MA, et al. Angiogenesis inhibitors endostatin or TNP-470 reduce intimal neovascularization and plaque growth in apolipoprotein E-deficient mice. Circulation. 1999;99:1726–1732.
- Stefanadis C, Toutouzas K, Stefanadi E, et al. Inhibition of plaque neovascularization and intimal hyperplasia by specific targeting vascular endothelial growth factor with bevacizumab-eluting stent: an experimental study. Atherosclerosis. 2007;195:269–276.
- Winnik S, Lohmann C, Siciliani G, et al. Systemic VEGF inhibition accelerates experimental atherosclerosis and disrupts endothelial homeostasis – implications for cardiovascular safety. Int J Cardiol. 2013;168:2453–2461.
- Mir O, Mouthon L, Alexandre J, et al. Bevacizumab-induced cardiovascular events: a consequence of cholesterol emboli syndrome? J Natl Cancer Inst. 2007;99:85–86.
- Luttun A, Tjwa M, Moons L, et al. Revascularization of ischemic tissues by PlGF treatment, and inhibition of tumor angiogenesis, arthritis and atherosclerosis by anti-Flt1. Nat Med. 2002;8:831–840.
- Khurana R, Moons L, Shafi S, et al. Placental growth factor promotes atherosclerotic intimal thickening and macrophage accumulation. Circulation. 2005;111:2828–2836.
- Fischer C, Jonckx B, Mazzone M, et al. Anti-PlGF inhibits growth of VEGF(R)-inhibitor-resistant tumors without affecting healthy vessels. Cell. 2007;131:463–475.
- Dashwood MR, Barker SG, Muddle JR, et al. [125I]-endothelin-1 binding to vasa vasorum and regions of neovascularization in human and porcine blood vessels: a possible role for endothelin in intimal hyperplasia and atherosclerosis. J Cardiovasc Pharmacol. 1993;22 Suppl 8:S343–S347.
- Herrmann J, Best PJ, Ritman EL, et al. Chronic endothelin receptor antagonism prevents coronary vasa vasorum neovascularization in experimental hypercholesterolemia. J Am Coll Cardiol. 2002;39:1555–1561.
- Kampschulte M, Gunkel I, Stieger P, et al. Thalidomide influences atherogenesis in aortas of ApoE(−/−)/LDLR (−/−) double knockout mice: a nano-CT study. Int J Cardiovasc Imaging. 2014;30:795–802.
- Bennett CL, Angelotta C, Yarnold PR, et al. Thalidomide- and lenalidomide-associated thromboembolism among patients with cancer. JAMA. 2006;296:2558–2560.
- Singh A, Gajra A. Thromboembolism with immunomodulatory agents in the treatment of multiple myeloma. CHAMC. 2011;9:7–13.
- Winter PM, Neubauer AM, Caruthers SD, et al. Endothelial alpha(v)beta3 integrin-targeted fumagillin nanoparticles inhibit angiogenesis in atherosclerosis. Arterioscler Thromb Vasc Biol. 2006;26:2103–2109.
- Kass DJ, Rattigan E, Kahloon R, et al. Early treatment with fumagillin, an inhibitor of methionine aminopeptidase-2, prevents pulmonary hypertension in monocrotaline-injured rats. PLoS One. 2012;7:e35388.
- Mollmark J, Ravi S, Sun B, et al. Antiangiogenic activity of rPAI-1(23) promotes vasa vasorum regression in hypercholesterolemic mice through a plasmin-dependent mechanism. Circ Res. 2011;108:1419–1428.
- Mulligan-Kehoe MJ. Anti-angiogenic activity of rPAI-1(23) and vasa vasorum regression. Trends Cardiovasc Med. 2013;23:114–120.
- Colon GP, Deveikis JP, Dickinson LD. Revascularization of occluded internal carotid arteries by hypertrophied vasa vasorum: report of four cases. Neurosurgery. 1999;45:634–637.
- Shanahan CM, Weissberg PL. Smooth muscle cell phenotypes in atherosclerotic lesions. Curr Opin Lipidol. 1999;10:507–513.
- Ma FX, Han ZC. Statins, nitric oxide and neovascularization. Cardiovasc Drug Rev. 2005;23:281–292.
- Chaldakov GN, Stankulov IS, Fiore M, et al. Nerve growth factor levels and mast cell distribution in human coronary atherosclerosis. Atherosclerosis. 2001;159:57–66.
- Asanome A, Kawabe J, Matsuki M, et al. Nerve growth factor stimulates regeneration of perivascular nerve, and induces the maturation of microvessels around the injured artery. Biochem Biophys Res Commun. 2014;443:150–155.
- Meurice T, Bauters C, Vallet B, et al. bFGF restores endothelium-dependent responses of hypercholesterolemic rabbit thoracic aorta. Am J Physiol. 1997;272:H613–H617.
- Six I, Mouquet F, Corseaux D, et al. Protective effects of basic fibroblast growth factor in early atherosclerosis. Growth Factors. 2004;22:157–167.
- Che J, Okigaki M, Takahashi T, et al. Endothelial FGF receptor signaling accelerates atherosclerosis. Am J Physiol Heart Circ Physiol. 2011;300:H154–H161.
- Williams JK. Endothelial FGF receptor signaling: angiogenic versus atherogenic effects. Am J Physiol Heart Circ Physiol. 2011;300:H27–H28.