
Abstract
Bipolar Disorder (BD) is a major psychiatric illness affecting up to 5% of the population. BD can progress over time to a chronic “neuroprogressive” course with cognitive and functional impairment. Currently, there are no validated predictors indicating which patients will develop a neuroprogressive course and there are no specific treatments.
This review presents data supporting a novel hypothesis on the mechanisms underlying bipolar neuroprogression. Insulin resistance (IR) is present in 52% of BD patients and is associated with chronic course, treatment nonresponse, adverse brain changes and cognitive impairment. Further, bipolar morbidity increases 12-fold following the onset of IR indicating that IR may modify disease progression. I review evidence that IR is a testable and treatable modifying factor in neuroprogression and that reversing IR may be an efficient (and perhaps the only) means of obtaining remission in some patients. I draw a parallel with Helicobacter pylori in peptic ulcer disease (a novel mechanism that brought together two previously unrelated phenomena that uncovered a new treatment approach).
This model of bipolar progression combines shared dysregulated mechanisms between IR and BD, allowing for early screening, case finding, and monitoring for neuroprogression, with the potential for intervention that could prevent advanced bipolar illness.
Neuroprogression in bipolar disorder is defined by a more severe form of illness and poor outcome. Currently, there are no validated predictors of neuroprogression, which could help inform treatment and improve prognosis.
Insulin resistance is present in more than half of all bipolar patients and is associated with a chronic course of illness, lack of response to mood stabilizing treatment, cognitive impairment and poor functional outcomes.
Insulin resistance may modify the course of bipolar disorder and promote neuroprogression. Insulin resistance may be a testable and potentially modifiable risk factor for neuroprogression in bipolar disorder.
KEY MESSAGES
Introduction
Clinical staging of bipolar disorder (BD) is a controversial and contentious area in psychiatry. As noted by Malhi et al., “accurate and reproducible clinical staging is only possible when an illness is known to progress along a specific and reasonably predictable course” [Citation1]. The difficulty with BD is that illness trajectories can be varied for a number of underlying reasons. Hence, staging on the basis of phenomenology alone is extremely difficult. Further, clinical staging needs to include objective testing of underlying pathophysiologic processes to help identify where a patient lies along a continuum of illness. Staging should indicate when risk factors are present, as well as which interventions may prevent or delay progression to advanced stages of illness or revert a patient to an earlier, less debilitating stage. Moreover, for a staging model to be helpful, effective, stage-specific treatments need to be available.
Staging models in any disease imply an understanding of the underlying pathophysiology. But what if a disease is heterogeneous, with varying presentations of the illness or more than one pathophysiologic process leading to the same symptoms? There are a few universal concepts that apply to medicine. First is the concept that one disease can cause symptoms in a variety of seemingly unrelated systems. An example of this is systemic lupus erythematosus (SLE). This is an autoimmune disorder that may present as myocarditis in one patient, glomerulonephritis in a second patient and optic neuritis in a third. The second is the concept that an observed illness can be caused by a number of etiologies. An example of this is peptic ulcer disease (PUD). It was once thought that PUD was the result of stress leading to hypersecretion of gastric acid. Anti-inflammatory induced gastritis was another etiology. Later, Barry Marshall discovered that PUD was the result of a bacterium called Helicobacter pylori – an infectious disease [Citation2]. This resulted in a paradigm shift in the understanding and treatment of PUD [Citation3]. Now all patients with PUD are tested for H. pylori infection, because if present, PUD will not remit unless the infection is treated.
In the same way that H. pylori turned out to be the etiologic process for many, but not all patients with PUD, this proposed model of progression of BD based on comorbid metabolic dysregulation (), likely does not describe all patients with BD. Insulin resistance (IR), however, is present in more than half of all bipolar patients [Citation4–7], is associated with poor outcome [Citation5,Citation6,Citation8] and may need to be treated if these patients are to remit – similar to H. pylori infection in PUD.
Figure 1. Proposed progression of bipolar disorder and metabolic dysregulation. BBB: blood-brain barrier; BDNF: brain-derived neurotrophic factor; CNS: central nervous system; CRP: C-reactive protein; CVA: cerebrovascular accident; CVD: cardiovascular disease; HOMA-IR: Homeostatic Model Assessment – Insulin Resistance; HPA: hypothalamic pituitary adrenal; IL: interleukin; IR: insulin resistance; SNS: sympathetic nervous system; TNF: tumor necrosis factor; T2DM: type 2 diabetes mellitus
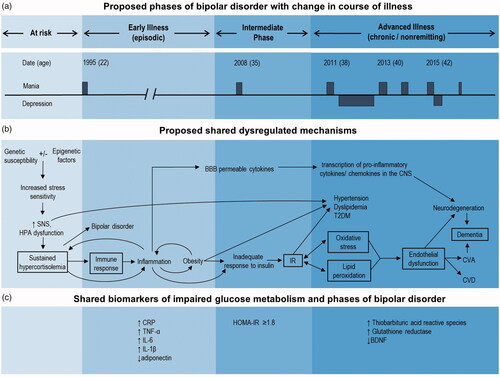
Evidence from studies informs the hypothesis that comorbid metabolic dysregulation may underlie the progression of BD
Clinical correlates of impaired glucose metabolism in BD
The association between metabolic dysregulation and course of bipolar illness
Earlier studies found that patients with BD and type 2 diabetes mellitus (T2DM) have a more chronic course, increased number of episodes (rapid cycling), increased number of psychiatric admissions to hospital, and increased mortality (30% shortened life expectancy) [Citation8–10]. Patients with BD and T2DM also have poorer quality of life and overall functioning and experience greater disability compared to those without T2DM [Citation8]. Additionally, hypertension is more prevalent among these patients and body mass index (BMI) is increased on average [Citation8]. Obesity is a risk factor for T2DM. When we studied obesity in relation to BD, we found that patients with elevated BMI also have a more chronic course, with longer duration of illness, more anxiety, poorer functioning, greater disability and higher rates of T2DM and hypertension [Citation11].
The high rates of obesity and T2DM in BD and their effect on outcome led us to study the association between IR (the earliest precursor to T2DM) and BD. In our cross-sectional study of 121 patients with BD, those with comorbid T2DM or IR had three times higher odds of a chronic course of BD compared to glucose tolerant (euglycemic) bipolar patients and three times higher odds of rapid cycling (more than 4 discrete mood episodes per year). All associations remained significant after controlling for age, sex, current and lifetime antipsychotic exposure and BMI in sensitivity analyses [Citation5,Citation12]. Similar results have recently been reported by Steardo et al. [Citation6], who found that patients with BD and comorbid IR/T2DM had a greater lifetime number of mood episodes than euglycemic patients. Lastly, in a prospective case series (n = 6) we found that psychiatric morbidity increases 12-fold following the onset of IR in bipolar patients [Citation13].
The association between metabolic dysregulation and mood stabilizing treatment response
The deleterious effects of many psychotropic medications on glucose metabolism are well-established [Citation14]. However, evidence indicates that prophylactic treatment with mood stabilizing medications may not be as effective for patients with BD and comorbid metabolic dysregulation as for euglycemic patients with BD [Citation5,Citation6]. We have studied response to lithium, the common maintenance treatment for BD, in relation to obesity, as measured by BMI. In a cross-sectional study of 276 patients with BD, those with healthier BMI (mean BMI 26.5) achieved complete remission on lithium, while those with BMI in the overweight range (mean BMI 30.0) had a partial response and those with BMI in the obese range (mean BMI 32.4) failed to respond to lithium [Citation11]. Further, Kemp et al. found that for every unit increase in BMI, the likelihood of response to any bipolar treatment decreased by 7.5% and the likelihood of remission decreased by 7.3% [Citation15].
We are the first group to study IR and T2DM and response to lithium. In our sample, euglycemic bipolar patients and those with IR/T2DM did not differ in the class of psychotropic medication they were taking, indicating that those with IR/T2DM were not more likely to be taking medications with the greatest impact on glucose metabolism [Citation14]. We found that bipolar patients with T2DM or IR were 8.4 times more likely to be refractory to lithium treatment compared to euglycemic patients. Furthermore, we found that the greater the IR (as measured by the Homeostatic Model Assessment – IR [HOMA-IR] equation [Citation16]), the less likely patients were to respond to lithium [Citation5]. Notably, bipolar patients with IR had equally poor psychiatric outcomes as those with T2DM. Steardo et al. [Citation6] have extended our findings, reporting that patients with IR/T2DM were 4.3 times more likely to have a poor response to any mood stabilizer.
The association between metabolic dysregulation, brain changes and cognitive functioning
Insulin resistance/T2DM may also be implicated in some of the brain changes seen in patients with BD. Our spectroscopic and structural magnetic resonance neuroimaging studies are the first to show that some of the neurochemical and morphological changes found in BD may actually be associated with impaired glucose metabolism. Patients with BD and IR or T2DM showed lower prefrontal N-acetyl aspartate (NAA) levels compared to euglycemic patients with BD, who had comparable NAA levels to euglycemic, non-psychiatric controls [Citation17]. The NAA levels were positively associated with total creatine (an energy metabolite), suggesting that the NAA changes are related to impaired energy metabolism, a hallmark of IR and T2DM. Patients with BD and IR or T2DM also had significantly smaller hippocampal and regional cortical volumes than euglycemic patients with BD or euglycemic, non-psychiatric controls [Citation18]. These results align with results of research in the general T2DM population demonstrating both metabolic and structural brain abnormalities [Citation19–21].
It remains unclear whether these brain abnormalities found in patients with BD and comorbid IR/T2DM adversely impact cognitive functioning. Few studies have been published that focus on the relationship between glucose levels and cognitive abilities in patients with BD. Of these studies, only one reported significantly poorer global cognitive functioning in bipolar patients with comorbid T2DM [Citation22]. Two other studies have not: Hubenak et al. did not find insulin or glucose levels to be related to lower global cognitive functioning [Citation23], and Depp et al. [Citation24] found that the use of T2DM medications was not significantly associated with global cognitive functioning. However, the study by Depp et al. did not assess patients’ glucose levels, meaning that patients classified as euglycemic may not truly have been [Citation5]; thus, this study may not be entirely representative of the cognitive abilities of bipolar patients with comorbid T2DM. Contrary to the results of the latter two studies, research in the general population supports a link between IR/T2DM and cognitive impairment [Citation25–28]. Therefore, impaired glucose metabolism may be contributing to the adverse brain changes and cognitive deficits in patients with BD.
In summary, evidence shows that comorbid IR and T2DM are associated with a chronic course, rapid cycling, poor response to lithium, possible neurocognitive deficits and adverse structural and chemical brain changes in bipolar patients.
I propose that IR may be a moderating factor affecting the course of bipolar illness, and it may be critical to intervene at the insulin resistant state, when reversing IR may still have some influence over the possible progression of bipolar illness.
Evidence for shared dysregulation in the development of insulin resistance and BD
An important indication that metabolic dysregulation may be a key underlying process in the progression of BD is the finding that patients with BD are at much greater risk of having metabolic dysfunction compared to the general population, independent of medication [Citation29–32]. Abnormalities in glucose metabolism may be seen as early as the first episode [Citation33], and the rate of T2DM is higher in children and adolescents with BD (5.8%) than in the pediatric/adolescent general population (1.9%) [Citation34]. In adults with BD, rates of T2DM are substantially higher than in children and adolescents. In a cross-sectional study, when systematically tested, we found that 32% of bipolar patients had IR and 22% had T2DM; rates that are 2–3 times that of the general population [Citation5]. Similar proportions have been found in studies by other research groups [Citation6,Citation7]. Therefore, more than half of all bipolar patients have dysregulated glucose metabolism (either IR or T2DM), suggesting that there is a possible connection between the two disorders [Citation35] ().
There are a number of proposed shared dysregulated mechanisms that underlie IR, T2DM and BD (see Calkin et al., for review [Citation35]), and these form the basis for this theoretical model of disease progression in a subset of bipolar patients. Common genetic abnormalities and shared susceptibility loci between BD and T2DM suggest shared mood and metabolic disease pathways [Citation36,Citation37]. When genetic susceptibility is combined with environmental factors affecting gene expression (epigenetics), hypothalamic-pituitary-adrenal (HPA) axis dysfunction can result, with sustained hypercortisolemia and activation of the sympathetic nervous system (SNS). (Hypercortisolemia is common in patients with BD, even when euthymic [Citation38]). Hypercortisolemia activates an immune response and an inflammatory cascade as an adaptive response to cellular damage. This leads to obesity (common in BD [Citation11,Citation39–41]) and an inadequate increase in insulin in response to plasma glucose, with progression from IR to glucose intolerance and eventually T2DM (present in more than half of bipolar patients [Citation5]). Insulin resistance has a bidirectional association with oxidative stress and lipid peroxidation [Citation42–44], and contributes to endothelial dysfunction [Citation45] (oxidative stress, lipid peroxidation and endothelial dysfunction are also found in bipolar patients [Citation46–51]). Endothelial dysfunction, resulting in microvascular and macrovascular damage, potentially leads to end-organ damage, including the heart and brain. This results in cardiovascular disease, cerebrovascular disease and neurodegeneration, and ultimately, dementia. Cardiovascular disease is the leading cause of death in BD [Citation52] and BD has also been associated with higher rates of dementia [Citation53].
Insulin resistance may modify the course of BD and promote disease progression. Insulin has been associated with neuroplasticity in critical regions of the brain, in particular, the hippocampus, which has a high concentration of insulin receptors [Citation54]. Insulin resistance results in chronic peripheral hyperinsulinemia and a relative decrease in central nervous system (CNS) insulin levels, as a result of compensatory down-regulation of the saturable insulin-receptor-mediated process by which insulin crosses the blood-brain barrier (BBB) [Citation55]. Further upstream in the cascade, another mechanism by which IR contributes to neurodegeneration is through the production of cytokines that can cross the BBB and activate pathways leading to transcription of pro-inflammatory cytokines and chemokines in the CNS [Citation56]. Therefore, neurodegeneration associated with advanced BD [Citation57] may be, at least in part, the result of progression along this metabolic cascade from comorbid IR to end-organ (brain) damage. This is in keeping with the impaired glucose metabolism and insulin dysregulation found in many other neurodegenerative disorders including Parkinson’s, Huntington’s and Alzheimer’s diseases [Citation26,Citation56,Citation58]. Theoretically, targeting insulin resistance in some brain disorders, including BD, may yield a new approach to treatment-refractory illness, via a more direct mechanism.
Biomarkers associated with each phase of illness: further evidence for the role of IR/T2DM in neuroprogression
Each step along the proposed physiological cascade leading to impaired glucose metabolism and BD presents measurable markers that provide clues to the underlying process by which IR/T2DM contributes to neuroprogression (see . Obesity is a chronic, proinflammatory state, with adipose tissue secreting cytokines and inflammatory mediators (including adipokines) [Citation59]. Here, we see a decrease in HDL cholesterol, as well as increases in CRP, triglycerides, the atherogenic index of plasma (markers for the development of T2DM and cardiovascular risk [Citation60–62]) and IL-1β, which induces proinflammatory cytokines (such as IL-6 and TNF-α) [Citation59]. These inflammatory markers are also found to be elevated in patients with BD [Citation63–66], but not in high-risk adolescents (offspring of a parent with BD) [Citation50] (although variations in certain immune growth factors may be present in high-risk adolescents [Citation67]). Adiponectin, an insulin sensitizer and anti-inflammatory cytokine that counteracts the pro-inflammatory effect of TNF-α, is decreased in obesity and IR/T2DM [Citation59], as it is in patients with a more severe course of BD [Citation68].
Obesity leads to IR in many patients, while other patients who are not obese may develop IR through other mechanisms [Citation42]. Either way, IR contributes to oxidative stress and lipid peroxidation [Citation44,Citation69]; processes reflected by elevated levels of thiobarbituric acid reactive species (TBARS) [Citation44,Citation69,Citation70] and glutathione reductase [Citation69,Citation71]. These markers are also found to be elevated in patients in the later stages of BD [Citation46,Citation72], but levels are normal in patients in the early stages [Citation46,Citation73,Citation74]. Overall, there are lower levels of anti-inflammatory and higher levels of pro-inflammatory cytokines, and the appearance of markers of oxidative stress, as one progresses from the early to the advanced phase of bipolar illness [Citation46,Citation72,Citation75]. As these late-phase markers are consistent with biomarkers found in T2DM [Citation44,Citation69,Citation71], I hypothesize that these markers are primarily elevated in bipolar patients with comorbid IR/T2DM or other inflammatory etiologies; however, this has not yet been studied.
An additional biomarker that has been identified with the different phases of bipolar illness in this proposed model is brain-derived neurotrophic factor (BDNF). Conflicting results have been found in studies of high-risk individuals [Citation67,Citation76], but in the early illness phase, BDNF levels appear to normalize, then decrease in the advanced phase [Citation75,Citation77]. Levels of BDNF are also decreased in individuals with T2DM who do not have a psychiatric illness [Citation78].
These biomarkers reflect the pathophysiology underlying the development of obesity, hypertension, dyslipidemia, T2DM, cerebrovascular and cardiovascular diseases, and likely BD. This may explain (at least in part) why we see BD so commonly comorbid with these medical illnesses: they may develop via shared dysregulated processes, similar to the multisystem effects of SLE.
Directly affecting the brain, IR may be one of the processes underlying the progression of BD to the advanced illness phase. There is overlap in the biomarkers that have been associated with early and late phases of BD and the biomarkers associated with the physiologic progression of the underlying multi-disease process, and this contributes to the plausibility of this proposed model.
Proposed model of progression of bipolar disorder based on metabolic dysregulation
Given this evidence, I propose a hypothetical model that divides the continuum of BD into three main phases (see ) based on the physiological cascade underlying IR; a process likely shared with BD [Citation35] (see . A summary of evidence for the model is provided in . Following an “at-risk” period, there is an early, episodic phase, an intermediate phase and an advanced, (chronic, accelerated) non-remitting phase which may also include cardiovascular disease, and/or dementia ().
Table 1. Evidence for a model of metabolic dysregulation-mediated progression of bipolar disorder and need for future research.
The “at-risk” period occurs for those with genetic and epigenetic risk for BD, including a family history of BD, major depressive disorder (MDD) and/or psychosis. These individuals are asymptomatic and may have a history of early life stress (an example of an epigenetic mechanism increasing risk for psychiatric and medical disorders [Citation79,Citation80]).
The early phase of bipolar illness is defined by an episodic course of illness, before the development of clinically significant metabolic dysregulation. This may begin with the index mood episode if it is manic (required for the diagnosis of BD).
The intermediate phase is marked by the development of IR (which increases allostatic load, possibly contributing to the progression of BD to the more advanced phase). Patients at this phase may still have an episodic course, or they may have already gone on to develop a more chronic course. It is not known how long a patient may be at this phase, as IR is not a static state (at least not initially). Patients may fluctuate between intermediate and early phases of BD with changes in weight, stress/cortisol levels and other possible factors as the state between euglycemia and IR fluctuates. Insulin resistance has direct effects on the brain; however, it may have to become persistent for a period of time before it affects course of illness, possibly converting the course from an episodic to a chronic/advanced one over time, with lack of response to treatment. This might be quite variable between patients and is yet to be determined.
The advanced phase of bipolar illness is defined by a chronic/non-remitting/accelerated course or rapid cycling with the development of IR. Many patients in this phase may have gone on to develop glucose intolerance or frank T2DM. They may also show evidence of cognitive impairment and/or other brain changes, including possible disruption of the BBB [Citation81,Citation82]. Patients who have entered the advanced phase – in whom metabolic dysregulation may be modifying their bipolar illness – may no longer respond to psychotropic treatments.
The importance of recognizing when patients have entered the intermediate phase is that IR is reversible (at least initially). There may be a window of opportunity to intervene and prevent further progression of BD to an advanced phase, or to revert a patient back to a more benign episodic phase. Delaying the onset of T2DM in this way would also decrease risk for cardiovascular disease, the most frequent cause of death in bipolar patients [Citation52]. We have recommended testing bipolar patients for IR (using fasting glucose and insulin levels and the HOMA-IR equation) to determine which patients may go on to an advanced phase [Citation83].
Anecdotal evidence from clinical practice suggests that treating IR, the earliest stage of metabolic dysregulation in BD, may be a novel strategy for altering unfavourable course and poor outcome. Empirical evidence to date from pioglitazone trials in BD and MDD suggests that this medication may ameliorate depressive symptoms [Citation84–86], although negative results have also been reported [Citation87]; these negative results may be influenced by patients’ degree of insulin resistance at study entry [Citation88]. To obtain further evidence that treating IR may improve outcomes in BD, we are currently conducting the TRIO-BD Study, a double-blinded randomized control trial of the efficacy of metformin in non-remitting bipolar patients with IR (NCT 02519543).
Since this model is based on only one of several possible mechanisms underlying BD, it is possible to have patients with advanced, chronic, non-remitting symptoms who are also euglycemic. As with any disease, there are also likely resilience factors present in some patients, which could be the focus of future research.
This model is likely applicable to bipolar patients of any age of onset. One study that compared medical comorbidities between patients with BD diagnosed before and after age 60 found no difference in rates of T2DM (the authors did not report whether the age of onset of T2DM was before or after the age of onset of BD [Citation89]). No additional research examining medical comorbidity in late-onset BD appears to have been conducted. There has been debate regarding whether early-onset and late-onset BD represent distinct subtypes of the disorder, as patients with an early onset tend to have a more severe course of illness than patients with a late onset [Citation90,Citation91]. However, the results of genetics studies suggest that early- and late-onset BD are not genetically distinct [Citation91,Citation92], and it has been proposed that the observed differences in clinical characteristics may be due to duration of illness [Citation93]. Thus, if early- and late-onset BD are not distinct subtypes, then one would expect this metabolic dysregulation model of BD progression to be relevant to both patients with early- or late-onset BD (though this model may still only be applicable to a subset of patients within both age groups).
Pathophysiology may inform treatment choices/suggest novel treatment approaches
One of the benefits of this hypothetical model of progression of BD, based on underlying shared dysregulation, is that treatment could be tailored to intervene at any point in the metabolic cascade to potentially halt the evolution of disease. This needs to be investigated. Instead of treating symptoms once they manifest, it might be possible to treat the pathological process in evolution. Based on underlying immune, inflammatory, oxidative stress, and lipid peroxidation processes, novel treatment strategies could prove beneficial (for example: immune modulators [Citation94], anti-inflammatories [Citation95], antioxidants [Citation96], vascular protective medications [Citation97]). It may be possible that intervening further upstream in the cascade could prevent both psychiatric and medical illness (BD, obesity, IR/T2DM, hypertension, dyslipidemia) in at-risk individuals. Further, early intervention for glucose dysregulation may prevent progression of illness that could otherwise result in cerebrovascular and cardiovascular diseases, as well as dementia.
Discussion
The ability to identify any potential prognostic factors in the progression of BD by which phase-appropriate intervention could possibly change illness trajectory and outcome would be helpful. Measuring inflammatory, immune and oxidative stress markers may become part of this proposed model for monitoring progression of BD as we test it and it evolves, but until there are specific proven interventions based on such markers, it is not clinically practical. Conversely, IR can be easily estimated using fasting glucose and concurrent insulin levels, and the HOMA-IR equation, and a specific and safe intervention is available: diet and exercise. While a single biomarker is not likely to entirely inform BD progression, IR could explain the changes in markers from anti-inflammatory to proinflammatory with increased oxidative stress/lipid peroxidation evident as bipolar illness progresses. Insulin resistance may be one factor of many in the progression of bipolar illness; however, it is one that can be easily tested for clinically, and for which treatment is available. Insulin resistance may be a piece of the puzzle that we have been missing – like H. pylori was in PUD.
Important questions arise from our earlier work and our new prospective studies and observations. Will reversing IR through diet and exercise or the use of insulin sensitizing medications be a helpful augmentation strategy in treating bipolar patients with an advanced refractory course? Will reversing IR with medication in otherwise healthy weight individuals revert a chronic course of illness back to an episodic course or facilitate remission? We hope that future research, including our prospective studies and the TRIO-BD Study, will address some of these questions.
When we continue to struggle to obtain remission in some bipolar patients, it may require a paradigm shift in our conceptualization of the illness process. Insulin resistance may underlie the progression of BD in more than half of all bipolar patients. This is not only meaningful, but also testable and treatable with the potential to affect prognosis and outcome. Further, this model may be generalizable to other major psychiatric illnesses such as schizophrenia, MDD and PTSD, for example. These illnesses also share higher rates of metabolic dysregulation [Citation98–102], which is associated with greater psychiatric morbidity [Citation103].
Conclusion
This hypothetical model of progression of BD, based on metabolic dysregulation, combines shared dysregulated mechanisms, allowing for early testing for IR, case finding, and monitoring for illness progression. Further, identifying and treating IR may not only facilitate remission in BD; it could also decrease risk for T2DM, cardiovascular disease and dementia, all comorbid in BD at higher rates compared to the general population.
This model offers an alternative to the current bipolar I versus bipolar II disorder dichotomy (which is largely based on severity of mania) that could help focus physicians on the need to test for IR and presents an avenue for future research directed at underlying dysregulated mechanisms versus syndromal symptoms. We may find that distinguishing between type of BD based on comorbid metabolic dysregulation – with or without – to be more informative in the future. This model requires further testing before it can be implemented as evidence-based practice.
Acknowledgements
The author would like to thank Kathleen Cairns for her assistance in preparing the manuscript.
Disclosure statement
The author has no conflicts of interest to report.
Additional information
Funding
Related Research Data
References
- Malhi GS, Rosenberg DR, Gershon S. Staging a protest! Bipolar Disord. 2014;16:776–779.
- Marshall BJ. One hundred years of discovery and rediscovery of helicobacter pylori and its association with peptic ulcer disease. In: Mobley H, Mendz G, Hazell S, editors. Helicobacter pylori: physiology and genetics. Washington, DC: ASM Press; 2001.
- Marshall B. Helicobacter pylori-a Nobel pursuit? Can J Gastroenterol. 2008;22:895–896.
- Calkin C, Ruzickova M, Slaney C, et al. Insulin resistance: an occult, modifiable risk factor for bipolar disorder? Br J Psychiatry. 2014;17:52–53.
- Calkin CV, Ruzickova M, Uher R, et al. Insulin resistance and outcome in bipolar disorder. Br J Psychiatry. 2015;206:52–57.
- Steardo L, Fabrazzo M, Sampogna G, et al. Impaired glucose metabolism in bipolar patients and response to mood stabilizer treatments. J Affect Disord. 2019;245:174–179. Available from: https://www-clinicalkey-com.libraryproxy.nshealth.ca/#!/content/playContent/1-s2.0-S0165032718313673?returnurl=null&referrer=null
- Mansur RB, Rizzo LB, Santos CM, et al. Impaired glucose metabolism moderates the course of illness in bipolar disorder. J Affect Disord. 2016;195:57–62. [cited 2017 Aug 16]Available from: http://ac.els-cdn.com.ezproxy.library.dal.ca/S0165032715312994/1-s2.0-S0165032715312994-main.pdf?_tid=c8d5fcc6-8287-11e7-b1d6-00000aab0f02&acdnat=1502890716_4a66bf74ac24446ede0cd626d899f491
- Ruzickova M, Slaney C, Garnham J, et al. Clinical features of bipolar disorder with and without comorbid diabetes mellitus. Can J Psychiatry. 2003;48:458–461.
- Colton CW, Manderscheid RW. Congruencies in increased mortality rates, years of potential life lost, and causes of death among public mental health clients in eight states. Prev Chronic Dis. 2006;3:A42.
- Charles EF, Lambert CG, Kerner B. Bipolar disorder and diabetes mellitus: evidence for disease-modifying effects and treatment implications. Int J Bipolar Disord.]. 2016;4:13.[cited 2017 Oct 6]. Available from: http://www.ncbi.nlm.nih.gov/pubmed/27389787
- Calkin C, Van de Velde C, Ruzickova M, et al. Can body mass index help predict outcome in patients with bipolar disorder? Bipolar Disord. 2009;11:650–656.
- Calkin C, Ruzickova M, Slaney C, et al. Are comorbid insulin resistance and type II diabetes risk factors for refractory bipolar illness? Eur Neuropsychopharmacol. 2013;23:S366–S7.
- Cairns K, McCarvill T, Ruzickova M, et al. Course of bipolar illness worsens after onset of insulin resistance. J Psychiatr Res. 2018;102:34–37.
- Bhuvaneswar CG, Baldessarini RJ, Harsh VL, et al. Adverse endocrine and metabolic effects of psychotropic drugs: selective clinical review. CNS Drugs. 2009;23:1003–1021.
- Kemp DE, Gao K, Chan PK, et al. Medical comorbidity in bipolar disorder: relationship between illnesses of the endocrine/metabolic system and treatment outcome. Bipolar Disord. 2010;12:404–413.
- Matthews DR, Hosker JP, Rudenski AS, et al. Homeostasis model assessment: insulin resistance and beta-cell function from fasting plasma glucose and insulin concentrations in man. Diabetologia. 1985;28:412–419.
- Hajek T, Calkin C, Blagdon R, et al. Type 2 diabetes mellitus: a potentially modifiable risk factor for neurochemical brain changes in bipolar disorders. Biol Psychiatry. 2015;77:295–303.
- Hajek T, Calkin C, Blagdon R, et al. Insulin resistance, diabetes mellitus, and brain structure in bipolar disorders. Neuropsychopharmacol. 2014;39:2910–2918.
- Wu G-Y, Zhang Q, Wu J-L, et al. Changes in cerebral metabolites in type 2 diabetes mellitus: a meta-analysis of proton magnetic resonance spectroscopy. J Clin Neurosci. 2017;45:9–13. [cited 2019 Jul 31].
- Jones N, Riby LM, Mitchell RLC, et al. Type 2 diabetes and memory: using neuroimaging to understand the mechanisms. CDR. 2014;10:118–123. [cited 2019 Aug 2]. Available from: http://www.ncbi.nlm.nih.gov/pubmed/24766069
- Lee JH, Choi Y, Jun C, et al. Neurocognitive changes and their neural correlates in patients with type 2 diabetes mellitus. Endocrinol Metab. 2014;29:112–121. Available from: http://www.ncbi.nlm.nih.gov/pubmed/25031883
- Tsai S-Y, Lee H-C, Chen C-C, et al. Cognitive impairment in later life in patients with early-onset bipolar disorder. Bipolar Disord. 2007;9:868–875.
- Hubenak J, Tuma I, Bazant J. Association of arterial hypertension and cognitive impairment in euthymic bipolar disorder. Neuroendocrinol Lett. 2015;36:294–300. Available from: http://www.ncbi.nlm.nih.gov/pubmed/26313398
- Depp CA, Strassnig M, Mausbach BT, et al. Association of obesity and treated hypertension and diabetes with cognitive ability in bipolar disorder and schizophrenia. Bipolar Disord. 2014;16:422–431. Available from: http://www.ncbi.nlm.nih.gov/pubmed/24725166
- Biessels GJ, Deary IJ, Ryan CM. Cognition and diabetes: a lifespan perspective. Lancet Neurol. 2008;7:184–190.
- de la Monte SM. Relationships between diabetes and cognitive impairment. Endocrinol Metab Clin North Am. 2014;43:245–267.
- Convit A. Links between cognitive impairment in insulin resistance: an explanatory model. Neurobiol Aging. 2005;26(SUPPL.):31–35.
- Cheng G, Huang C, Deng H, et al. Diabetes as a risk factor for dementia and mild cognitive impairment: a meta-analysis of longitudinal studies. Intern Med J. 2012;42:484–491.
- Regenold WT, Thapar RK, Marano C, et al. Increased prevalence of type 2 diabetes mellitus among psychiatric inpatients with bipolar I affective and schizoaffective disorders independent of psychotropic drug use. J Affect Disord. 2002;70:19–26.
- Kooy F. Hyperglycemia in mental disorders. Brain. 1919;42:214–289.
- Henneman DH, Altschule MD, Goncz RM. Carbohydrate metabolism in brain disease. II. Glucose metabolism in Schizophrenic, manic-depressive, and involutional psychoses. AMA Arch Intern Med. 1954;94:402–416.
- Kretschmer E. Physique and character. In Kegan, P, Trench, T, editors. London: Kegan, Paul, Trench, Trubner; 1936.
- Kucukgoncu S, Kosir U, Zhou E, et al. Glucose metabolism dysregulation at the onset of mental illness is not limited to first episode psychosis: a systematic review and meta-analysis. Early Interv Psychiatry. 2018. DOI:https://doi.org/10.1111/eip.12749
- Jerrell JM, McIntyre RS, Tripathi A. A cohort study of the prevalence and impact of comorbid medical conditions in pediatric bipolar disorder. J Clin Psychiatry. 2010;71:1518–1525.
- Calkin CV, Gardner DM, Ransom T, et al. The relationship between bipolar disorder and type 2 diabetes: more than just co-morbid disorders. Ann Med. 2013;45:171–181.
- Torkamani A, Topol EJ, Schork NJ. Pathway analysis of seven common diseases assessed by genome-wide association. Genomics. 2008;92:265–272.
- Amare AT, Schubert KO, Klingler-Hoffmann M, et al. The genetic overlap between mood disorders and cardiometabolic diseases: a systematic review of genome wide and candidate gene studies. Transl Psychiatry. 2017;7:e1007. Available from: http://www.ncbi.nlm.nih.gov/pubmed/28117839
- Watson S, Gallagher P, Ritchie JC, et al. Hypothalamic-pituitary-adrenal axis function in patients with bipolar disorder. Br J Psychiatry. 2004;184:496–502.
- McElroy SL, Keck PE. Jr. Obesity in bipolar disorder: an overview. Curr Psychiatry Rep. 2012;14:650–658.
- Fagiolini A, Kupfer DJ, Houck PR, et al. Obesity as a correlate of outcome in patients with bipolar I disorder. Am J Psychiatry. 2003;160:112–117.
- Elmslie JL, Mann JI, Silverstone JT, et al. Determinants of overweight and obesity in patients with bipolar disorder. J Clin Psychiatry. 2001;62:486–491.
- Shoelson SE, Lee J, Goldfine AB. Inflammation and insulin resistance. J Clin Invest. 2006;116:1793–1801. Available from: http://www.ncbi.nlm.nih.gov/pubmed/16823477
- Rask-Madsen C, King GL. Mechanisms of disease: endothelial dysfunction in insulin resistance and diabetes. Nat Rev Endocrinol. 2007;3:46–56.
- Shah S, Iqbal M, Karam J, et al. Oxidative stress, glucose metabolism, and the prevention of type 2 diabetes: pathophysiological insights. Antioxid Redox Signal. 2007;9:911. Available from: http://www.ncbi.nlm.nih.gov/pubmed/17508914
- Katakam PVG, Ujhelyi MR, Hoenig ME, et al. Endothelial dysfunction precedes hypertension in diet-induced insulin resistance. Am J Physiol. 1998;275:R788–92. Available from: http://www.ncbi.nlm.nih.gov/pubmed/9728076
- Andreazza AC, Kapczinski F, Kauer-Sant’anna M, et al. 3-Nitrotyrosine and glutathione antioxidant system in patients in the early and late stages of bipolar disorder. J Psychiatry Neurosci. 2009;34:263–271.
- Andreazza AC, Kauer-Sant’anna M, Frey BN, et al. Oxidative stress markers in bipolar disorder: a meta-analysis. J Affective Disorders. 2008;111:135–144.
- Brown NC, Andreazza AC, Young LT. An updated meta-analysis of oxidative stress markers in bipolar disorder. Psychiatry Res. 2014;218:61–68. Available from: http://www.ncbi.nlm.nih.gov/pubmed/24794031
- Frey BN, Andreazza AC, Houenou J, et al. Biomarkers in bipolar disorder: a positional paper from the International Society for Bipolar Disorders Biomarkers Task Force. Aust N Z J Psychiatry. 2013;47:321–332.
- Scola G, Mcnamara RK, Croarkin PE, et al. Lipid peroxidation biomarkers in adolescents with or at high-risk for bipolar disorder. J Affect Disord. 2016;192:176–183. Available from: https://www-ncbi-nlm-nih-gov.libraryproxy.nshealth.ca/pmc/articles/PMC5549852/pdf/nihms885439.pdf
- Scola G, Andreazza AC. Current state of biomarkers in bipolar disorder. Curr Psychiatry Rep. 2014;16:514.
- Osby U, Brandt L, Correia N, et al. Excess mortality in bipolar and unipolar disorder in Sweden. Arch Gen Psychiatry. 2001;58:844–850.
- da Silva J, Gonçalves-Pereira M, Xavier M, et al. Affective disorders and risk of developing dementia: systematic review. Br J Psychiatry. 2013;202:177–186.
- van der Heide LP, Kamal A, Artola A, et al. Insulin modulates hippocampal activity-dependent synaptic plasticity in a N-methyl-d-aspartate receptor and phosphatidyl-inositol-3-kinase-dependent manner. J Neurochem. 2005;94:1158–1166.
- Wallum BJ, Taborsky GJ, Jr, Porte D, Jr, et al. Cerebrospinal fluid insulin levels increase during intravenous insulin infusions in man. J Clin Endocrinol Metab. 1987;64:190–194.
- Craft S, Watson GS. Insulin and neurodegenerative disease: shared and specific mechanisms. Lancet Neurol. 2004;3:169–178.
- Passos IC, Mwangi B, Vieta E, et al. Areas of controversy in neuroprogression in bipolar disorder. Acta Psychiatr Scand. 2016;134:91–103.
- Sandyk R. The relationship between diabetes mellitus and Parkinson’s disease. Int J Neurosci. 1993;69:125–130.
- Bastard J-P, Maachi M, Lagathu C, et al. Recent advances in the relationship between obesity, inflammation, and insulin resistance. Eur Cytokine Netw. 2006;17:4–12.
- Wang X, Bao W, Liu J, et al. Inflammatory markers and risk of type 2 diabetes: a systematic review and meta-analysis. Diabetes Care. 2013;36:166. Available from: http://www.ncbi.nlm.nih.gov/pubmed/23264288
- Onat A, Can G, Kaya H, et al. Atherogenic index of plasma”(log10 triglyceride/high-density lipoprotein-cholesterol) predicts high blood pressure, diabetes, and vascular events. J Clin Lipidol. 2010;4:89–98. Available from: https://linkinghub.elsevier.com/retrieve/pii/S1933287410000528
- Alberti K, Eckel RH, Grundy SM, et al. Harmonizing the metabolic syndrome: a joint interim statement of the International Diabetes Federation Task Force on Epidemiology and Prevention; National Heart, Lung, and Blood Institute; American Heart Association; World Heart Federation; International. Circulation. 2009;120:1640–1645.
- Ortiz-Dominguez A, Hernandez ME, Berlanga C, et al. Immune variations in bipolar disorder: phasic differences. Bipolar Disord. 2007;9:596–602.
- Brietzke E, Stertz L, Fernandes BS, et al. Comparison of cytokine levels in depressed, manic and euthymic patients with bipolar disorder. J Affect Disord. 2009;116:214–217.
- Maes M, Bosmans E, Calabrese J, et al. Interleukin-2 and interleukin-6 in schizophrenia and mania: effects of neuroleptics and mood stabilizers. J Psychiatr Res. 1995;29:141–152.
- Cunha AB, Andreazza AC, Gomes FA, et al. Investigation of serum high-sensitive C-reactive protein levels across all mood states in bipolar disorder. Eur Arch Psychiatry Clin Neurosc.. 2008;258:300–304.
- Snijders G, Mesman E, de Wit H, et al. Immune dysregulation in offspring of a bipolar parent. Altered serum levels of immune growth factors at adolescent age. Brain Behav Immun. 2017;64:116–123.
- Mansur RB, Rizzo LB, Santos CM, et al. Adipokines, metabolic dysfunction and illness course in bipolar disorder. J Psychiatr Res. 2016;74:63–69. Available from: http://www.sciencedirect.com/science/article/pii/S0022395615300194?via%3Dihub
- Maritim AC, Sanders RA, Watkins JB. Diabetes, oxidative stress, and antioxidants: a review. J Biochem Mol Toxicol. 2003;17:24–38.
- Folmer V, Soares JCM, Rocha J. Oxidative stress in mice is dependent on the free glucose content of the diet. Int J Biochem Cell Biol.]. 2002;34:1279–1285. Available from: http://www.ncbi.nlm.nih.gov/pubmed/12127578
- Gupta BL, Azam M, Baquer NZ. Changes in erythrocyte glutathione peroxidase and glutathione reductase in alloxan diabetes. Biochem Int. 1990;21:725–731. Available from: http://www.ncbi.nlm.nih.gov/pubmed/2241998
- Andreazza AC, Gildengers A, Rajji T, et al. Oxidative stress in older patients with bipolar disorder. Am J Geriatr Psychiatry. 2015;23:314–319. Available from: https://www.ncbi.nlm.nih.gov/pmc/articles/PMC4247347/pdf/nihms609561.pdf
- de Sousa RT, Zarate CA, Zanetti MV, et al. Oxidative stress in early stage Bipolar Disorder and the association with response to lithium. J Psychiatr Res. 2014;50:36–41. Available from: http://www.ncbi.nlm.nih.gov/pubmed/24332923
- Magalhães PVS, Jansen K, Pinheiro RT, et al. Peripheral oxidative damage in early-stage mood disorders: a nested population-based case-control study. Int J Neuropsychopharm. 2012;15:1043–1050. Available from: http://www.ncbi.nlm.nih.gov/pubmed/22008234
- Kauer-Sant'Anna M, Kapczinski F, Andreazza AC, et al. Brain-derived neurotrophic factor and inflammatory markers in patients with early- vs. late-stage bipolar disorder. Int J Neuropsychopharm.. 2009;12:447–458.
- Duffy A, Horrocks J, Doucette S, et al. Immunological and neurotrophic markers of risk status and illness development in high-risk youth: understanding the neurobiological underpinnings of bipolar disorder. Int J Bipolar Disord. 2014;2:29. Available from: https://doi.org/https://doi.org/10.1186/2194-7511-2-4
- Fries GR, Pfaffenseller B, Stertz L, et al. Staging and neuroprogression in bipolar disorder. Curr Psychiatry Rep. 2012;14:667–675. Available from: http://www.ncbi.nlm.nih.gov/pubmed/23090632
- Eyileten C, Kaplon-Cieslicka A, Mirowska-Guzel D, et al. Antidiabetic effect of brain-derived neurotrophic factor and its association with inflammation in type 2 diabetes mellitus. J Diabetes Res. 2017;2017:1.
- Bifulco A, Moran PM, Baines R, et al. Exploring psychological abuse in childhood: II. Association with other abuse and adult clinical depression. Bull Menninger Clin. 2002;66:241–258.
- Goodwin RD, Stein MB. Association between childhood trauma and physical disorders among adults in the United States. Psychol Med. 2004;34:509–520.
- Calkin CV, Kamintsky L, Cairns K, et al. Blood-brain barrier dysfunction may be predictive of neuroprogression in bipolar disorder. Eur Psychiatry. 2018;48:356.
- Calkin C, Kamintsky L, Cairns K, et al. Brain microvascular pathology is associated with bipolar neuroprogression. Biol Psychiatry. 2019;85:S66. Available from: https://linkinghub.elsevier.com/retrieve/pii/S0006322319303233
- Calkin CV, Alda M. Insulin resistance in bipolar disorder: relevance to routine clinical care. Bipolar Disord. 2015;17:683–688.
- Zeinoddini A, Sorayani M, Hassanzadeh E, et al. Pioglitazone adjunctive therapy for depressive episode of bipolar disorder: a randomized, double-blind, placebo-controlled trial. Depress Anxiety. 2015;32:167–173.
- Kemp DE, Schinagle M, Gao K, et al. PPAR-γ Agonism as a modulator of mood: proof-of-concept for pioglitazone in bipolar depression. CNS Drugs. 2014;28:571–581.
- Colle R, De Larminat D, Rotenberg S, et al. Pioglitazone could induce remission in major depression: a meta-analysis. NDT. 2016;13:9–16.
- Aftab A, Kemp DE, Ganocy SJ, et al. Double-blind, placebo-controlled trial of pioglitazone for bipolar depression. J Affect Disord. 2019;245:957–964.
- Lin KW, Wroolie TE, Robakis T, et al. Adjuvant pioglitazone for unremitted depression: clinical correlates of treatment response. Psychiatry Res. 2015;230:846–852. Available from: http://www.ncbi.nlm.nih.gov/pubmed/26602230
- Almeida OP, Hankey GJ, Yeap BB, et al. Older men with bipolar disorder diagnosed in early and later life: physical health morbidity and general hospital service use. J Affect Disord. 2018;241:269–274.
- Coryell W, Fiedorowicz J, Leon AC, et al. Age of onset and the prospectively observed course of illness in bipolar disorder. J Affect Disord. 2013;146:34–38. Available from: http://www.ncbi.nlm.nih.gov/pubmed/23062746
- Kennedy KP, Cullen KR, Deyoung CG, et al. The genetics of early-onset bipolar disorder: a systematic review. J Affect Disord. 2015;184:1–12. Available from: https://www-ncbi-nlm-nih-gov.libraryproxy.nshealth.ca/pmc/articles/PMC5552237/pdf/nihms885432.pdf
- Kalman JL, Papiol S, Forstner AJ, et al. Investigating polygenic burden in age at disease onset in bipolar disorder: findings from an international multicentric study. Bipolar Disord. 2018;62:68–75. Available from: https://www-ncbi-nlm-nih-gov.libraryproxy.nshealth.ca/pmc/articles/PMC6585855/pdf/BDI-21-68.pdf
- Almeida OP, Fenner S. Bipolar disorder: similarities and differences between patients with illness onset before and after 65 years of age. Int Psychogeriatr.. 2002;14:311–322. Available from: http://www.ncbi.nlm.nih.gov/pubmed/12475092
- Soczynska JK, Kennedy SH, Goldstein BI, et al. The effect of tumor necrosis factor antagonists on mood and mental health-associated quality of life: novel hypothesis-driven treatments for bipolar depression? Neurotoxicology. 2009;30:497–521.
- Rosenblat JD, Kakar R, Berk M, et al. Anti-inflammatory agents in the treatment of bipolar depression: a systematic review and meta-analysis. Bipolar Disord. 2016;18:89–101.
- Henter ID, De Sousa RT, Gold PW, et al. Mood therapeutics: novel pharmacological approaches for treating depression. Expert Rev Clin Pharmacol. 2017;10:153–166. Available from: https://www-ncbi-nlm-nih-gov.libraryproxy.nshealth.ca/pmc/articles/PMC5552063/pdf/nihms883292.pdf
- Su JB. Vascular endothelial dysfunction and pharmacological treatment. WJC. 2015;7:719–741. Available from: http://www.ncbi.nlm.nih.gov/pubmed/26635921
- Dasgupta A, Singh OP, Rout JK, et al. Insulin resistance and metabolic profile in antipsychotic naïve schizophrenia patients. Prog Neuro-Psychopharmacol Biol Psychiatry. 2010;34:1202–1207. Available from: http://linkinghub.elsevier.com/retrieve/pii/S0278584610002289
- Trief PM, Ouimette P, Wade M, et al. Post-traumatic stress disorder and diabetes: co-morbidity and outcomes in a male veterans sample. J Behav Med. 2006;29:411–418. Available from: https://doi.org/https://doi.org/10.1007/s10865-006-9067-2
- Ahmadi N, Arora R, Vaidya N, et al. Post-traumatic stress disorder is associated with increased incidence of insulin resistance and metabolic syndrome. J Am Coll Cardiol. 2013;61:E1347. Available from: http://linkinghub.elsevier.com/retrieve/pii/S0735109713613479
- Okamura F, Tashiro A, Utumi A, et al. Insulin resistance in patients with depression and its changes during the clinical course of depression: minimal model analysis. Metabolism. 2000;49:1255–1260. Available from: http://ac.els-cdn.com.ezproxy.library.dal.ca/S0026049500274179/1-s2.0-S0026049500274179-main.pdf?_tid=384a1ac0-8345-11e7-9ff4-00000aacb362&acdnat=1502972078_68f1ae22e1db6bced3de3fcc294a4e51
- Vancampfort D, Correll CU, Galling B, et al. Diabetes mellitus in people with schizophrenia, bipolar disorder and major depressive disorder: a systematic review and large scale meta-analysis. World Psychiatry. 2016;15:166–174.
- Egede LE. Diabetes, major depression; and functional disability among U.S. adults. Diabetes Care. 2004;27:421–428. Available from: www.who.int/msa/cidi/index.htm