
Abstract
Objective: Mechanisms behind sustained inflammation in patients with coronary artery disease (CAD) are not clarified but hypothalamus-pituitary-adrenal (HPA) axis dysfunction may have a role. Here, we investigated whether inflammatory status of peripheral blood mononuclear cells (PBMCs) was associated with altered glucocorticoid sensitivity in CAD patients.
Methods: In 55 CAD patients and 30 controls, mRNA levels of GR-α, GR-β, NF-κB, IκBα, MMP-9 and TIMP-1 were measured in PBMCs. Suppressive effects of dexamethasone on GR-α, GR-β, NF-κB, IκBα, MMP-9 and TIMP-1 mRNA levels were assessed in PBMCs ex vivo. Salivary cortisol was repeatedly measured over 3 days.
Results: GR-α mRNA levels were higher in CAD patients than in controls, 0.50 (0.38–0.59) versus 0.26 (0.18–0.37), p < .001, while GR-β mRNA levels were equally low in both groups. GR-α mRNA expression was associated with inflammatory gene expression and, also, with flatter diurnal cortisol rhythm. In both patients and controls, dexamethasone suppressed gene expression of NF-κB, IκBα, MMP-9 and TIMP-1 (p < .001). Dexamethasone also reduced GR-α mRNA levels (p < .001), while LPS increased it (p < .001).
Conclusions: PBMCs from CAD patients displayed an inflammatory gene expression profile. This was not explained by reduced glucocorticoid sensitivity. Instead, inflammation was associated with increased expression of GR-α mRNA, suggesting a hypocortisolemic state.
• Peripheral blood mononuclear cells from patients with coronary artery disease (CAD) display an inflammatory gene expression profile.
• This inflammatory state cannot be explained by reduced glucocorticoid sensitivity in CAD patients.
• Instead, the inflammatory gene expression profile is associated with upregulated levels of glucocorticoid receptor-α mRNA, suggesting a hypocortisolemic state.
Key messages
Introduction
The presence of inflammation in atherosclerosis is a strong prognostic determinant and also a target of therapy [Citation1,Citation2]. One major feature of this inflammatory process is activation of the nuclear factor κB (NF-κΒ) pathway [Citation3]. As a transcription factor, NF-κΒ plays a pivotal role in the expression of proinflammatory factors, such as matrix-degrading enzyme matrix metalloproteinase (MMP)-9. Although causality has not been fully established, much evidence point towards this enzyme playing an active role in atherosclerosis, from plaque development to plaque destabilization and rupture. It is abundantly expressed in human atherosclerotic lesions, in particular those with unstable morphology [Citation4]. The expression of MMP-9 is also higher in peripheral blood mononuclear cells (PBMCs) from patients with coronary artery disease (CAD) and carotid atherosclerosis, as compared with healthy controls [Citation5–7]. In addition, the main inhibitor of MMP-9, tissue inhibitor of metalloproteinases (TIMP)-1, is upregulated in PBMCs from patients [Citation7]. Simultaneous overexpression of MMP-9 and TIMP-1 in PBMCs has been described in autoimmune diseases [Citation8,Citation9], likely reflecting a state of sustained and/or dysregulated inflammation.
A crucial question is why inflammation is sustained in CAD patients. One potential explanation is hypothalamus–pituitary–adrenal (HPA) axis dysfunction. The HPA axis and its end-product glucocorticoids, predominantly cortisol, are essential in resolving the inflammatory process and maintaining homeostasis [Citation10,Citation11]. This occurs mainly through the binding of glucocorticoids to glucocorticoid receptor (GR)-α in the cytosol. GR-α is known to be the predominant and functionally active receptor while an alternative splice variant of the GR gene, the GR-β isoform, is considered a negative regulator of GR-α-mediated effects [Citation12]. After translocation to the nucleus, the glucocorticoid/GR-α complex exerts its anti-inflammatory effects by DNA binding and interaction with transcription factors, such as NF-κB. A number of in vitro studies have also consistently shown that glucocorticoids suppress the inflammation-induced synthesis and release of MMP-9 and TIMP-1 in various cell types, including human PBMCs [Citation13–15].
Inappropriate cortisol output as well as reduced sensitivity to glucocorticoids has been demonstrated in a variety of chronic inflammatory disorders, including rheumatoid arthritis and asthma [Citation10,Citation11]. Interestingly, there is also increasing evidence for a link between sustained inflammatory activity including stress-induced release of inflammatory mediators, for example, MMP-9, and dysfunctional cortisol response in patients with CAD [Citation16–19]. Moreover, a common GR gene haplotype, related to diminished glucocorticoid sensitivity, has been associated with low-grade inflammation and increased risk of cardiovascular disease in a population-based cohort [Citation20]. In the present study, we investigated whether PBMCs in CAD patients were less sensitive to glucocorticoids compared with those in healthy controls. First, as an indirect measure of glucocorticoid sensitivity, gene expression of GR-α and GR-β was determined in freshly isolated PBMCs from CAD patients and controls and related to NF-κB, NF-κB inhibitor α (ΙκBα), MMP-9 and TIMP-1 expression. The glucocorticoid sensitivity in patients and controls was then functionally assessed by measuring suppressive effects of the synthetic glucocorticoid dexamethasone on NF-κB, IκBα, MMP-9 and TIMP-1 gene expression in PBMCs in an ex vivo model.
Materials and methods
Study population
Fifty-seven CAD patients with a history of ACS (ST- or non-ST elevation myocardial infarction) were consecutively recruited from the Outpatients´ Cardiology Clinic at the University Hospital in Linköping, Sweden. Exclusion criteria were age >75 years, severe heart failure, neoplastic disease, major clinical depression, chronic immunologic disorders or treatment with immunosuppressive/anti-inflammatory agents. Blood sampling was performed in the morning after an overnight fast between 6 and 18 months after the index event when the patients were in a metabolically stable condition. The questionnaire of Center for Epidemiological Studies – Depression (CES-D) was used to assess depressive symptoms, as previously described [Citation7]. In parallel with recruitment of patients, 30 control subjects from the region were randomly invited from the Swedish Population Register. Subjects who accepted the invitation were included as controls if they were anamnestically healthy. Use of statins or antihypertensive drugs for primary prevention was allowed. The study was conducted in accordance with the ethical guidelines of Declaration of Helsinki, and the research protocols were approved by the Ethical Review Board of Linköping University. Written informed consent was obtained from all patients and controls.
Salivary cortisol
Salivary cortisol was collected with Salivette cotton swabs (Sarstedt, Nümbrecht, Germany), placed under the tongue for 2 min, during three consecutive days (not the day after Sunday or holiday, or the day before weekend). The morning sample was taken 30 min after awakening and the evening sample before bedtime. Participants were instructed not to eat, drink or use tobacco for at least 60 min prior to saliva sampling. The Salivettes were immediately frozen at −20 °C by the participants.
Levels of free cortisol in saliva were determined by a commercial radioimmunoassay assay, CORT-CT2 (Cisbio Bioanalyser, Codolet, France) at the accredited Clinical Chemical Laboratory at Karolinska University Hospital, Stockholm, Sweden. According to repeatedly performed quality assessments, the interassay CV was less than 10%. The diurnal cortisol decline (slope) was calculated using mean values of 3 consecutive 30-min post-awakening and bedtime measurements.
Isolation of peripheral blood mononuclear cells and cell culture
PBMCs were isolated by density centrifugation (Ficoll–Paque) 400 g × g for 40 min at room temperature (RT). PBMCs were then washed twice in phosphate-buffered saline (PBS) with 0.5% foetal calf serum (FCS). Freshly isolated cells were immediately snap frozen in liquid nitrogen and stored in −80 °C or resuspended in RPMI 1640 medium supplemented with L-glutamine (Life Technologies, Carlsbad, CA), 10% FCS, 100 U/ml penicillin and 100 µg/ml streptomycin (Life Technologies) to a concentration of 106 cells/ml. The cells were incubated with or without dexamethasone (Sigma-Aldrich, St Louis, MO), 10−7 and 10−8 mol/l, for 19 h at 37 °C in a humidified atmosphere with 5% of CO2. In some experiments, cells were also incubated with 100 ng/mL lipopolysaccharide (LPS) from E. coli (Sigma-Aldrich, St Louis, MO). Cell supernatants and cells were collected and stored at −80 °C.
Measurements in plasma and cell supernatants
MMP-9 and TIMP-1 were analysed in plasma by Luminex® Performance Assay Human MMP-9 and Human TIMP Luminex Performance Assay, respectively, (R&D Systems, Minneapolis MN). IL-6 in plasma and in supernatants from PBMCs incubated with LPS was determined using Quantikine® High sensitivity ELISA kit (R&D Systems).The interassay CVs were 13% for MMP-9, 6.5% for TIMP1 and 7% for IL-6.
Quantitative real-time rt-PCR
The following TaqMan Gene Expression Assay kits (Life Technologies) were used: MMP-9, Hs00957562_m1; TIMP-1, Hs00171558_m1; GR-β, Hs00354508_m1, NF-κB, Hs00765730_m1, GR-α Custom TaqMan Assays, (forward primer 5′ GAAGGAAACTCCAGCCAGAA 3′ and reverse primer 5′ CAGCTAACATCTCGGGGAAT 3′), as previously described [Citation21]. For detection of IκB-α the following primers were used; forward primer 5′ GCTCAGGAGCCCTGTAATGG and reverse primer 5′ TCTGTTGACATCAGCCCCAC. Eukaryotic 18S rRNA (Part number: 4352930E) with an amplicon length of 187 bp served as endogenous control. The amount of expressed genes was calculated relative to the amount of rRNA in each sample. Standards were used to create a standard curve in each run for MMP-9 and TIMP-1 according to the standard curve method in user bulletin no 2 (Life Technologies) and normalized values are presented as arbitrary units. GR-α, GR-β, NF-κB and IκB-α were calculated with the comparative CT method where the amount of target, normalized to an endogenous reference and relative to a calibrator, is given by 2−ΔΔCT according to the user bulletin no 2 (Life Technologies). To detect GR-β, 6 µl cDNA instead of 1µl was used. The CT values for GR-β were still much higher than GR-α, 33.6 (33-34.5) versus 27.3 (26.7–28.2). Each sample was run in duplicates and a maximum deviation of 15% was allowed.
Statistics
SPSS 21 was used for statistical analyses. For clinical and laboratory characteristics, data are presented as median (inter-quartile range). The statistical significance of any difference between two groups was tested by using Mann–Whitney U-test. Chi-square test was used for nominal data. Friedman´s test was used to compare more than two observations repeated on the same subjects and Wilcoxon signed-ranks test was used for pairwise comparisons. Spearman’s rank correlation coefficient was used for correlation analyses. A p value <.05 was considered statistically significant.
Results
Characteristics of study participants
Clinical and biological characteristics of patients and controls are presented in . A number of parameters suggested that freshly isolated PBMCs from patients were in an inflammatory predisposition than PBMCs from healthy controls. As previously reported [Citation7], MMP-9 and TIMP-1 mRNA levels were significantly higher in PBMCs from patients. To further evaluate the inflammatory status, we measured mRNA expression of NF-κB and IκB-α. Quantification of IκB-α mRNA has been shown to be a sensitive method for measuring the activation status of NF-κB [Citation22]. Both NF-κB and IκBα mRNA levels were significantly increased in PBMCs from CAD patients relative to controls (). Moreover, when PBMCs were treated with LPS for 19 h, the secretion of IL-6 appeared to be significantly higher in CAD patients than in controls.
Table 1. Clinical and laboratory characteristics of CAD patients and controls.
As also shown in , the diurnal cortisol slope was significantly flatter in the patient group. Moreover, medication and lipid profile differed between groups while no significant differences were seen in regard to age, sex, body mass index, current smoking, depressive symptoms, leukocyte counts or plasma levels of MMP-9, TIMP-1 and IL-6.
Diabetes may have a major impact on inflammatory status. When the 12 CAD patients with diabetes were excluded from the analyses, the mRNA levels of GR-α, MMP-9, and TIMP-1, NF-κB and IκBα as well as secreted amounts of IL-6 remained significantly higher in patients without diabetes (n = 45) compared with controls (data not shown).
Expression of GR isoforms and its relation to inflammatory status in PBMCs from CAD patients and controls
The levels of GR-α mRNA in freshly isolated PBMCs were significantly increased in patients compared with controls, p < .001 (). The levels of GR-β mRNA were, on the other hand, very low relative to those of GR-α mRNA without any significant differences between patients and controls, 0.06 (0.03–0.11) versus 0.02 (0.00–0.13), p = .076.
Figure 1. Expression of GR-α mRNA levels in freshly isolated PBMCs from 29 controls and 49 CAD patients. Values are expressed as 2−ΔΔ CT-values. Box and Whisker plots show median and interquartile range.
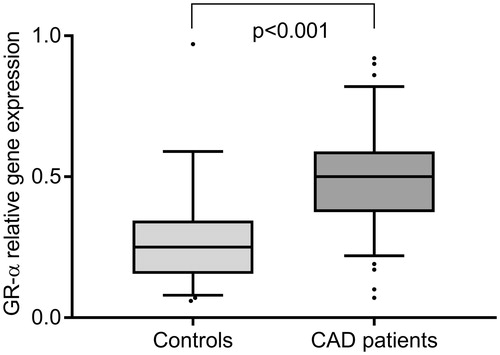
The gene expression of GR-α correlated positively and significantly with the inflammatory status of PBMCs, that is, with gene expression levels of NF-κB, IκBα, TIMP-1 (all p < .001) and MMP-9 (p = .034) when both groups were combined. GR-α mRNA levels also correlated with the diurnal slope of cortisol, r = 0.358, p = .002, and evening cortisol levels, r = 0.268, p = .012, but not with morning cortisol levels. There were no correlations between GR-α mRNA expression and other characteristics, such as age, sex, body mass index or depressive symptoms. GR-α and GR-β mRNA levels were strongly intercorrelated, r = 0.661, p < .000, and GR-β mRNA levels showed similar correlation results as GR-α mRNA regarding inflammatory status of PBMCs and cortisol deviation (data not shown).
Glucocorticoid sensitivity of PBMCs from CAD patients and controls
We compared the glucocorticoid sensitivity in CAD patients and controls by assessing the dexamethasone-mediated suppression of NF-κB, IκBα, MMP-9 and TIMP-1 expression in PBMCs cultured for 19 h. As shown in , NF-κB, MMP-9 and TIMP-1 mRNA levels were significantly suppressed by dexamethasone 10−7 M and 10−8 M in a dose-dependent way, without any differences between patients and controls. IκBα mRNA levels in PBMCs from patients were suppressed by dexamethasone at both doses (10−7 M and 10−8 M), while IκBα mRNA levels in PBMCs from controls were significantly suppressed by the higher dose of dexamethasone. The data indicate that PBMCs from patients were at least as sensitive to glucocorticoids as PBMCs from controls.
Table 2. Expression of NF-κB, IκBα, MMP-9 and TIMP-1 mRNA in PBMCs (from 30 controls and 57 CAD patients) cultured for 19 h in medium only or with dexamethasone (dex) at 10−8M or 10−7 M.
Modulation of GR-α mRNA expression in PBMCs from CAD patients and controls
We then investigated the effect of dexamethasone on the GR-α gene expression in PBMCs cultured for 19 hours. Since GR-β mRNA levels were so low relative to GR-α mRNA levels in freshly isolated PBMCs, we chose to focus on GR-α in these studies. Incubation with dexamethasone at both concentrations (10−7 M and 10−8 M) resulted in significant reductions of GR-α mRNA in PBMCs from both patients and controls, all p < .001 ().
Figure 2. The effect of dexamethasone (Dex) at 10−8 M (29 controls and 54 CAD patients) or 10−7 M (30 controls and 54 CAD patients) or LPS 100 ng/mL (28 controls and 52 CAD patients) on GR-α gene expression in PBMCs, isolated and thereafter cultured for 19 h. Results are given as percentage change of GR-α mRNA levels, calculated as 2−ΔΔ CT, in cells cultured in medium only. Incubation with dexamethasone at both concentrations (10−7 M and 10−8 M) resulted in significant reductions of GR-α mRNA in both patients and controls, all p < .001, whereas incubation with LPS resulted in significantly higher levels of GR-α mRNA in both groups, both p < .001. There were no significant differences in the response to Dex or LPS between patients and controls. Boxes represent median values and bars inter-quartile ranges.
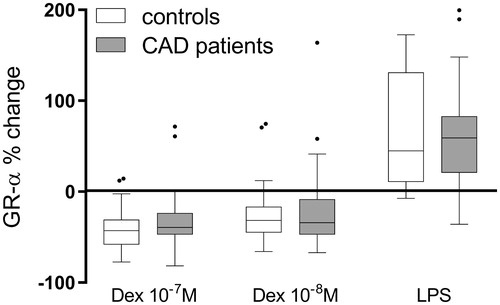
We also investigated the effects of inflammatory stimuli (LPS) on GR-α gene expression in cultured PBMCs from patients and controls. After 19 h of culture, the levels of GR-α mRNA were significantly higher in LPS-treated cells compared with untreated cells, both p < .00, without any significant difference between patients and controls ().
In a subgroup of 10 controls, the levels of NF-κB and IκBα mRNA were measured before and after 19 h of LPS treatment. Both were significantly increased, 58 (36–95) %, p = .003, and 181 (79–223) %, p = .023, respectively.
Discussion
The main findings of the present study were that CAD patients displayed an inflammatory gene expression profile in PBMCs compared with healthy controls. Moreover, when PBMCs from patients were stimulated with LPS, they released IL-6 to a greater extent than PBMCs from controls supporting the presence of a proinflammatory state in the patient group. However, this was not associated with reduced glucocorticoid sensitivity. Instead, somewhat unexpectedly, the inflammatory status in patients was associated with upregulated levels of GR-α mRNA.
The increased gene expression of NF-κB and IκBα in PBMCs is thus indicative of a low-grade chronic inflammatory state in CAD patients and may also explain why MMP-9 and TIMP-1 gene expression is amplified. This gene expression profile can be part of the “residual inflammatory risk”, a term proposed by Ridker [Citation2,Citation23] describing the cardiovascular risk among CAD patients despite extensive medication with, for example, cholesterol-lowering drugs. The mechanisms behind the maintenance of low-grade chronic inflammation in CAD are not clarified but lately, HPA axis dysfunction has emerged as a critical component potentially affecting the ability to resolve inflammation. In the present study, we therefore investigated if the inflammatory status of PBMCs in CAD patients was associated with HPA axis dysfunction, more precisely reduced glucocorticoid sensitivity. Reduced sensitivity to glucocorticoids has been linked to chronic inflammation in other pathological conditions, such as autoimmune diseases and depression [Citation10,Citation11]. The amount of GR-α mRNA in cells can be used as an indirect measure of glucocorticoid sensitivity [Citation24,Citation25] and consequently, downregulation of GR-α mRNA has been described in both autoimmune diseases and depression [Citation25–27]. In contrast, we found that PBMCs from CAD patients had significantly higher levels of GR-α mRNA compared to PBMCs from controls. In addition, the GR-α mRNA levels were positively correlated with the inflammatory gene expression profile in PBMCs suggesting that GR-α mRNA expression was enhanced by inflammation. This idea was supported by our ex vivo data demonstrating that GR-α mRNA expression was upregulated upon LPS treatment. The results are in agreement with a study by Webster et al [Citation28] in which activation of NF-κB was followed by upregulation of GR-α mRNA in human cell lines. A close relationship between GR-α expression and inflammatory status has also been reported in previous clinical studies. In PBMCs from patients with rheumatoid arthritis, the GR and NF-κB subunit p50 levels were increased in those who had never received glucocorticoid treatment while decreased levels were observed in those who received treatment [Citation29]. Moreover, in a randomized intervention study on healthy sedentary men, a concomitant significant reduction in GR-α, NF-κB and IκBα mRNA was seen in PBMCs after a period of physical exercise [Citation30].
Thereafter, we measured the glucocorticoid sensitivity of PBMCs from CAD patients and controls in an ex vivo model. Dexamethasone suppressed the gene expression of NF-κB, IκBα, MMP-9 and TIMP-1 to the same extent in patients and controls, indicating that PBMCs from patients were at least as sensitive to glucocorticoids as PBMCs from controls. Dexamethasone-induced suppression of the inhibitor of NF-κB (IκBα) may seem paradoxical but likely reflects a suppression of the IκB kinase/NF-κB signalling pathway. It is well known that NF-κB activation is followed by a dramatic increase in IκBα mRNA in cell lines as well as in human PBMCs [Citation22,Citation31,Citation32]. IκBα mRNA has also been proposed as a tool to quantify the transcriptional power of NF-κB [Citation22]. In a similar way, inhibitors of proteases, like TIMP-1, are concomitantly upregulated with their corresponding proteases and in line with our results, one previous study has shown that dexamethasone is capable of suppressing both MMP-9 and TIMP-1 in human PBMCs [Citation15].
The mRNA levels of GR-β, as opposed to GR-α, were extremely low in both CAD patients and controls. This is in agreement with previous studies showing that GR-β mRNA levels in PBMCs are up to 2000 times lower than GR-α mRNA levels [Citation33,Citation34]. Upregulated levels of GR-β have been associated with glucocorticoid unresponsiveness in cell line models and glucocorticoid-resistant forms of diseases, such as asthma, rheumatoid arthritis and inflammatory bowel disease [Citation12,Citation35–37]. Still, the role of GR-β mRNA in glucocorticoid sensitivity remains controversial. Similar to our results, several studies have not been able to indicate that the expression of GR-β mRNA is altered in inflammatory disease [Citation27,Citation33,Citation38,Citation39].
Increased expression of GR-α mRNA in PBMC has been associated with hypocortisolism in a clinical population [Citation40]. Conversely, glucocorticoids have been shown to downregulate GR-α mRNA in various human cell lines [Citation41,Citation42]. This is also what we observed when PBMCs were exposed to dexamethasone ex vivo. Hence, an intriguing question is whether enhanced expression of GR-α mRNA in CAD patients reflects a state of subclinical hypocortisolism which, in turn, may be a consequence of prolonged physiological stress. The development of hypocortisolism has been linked to impaired HPA axis function as evidenced by flat diurnal cortisol rhythm and reduced cortisol reactivity to stress [Citation43]. Here, we found that high GR-α mRNA levels were associated with a flat diurnal cortisol slope and elevated evening cortisol. This may be of particular interest in view of cardiovascular risk as flat diurnal slopes and raised evening levels of cortisol have been shown to predict cardiovascular mortality in a community-dwelling population [Citation44]. Previous studies have also reported flatter cortisol slopes, as well as blunted cortisol responses to stress, in patients with manifest CAD, and even more so in those with CAD and depression [Citation17,Citation19,Citation45,Citation46]. In the present study, we did not find any correlation between GR-α mRNA levels and depressive symptoms but the sample may have been too small to evaluate the impact of clinical depression.
Besides the restricted sample size, other limitations of our study need to be acknowledged. First, we measured cortisol in morning and evening saliva samples collected on three consecutive days. This strategy has been shown to strongly correlate with estimates using several samples per day [Citation47]. Still, collecting samples over more days might have improved the reliability of diurnal cortisol estimates. We also lack data on long-term exposure to cortisol. In that respect, other techniques like analysis of hair cortisol might have been useful. Secondly, we did not perform any measurements of GR at the protein level. The reason is the lack of appropriate methods for detecting nuclear translocation of GR. In a previous study [Citation48], we demonstrated that GR-α protein was ubiquitously expressed in blood mononuclear cells without any differences between CAD patients and controls. However, we used whole blood flow cytometry and were not able to discriminate between the inactive cytoplasmic form and the active nuclear form of GR.
Several cardiovascular drugs, including statins and angiotensin II modulators, have been associated with anti-inflammatory effects, assessed by measurements of inflammatory markers in plasma [Citation49]. The extensive use of these drugs may explain why systemic levels of IL-6, MMP-9 or TIMP-1 were not increased in our patient group. However, it should be noted that PBMCs are not a major contributor to MMP-9 and TIMP-1 levels in plasma [Citation50]. Instead, their secretion of MMP-9 and its inhibitor takes place mainly when they have migrated from blood into the tissue.
The assessment of glucocorticoid sensitivity was done in PBMCs and therefore, we can only speculate whether findings apply to other cells or tissues. The ex vivo dexamethasone suppression assay has been shown to be concordant with in vivo glucocorticoid sensitivity [Citation11]. However, glucocorticoid sensitivity can still vary among tissues and cells. Interestingly, a number of experimental studies have reported that glucocorticoids are able to exert direct anti-atherosclerotic effects in the arterial wall [Citation51]. Recently, endogenous glucocorticoids were shown to suppress arterial inflammation and thereby limit progression of atherosclerosis through binding to endothelial GRs in a mouse model [Citation52]. The intriguing possibility that glucocorticoids have a role in stabilizing human atherosclerotic plaques remains to be investigated.
To summarize, there was no evidence that the proinflammatory status of PBMCs in CAD patients was explained by reduced glucocorticoid sensitivity, as assessed by GR gene expression in vivo and functional glucocorticoid sensitivity ex vivo. Instead, the overexpression of GR-α mRNA in CAD patients allowed us to speculate that a state of hypocortisolism can contribute to defective inflammation resolution. Although this theory needs to be confirmed in future studies, it may provide new and important insights into the mechanisms behind “residual inflammatory risk” in CAD patients and thereby open up for novel treatment strategies.
Disclosure statement
The authors report no conflicts of interest.
Additional information
Funding
References
- Libby P, Ridker PM, Hansson GK. Progress and challenges in translating the biology of atherosclerosis. Nature. 2011;473:317–325.
- Ridker PM, Everett BM, Thuren T, et al. Antiinflammatory therapy with canakinumab for atherosclerotic disease. N Engl J Med. 2017;377:1119–1131.
- de Winther MP, Kanters E, Kraal G, et al. Nuclear factor kappaB signaling in atherogenesis. Arterioscler Thromb Vasc Biol. 2005;25:904–914.
- Newby AC. Metalloproteinase expression in monocytes and macrophages and its relationship to atherosclerotic plaque instability. Arterioscler Thromb Vasc Biol. 2008;28:2108–2114.
- Brunner S, Kim JO, Methe H. Relation of matrix metalloproteinase-9/tissue inhibitor of metalloproteinase-1 ratio in peripheral circulating CD14+ monocytes to progression of coronary artery disease. Am J Cardiol. 2010;105:429–434.
- Sternberg Z, Ghanim H, Gillotti KM, et al. Flow cytometry and gene expression profiling of immune cells of the carotid plaque and peripheral blood. Atherosclerosis. 2013;229:338–347.
- Jonsson S, Lundberg AK, Jonasson L. Overexpression of MMP-9 and its inhibitors in blood mononuclear cells after myocardial infarction–is it associated with depressive symptomatology? PLoS One. 2014;9:e105572.
- Bar-Or A, Nuttall RK, Duddy M, et al. Analyses of all matrix metalloproteinase members in leukocytes emphasize monocytes as major inflammatory mediators in multiple sclerosis. Brain. 2003;126:2738–2749.
- Matache C, Stefanescu M, Dragomir C, et al. Matrix metalloproteinase-9 and its natural inhibitor TIMP-1 expressed or secreted by peripheral blood mononuclear cells from patients with systemic lupus erythematosus. J Autoimmun. 2003;20:323–331.
- Silverman MN, Sternberg EM. Glucocorticoid regulation of inflammation and its functional correlates: from HPA axis to glucocorticoid receptor dysfunction. Ann N Y Acad Sci. 2012;1261:55–63.
- Quax RA, Manenschijn L, Koper JW, et al. Glucocorticoid sensitivity in health and disease. Nat Rev Endocrinol. 2013;9:670–686.
- Lewis-Tuffin LJ, Cidlowski JA. The physiology of human glucocorticoid receptor beta (hGRbeta) and glucocorticoid resistance. Ann N Y Acad Sci. 2006;1069:1–9.
- Harkness KA, Adamson P, Sussman JD, Davies-Jones GA, Greenwood J, Woodroofe MN. Dexamethasone regulation of matrix metalloproteinase expression in CNS vascular endothelium. Brain. 2000;123:698–709.
- Russell RE, Culpitt SV, DeMatos C, et al. Release and activity of matrix metalloproteinase-9 and tissue inhibitor of metalloproteinase-1 by alveolar macrophages from patients with chronic obstructive pulmonary disease. Am J Respir Cell Mol Biol. 2002;26:602–609.
- Park CS, Lee YS, Kwon HS, et al. Chlamydophila pneumoniae inhibits corticosteroid-induced suppression of metalloproteinase-9 and tissue inhibitor metalloproteinase-1 secretion by human peripheral blood mononuclear cells. J Med Microbiol. 2012;61:705–711.
- Fantidis P, Perez De Prada T, Fernandez-Ortiz A, et al. Morning cortisol production in coronary heart disease patients. Eur J Clin Invest. 2002;32:304–308.
- Nijm J, Kristenson M, Olsson AG, et al. Impaired cortisol response to acute stressors in patients with coronary disease. Implications for inflammatory activity. J Intern Med. 2007;262:375–384.
- Szymanowski A, Nijm J, Kristenson M, et al. Elevated levels of circulating matrix metalloproteinase-9 are associated with a dysregulated cortisol rhythm-A case-control study of coronary artery disease. Psychoneuroendocrinology. 2011;36:139–143.
- Nijm J, Jonasson L. Inflammation and cortisol response in coronary artery disease. Ann Med. 2009;41:224–233.
- van den Akker EL, Koper JW, van Rossum EF, et al. Glucocorticoid receptor gene and risk of cardiovascular disease. Arch Intern Med. 2008;168:33–39.
- Melo MR, Faria CD, Melo KC, et al. Real-time PCR quantitation of glucocorticoid receptor alpha isoform. BMC Mol Biol. 2004;5:19.
- Bottero V, Imbert V, Frelin C, et al. Monitoring NF-kappa B transactivation potential via real-time PCR quantification of I kappa B-alpha gene expression. Mol Diagn. 2003;7:187–194.
- Ridker PM. Residual inflammatory risk: addressing the obverse side of the atherosclerosis prevention coin. Eur Heart J. 2016;37:1720–1722.
- Pujols L, Xaubet A, Ramirez J, et al. Expression of glucocorticoid receptors alpha and beta in steroid sensitive and steroid insensitive interstitial lung diseases. Thorax. 2004;59:687–693.
- Koga Y, Matsuzaki A, Suminoe A, et al. Differential mRNA expression of glucocorticoid receptor alpha and beta is associated with glucocorticoid sensitivity of acute lymphoblastic leukemia in children. Pediatr Blood Cancer. 2005;45:121–127.
- Carvalho LA, Bergink V, Sumaski L, et al. Inflammatory activation is associated with a reduced glucocorticoid receptor alpha/beta expression ratio in monocytes of inpatients with melancholic major depressive disorder. Transl Psychiatry. 2014;4:e344.
- Hori T, Watanabe K, Miyaoka M, et al. Expression of mRNA for glucocorticoid receptors in peripheral blood mononuclear cells of patients with Crohn’s disease. J Gastroenterol Hepatol. 2002;17:1070–1077.
- Webster JC, Oakley RH, Jewell CM, et al. Proinflammatory cytokines regulate human glucocorticoid receptor gene expression and lead to the accumulation of the dominant negative beta isoform: a mechanism for the generation of glucocorticoid resistance. Proc Natl Acad Sci USA. 2001;98:6865–6870.
- Eggert M, Kluter A, Rusch D, et al. Expression analysis of the glucocorticoid receptor and the nuclear factor-kB subunit p50 in lymphocytes from patients with rheumatoid arthritis. J Rheumatol. 2002;29:2500–2506.
- Silva TS, Longui CA, Faria CD, et al. Impact of prolonged physical training on the pituitary glucocorticoid sensitivity determined by very low dose intravenous dexamethasone suppression test. Horm Metab Res. 2008;40:718–721.
- Brown K, Park S, Kanno T, et al. Mutual regulation of the transcriptional activator NF-kappa B and its inhibitor, I kappa B-alpha. Proc Natl Acad Sci USA. 1993;90:2532–2536.
- Sharif O, Bolshakov VN, Raines S, et al. Transcriptional profiling of the LP0S induced NF-kappaB response in macrophages. BMC Immunol. 2007;8:1.
- Torrego A, Pujols L, Roca-Ferrer J, et al. Glucocorticoid receptor isoforms alpha and beta in in vitro cytokine-induced glucocorticoid insensitivity. Am J Respir Crit Care Med. 2004;170:420–425.
- Gola H, Engler A, Morath J, et al. Reduced peripheral expression of the glucocorticoid receptor alpha isoform in individuals with posttraumatic stress disorder: a cumulative effect of trauma burden. PLoS One. 2014;9:e86333.
- Hamid QA, Wenzel SE, Hauk PJ, et al. Increased glucocorticoid receptor beta in airway cells of glucocorticoid-insensitive asthma. Am J Respir Crit Care Med. 1999;159:1600–1604.
- Orii F, Ashida T, Nomura M, et al. Quantitative analysis for human glucocorticoid receptor alpha/beta mRNA in IBD. Biochem Biophys Res Commun. 2002;296:1286–1294.
- Kozaci DL, Chernajovsky Y, Chikanza IC. The differential expression of corticosteroid receptor isoforms in corticosteroid-resistant and -sensitive patients with rheumatoid arthritis. Rheumatology. (Oxford). 2007;46:579–585.
- Goleva E, Li LB, Eves PT, et al. Increased glucocorticoid receptor beta alters steroid response in glucocorticoid-insensitive asthma. Am J Respir Crit Care Med. 2006;173:607–616.
- Hausmann M, Herfarth H, Scholmerich J, et al. Glucocorticoid receptor isoform expression does not predict steroid treatment response in IBD. Gut. 2007;56:1328–1329.
- Hagendorf A, Koper JW, de Jong FH, et al. Expression of the human glucocorticoid receptor splice variants alpha, beta, and P in peripheral blood mononuclear leukocytes in healthy controls and in patients with hyper- and hypocortisolism. J Clin Endocrinol Metab. 2005;90:6237–6243.
- Rosewicz S, McDonald AR, Maddux BA, et al. Mechanism of glucocorticoid receptor down-regulation by glucocorticoids. J Biol Chem. 1988;263:2581–2584.
- Silva CM, Powell-Oliver FE, Jewell CM, et al. Regulation of the human glucocorticoid receptor by long-term and chronic treatment with glucocorticoid. Steroids. 1994;59:436–442.
- Heim C, Ehlert U, Hellhammer DH. The potential role of hypocortisolism in the pathophysiology of stress-related bodily disorders. Psychoneuroendocrinology. 2000;25:1–35.
- Kumari M, Shipley M, Stafford M, et al. Association of diurnal patterns in salivary cortisol with all-cause and cardiovascular mortality: findings from the Whitehall II study. J Clin Endocrinol Metab. 2011;96:1478–1485.
- Bhattacharyya MR, Molloy GJ, Steptoe A. Depression is associated with flatter cortisol rhythms in patients with coronary artery disease. J Psychosom Res. 2008;65:107–113.
- Waller C, Bauersachs J, Hoppmann U, et al. Blunted Cortisol Stress Response and Depression-Induced Hypocortisolism Is Related to Inflammation in Patients With CAD. J Am Coll Cardiol. 2016;67: 1124–1126.
- Segerstrom SC, Boggero IA, Smith GT, et al. Variability and reliability of diurnal cortisol in younger and older adults: implications for design decisions. Psychoneuroendocrinology. 2014;49:299–309.
- Bergstrom I, Lundberg AK, Jonsson S, et al. Annexin A1 in blood mononuclear cells from patients with coronary artery disease: Its association with inflammatory status and glucocorticoid sensitivity. PLoS One. 2017;12:e0174177.
- Takeda T, Hoshida S, Nishino M, et al. Relationship between effects of statins, aspirin and angiotensin II modulators on high-sensitive C-reactive protein levels. Atherosclerosis. 2003;169:155–158.
- Jonsson S, Lundberg A, Kalvegren H, et al. Increased levels of leukocyte-derived MMP-9 in patients with stable angina pectoris. PLoS One. 2011;6:e19340.
- Hadoke PW, Iqbal J, Walker BR. Therapeutic manipulation of glucocorticoid metabolism in cardiovascular disease. Br J Pharmacol. 2009;156:689–712.
- Goodwin JE, Zhang X, Rotllan N, et al. Endothelial glucocorticoid receptor suppresses atherogenesis–brief report. Arterioscler Thromb Vasc Biol. 2015;35:779–782.