
Abstract
Lipoprotein(a) (Lp(a)) was discovered more than 50 years ago, and a decade later, it was recognized as a risk factor for coronary artery disease. However, it has gained importance only in the past 10 years, with emergence of drugs that can effectively decrease its levels. Lp(a) is a low-density lipoprotein (LDL) with an added apolipoprotein(a) attached to the apolipoprotein B component via a disulphide bond. Circulating levels of Lp(a) are mainly genetically determined. Lp(a) has many functions, which include proatherosclerotic, prothrombotic and pro-inflammatory roles. Here, we review recent data on the role of Lp(a) in the atherosclerotic process, and treatment options for patients with cardiovascular diseases. Currently ‘Proprotein convertase subtilisin/kexin type 9’ (PCSK9) inhibitors that act through non-specific reduction of Lp(a) are the only drugs that have shown effectiveness in clinical trials, to provide reductions in cardiovascular morbidity and mortality. The effects of PCSK9 inhibitors are not purely through Lp(a) reduction, but also through LDL cholesterol reduction. Finally, we discuss new drugs on the horizon, and gene-based therapies that affect transcription and translation of apolipoprotein(a) mRNA. Clinical trials in patients with high Lp(a) and low LDL cholesterol might tell us whether Lp(a) lowering per se decreases cardiovascular morbidity and mortality.
Lipoprotein(a) is an important risk factor in patients with cardiovascular diseases.
Lipoprotein(a) has many functions, which include proatherosclerotic, prothrombotic and pro-inflammatory roles.
Treatment options to lower lipoprotein(a) levels are currently scarce, but new drugs are on the horizon.
KEY MESSAGES
Introduction
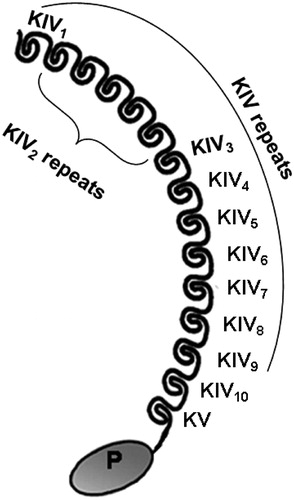
Lipoprotein(a) (Lp(a)) was discovered in 1963 [Citation1]; however, it appeared to be of little importance until it was shown that this particular lipoprotein is an independent causal risk factor for cardiovascular disease (CVD) [Citation2]. Lp(a) consists of a low-density lipoprotein (LDL)-like particle that is bound to apolipoproteinB100 (apoB), which is then linked with apolipoprotein(a) (apo(a)). Apo(a) has a very similar structure to plasminogen, although while apo(a) has only two types of kringle domains, as IV and V, plasminogen has five types (I–V). The molecular structure of apo(a) is shown in . These kringle domains are protein domains with large loops that are stabilised by disulphide bridges and that contain lysine binding sites, which allow plasminogen to bind fibrin. Another difference here is the numbers of kringle IV (KIV) subtypes; apo(a) has 10 KIV subtypes, while plasminogen has only one KIV subtype [Citation3]. However, as apo(a) has a similar overall structure to plasminogen, it can interfere with fibrinolysis [Citation4]. Due to these similarities, apo(a) either binds to fibrin or forms a complex with fibrin, plasminogen and tissue plasminogen activator (t-PA). The result of these two actions is decreased fibrinolysis, and thus increasing risk for thrombotic events.
Figure 1. Molecular structure of apolipoprotein(a). Apolipoprotein(a) contains 10 subtypes of KIV repeats, as one copy of KIV1, multiple copies of KIV2 and one copy each of KIV3 to KIV10, KV and inactive protease-like (P) domain.
Despite this potential action of apo(a), Lp(a) itself is better known for its pathological roles in thrombosis and atherosclerosis. Lp(a) contributes to the development of atherosclerosis in several ways: it increases expression of vascular cell adhesion molecule-1 and E-selectin in endothelial cells; it increases accumulation of peripheral blood mononuclear cells in vessels; and it acts as a pro-inflammatory through its oxidised phospholipids [Citation3]. Also, staining of atherosclerotic plaques has revealed a high abundance of apo(a) [Citation5].
Synthesis, metabolism and measurement of Lp(a)
The biosynthesis and metabolism of Lp(a) are not completely known, although these pathways are known to be independent of the synthesis and metabolism of LDL cholesterol. Biosynthesis of Lp(a) takes place almost exclusively in the liver. It begins with synthesis of apo(a) in the hepatocytes, while apoB binding occurs at the cell surface [Citation6]. Lp(a) does not arise from catabolism of any other lipoproteins [Citation7]. Opposite to LDL cholesterol, plasma levels of Lp(a) correlate very strongly with the production rates of Lp(a), with little or no relationship between levels and the fractional catabolic rate of Lp(a) [Citation8]. Moreover, Ma et al. [Citation9] showed that this is the case in statin-treated or untreated patients with elevated or normal Lp(a) levels.
Due to the complex molecular structure of Lp(a) and the variation in the size of apo(a), there are particular problems for the measurement of Lp(a) concentrations. There are several assays available [Citation10], which are, to varying degrees, influenced by the apo(a) isoform [Citation11]. Another problem is the reporting of the concentration of Lp(a), which can be expressed as molar concentrations (nmol/L) or as mass concentrations (mg/dL). The conversion between those two has been shown to be both size and concentration dependent, and is far from accurate [Citation11–13]. In 2019, the HEART UK consensus statement of Lp(a) measurement suggested that Lp(a) concentrations should be expressed in nmol/L [Citation13] and the concentration of Lp(a) should be measured using appropriate antibodies, to minimise the effects of isoform size. As the Lp(a) concentration in mass units encompasses the mass of the entire particle (i.e. apo(a), apoB100, cholesterol, cholesteryl esters, phospholipids, triglycerides), and due to the heterogeneity in the size of apo(a), standardisation using a single calibration method is still not possible [Citation13]. Lp(a) should be measured using an immunoassay that minimises sensitivity to apolipoprotein(a) isoform size and is calibrated against the World Health Organization/International Federation of Clinical Chemistry and Laboratory Medicine reference material; results should be reported in nmol/L of Lp(a) particles [Citation14].
Therefore, despite these recent efforts, Lp(a) measurement remains a major problem, and a better route to its standardisation is awaited.
Familial hypercholesterolaemia (FH) is an autosomal dominant genetic disorder of lipoprotein metabolism that is due to function-loss mutations in the LDL receptor gene. FH is the second most common, monogenic cause of inherited heart disease worldwide, behind Lp(a) [Citation15]. The mutations in FH can lead to uncontrolled and high plasma levels of LDL cholesterol. Even moderate increases in Lp(a) concentrations from 30 to 50 mg/dL in patients with FH can significantly increase their risk for advanced CVD, as even without elevated Lp(a), these patients are at high risk. It has also been shown that plasma Lp(a) concentrations are elevated in FH compared to the general population [Citation16]. In subjects with FH, these higher Lp(a) levels are associated with both small and large apo(a) isoforms [Citation3].
In patients with FH who have experienced an acute cardiovascular (CV) event, Lp(a) levels are significantly higher than in patients with FH and no history of coronary artery disease (CAD) [Citation17].The same findings were reported in a much smaller study with FH patients [Citation18]. In addition to this finding, this small study also showed increased levels of t-PA antigen and plasminogen activator inhibitor (PAI) antigen, and increased activity that indicated that Lp(a) also has anti-fibrinolytic effects in these patients [Citation18].
In subjects with hypertriglyceridaemia, Lp(a) is reduced, possibly due to an increase in plasma clearance of lipoproteins [Citation19]. It has been predicted that Lp(a) binds to specific LDL receptors, although as this binding has lower affinity than that for LDL cholesterol, LDL receptors are not as important for the removal of plasma Lp(a).
Genetic and ethnic background of Lp(a)
The gene that encodes the apo(a) component of the Lp(a) particle, LPA, has up to 70% homology with the human plasminogen gene. LPA is located on the long arm of chromosome 6, within 6q2.6-2.7 [Citation6].
Lp(a) plasma levels can vary very widely, with up to 1000-fold differences seen between individuals in the same population. These variable Lp(a) levels arise due to genetic diversity, with 10 different variants in the KIV domain, and different numbers of repeats [Citation20]. LPA appears to be responsible for 91% of the variation in Lp(a) levels. For this variation, 69% is due to the number of KIV type 2 repetitions, and 22% to other factors [Citation21]. In another study, the number of KIV type 2 repeats explained up to 30% of the variation in Lp(a) levels () [Citation22].
Most of the variations in Lp(a) levels are reflected in the effects of genetic variation of the LPA gene. Variations in the copy numbers of LPA determine the number of KIV type 2 repeats and the isoform of apo(a). There is an inverse relationship between the number of LPA repetitions and the Lp(a) levels [Citation23].
In a multicenter study by Clarke et al. [Citation24], three chromosomal regions were associated with risk of CAD; 6q26–27, 9p21, and 1p13. The LPA locus at region 6q26–27 has the strongest relationship between increased Lp(a) levels and risk of CAD. Two common independent LPA variants, as the single nucleotide polymorphisms (SNPs) rs10455872 and rs3798220 within the LPA site, have been shown to be associated with increased Lp(a) levels, a smaller size of apo (a), and reduced copy numbers of LPA, which determines the number of repetitions of KIV type 2. These two common variants together explain 36% of the variation in Lp(a) levels. Both of these SNPs are independently associated with increased Lp(a) levels and increased risk of CVD.
The SNP rs3798220 encodes a nonsynonymous variant of LPA, and it has been reported to have strong association with Lp(a) levels [Citation25], moderate association with LDL cholesterol levels [Citation26], and a connection with risk of CVD [Citation25]. Clarke et al. [Citation24] reported on a more definitive assessment of the strength of association between rs3798220 and CAD risk. In their study, which involved almost 8000 patients with CVD, they found four haplotypes of rs3798220, which might explain and improve part of the association between the SLC22A3–LPAL2–LPA gene cluster and CVD [Citation24]. The second SNP, rs10455872, indicated intron 25 in the LPA gene, where the allele frequency of the high-risk variant was about 7% [Citation27]. Clarke et al. [Citation24] did not find any associations between either rs10455872 or rs3798220 and plasma levels of apoB, fibrinogen or C-reactive protein.
The frequency of rs3798220 is increased in subjects with elevated Lp(a) levels, and 24% of Europeans carry this SNP. LPA SNP rs10455872 is present in 7–15% of the general population in Europeans. Its prevalence increases with increasing Lp(a) levels, and is present in 47% of northern Europeans with Lp(a) levels >95th percentile. Its prevalence was reported as 37.2% of patients with progressive CAD who were undergoing apheresis. In non-European populations, in Japan or other similar east Asian populations, this SNP has not been associated with CVD or small apo(a) isoform size [Citation28].
In the Pakistan Risk of Myocardial Infarction Study, this genome-wide analysis identified rs2457564 as a variant associated with smaller apo(a) isoform size, but not Lp(a) concentration, and rs3777392 as a variant associated with Lp(a) concentration, but not apo(a) isoform size. This study suggested that for the human genetic data, both smaller apo(a) isoform size and increased Lp(a) concentrations are independent and causal risk factors for CAD [Citation29].
Race (i.e. ethnicity) has a significant impact on Lp(a) levels, as variable frequencies of alleles have been reported within ethnic groups [Citation30]. In one study with multiple ethnic groups, polymorphisms for Lp(a) had an influence on 17–77% of the variation in Lp(a) levels, 80% of which was due to the number of kringle domains in Lp(a) [Citation31]. The genetic factor is a major cause of diversity in Lp(a) levels, whereas gender and age do not affect these [Citation6]. Jenner at al. [Citation32] reported no difference in Lp(a) levels between age-adjusted premenopausal and postmenopausal women, and none between men and women. Lp(a) levels also do not depend on environmental factors, or dietary or lifestyle factors. Throughout life, Lp(a) levels should remain the same [Citation33].
The prevalence and association of LPA SNPs with apo(a) isoform size and with the levels of Lp(a) and its associated oxidized phospholipids on apolipoprotein B100 are highly variable and ethnicity specific [Citation28]. Lp(a) and its associated oxidized phospholipids on apolipoprotein B100 have been proposed to have multiple atherogenic effects, including proliferation of vascular smooth muscle cells and foam-cell formation, which promote the release of pro-inflammatory mediators and anti-fibrinolytic effects [Citation29]. Lee at al. [Citation28] explained the role of the LPA gene and the elevated Lp(a) levels in the prediction of CVD risk through the demonstration that elevated circulating Lp(a) levels are the key variable in predicting CVD risk, irrespective of ethnicity. Blacks have the highest mean Lp(a) levels, but they have larger isoforms that are associated with these levels, rather than small isoforms, as has been shown in subjects of European descent. Small apo(a) isoforms are primarily important for CVD risk prediction, in that they contribute to higher Lp(a)/oxidized phospholipids on apolipoprotein B100 levels via shorter synthesis time in the hepatocyte. This has been noted for Whites and Hispanics, and not for Blacks, which is consistent with the relationship that apo(a) isoform size explains <50% of circulating Lp(a) levels. LPA SNPs appear to be tagging SNPs for apo(a) isoform size, although not only for small isoforms. The SNP rs3798220 is present in 42.8% of Hispanics, but only in 4.3% of Whites. It is associated with large apo(a) isoforms and lower Lp(a) levels in Hispanics, but with very small apo(a) isoforms and higher Lp(a) levels in Whites. These data suggest that circulating Lp(a) levels are the main variable in the quantification of CV risk, rather than LPA SNPs or apo(a) isoform size. In European populations, rs3798220 is associated with small apo(a) isoform size and markedly elevated Lp(a) levels, particularly in homozygotes. In contrast, this rs3798220 is not present at all in Africans, is present in <10% of Asian Indians, and is more prevalent (11.6%) in east Asians, such as the Chinese in Hong Kong. The prevalence of rs9457951 is primarily associated with Black subjects, but it only explains approximately 5% of the contribution to Lp(a) levels [Citation28].
The role of Lp(a) in atherosclerosis and thrombosis
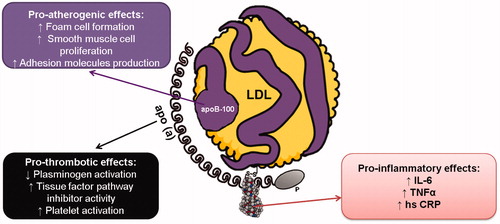
The physiological role of Lp(a) is not yet fully understood. However, elevated Lp(a) levels are known to be independently associated with increased risk of CAD. Lp(a) is believed to have many possible functions, which include proatherosclerotic, prothrombotic and pro-inflammatory roles () [Citation3].
Figure 2. Role of lipoprotein(a) in the atherosclerotic process. The three defined roles of lipoprotein(a) in progression of the atherosclerotic process include its proatherogenic, prothrombotic, and pro-inflammatory effects. hsCRP, high sensitivity C-reactve protein; IL-6, interleukin 6; TNFα, tumour necrosis factor α.
Lp(a) levels are transiently increased by inflammatory processes and by tissue damage caused by acute-phase proteins (e.g. haptoglobin, α-1-antitrypsin, C-reactive protein) [Citation6]. Lp(a) stimulates vascular cell adhesion molecule-1 and E-selectin expression in cultured human coronary artery endothelial cells [Citation34]. High Lp(a) levels have been reported for coronary atheroma in patients with unstable angina, compared to patients with stable angina. Here, 90% of the Lp(a) area in coronary atheromas was co-localised with plaque macrophages, and 30% was correlated with plaque α-actin, which might be related to the role of Lp(a) in plaque enlargement [Citation5]. Similarly, in acute myocardial infarction (MI), Lp(a) levels increase significantly within the first 24 h, and normalise within about 30 days [Citation35].
The mechanisms underlying the influence of Lp(a) in atherogenesis are not yet completely clear. One of the proposed mechanisms is direct deposition of Lp(a) on arterial walls. Lp(a) is more susceptible to oxidation than LDL, which will thus accelerate the uptake of Lp(a) into macrophages via scavenger receptors. This impacts on the most universal mechanism of atherogenesis, in which macrophages change over time into foam cells, the precursors of atherosclerosis [Citation36]. Another possible proatherogenic mechanism of Lp(a) is the inverse relationship between Lp(a) levels and vascular reactivity. In this case, increased plasma Lp(a) levels induce endothelial dysfunction [Citation37]. Lp(a) is more atherogenic than LDL cholesterol. In addition to containing all of the proatherogenic properties of LDL cholesterol, apo(a) also acts as a sink for inflammatory molecules, such as oxidised phospholipids. Oxidised phospholipids can affect many of the pro-atherogenic properties of Lp(a), including its inflammatory function [Citation38].
Elevated Lp(a) levels have also been reported for patients with chronic inflammatory diseases (e.g. rheumatoid arthritis [Citation39], systemic lupus erythematosus [Citation40], acquired immunodeficiency syndrome [Citation41]), chronic renal failure, and pulmonary hypertension. On the other hand, with liver disease and steroid intake, Lp(a) levels are decreased [Citation6].
There are conflicting data from studies of the associations between increased Lp(a) levels and diabetes. In a study by Singla et al. [Citation33], increased Lp(a) levels were reported for patients with type 2 diabetes. On the other hand, several other studies have observed lower plasma Lp(a) levels in diabetic patients. Epidemiology studies of Lp(a) and risk of CVD in diabetic patients have also generated opposing data [Citation42]. It was reported that Lp(a)-related genetic markers do not predict CVD in two diabetic cohorts [Citation43]. A cross-sectional analysis of plasma Lp(a) levels from 36 studies showed that Lp(a) levels were 11% (95% CI: 4–17%) lower in people with diabetes than for those without [Citation2].
Connections between Lp(a) and inflammatory cytokines have also been reported, which have included tumour necrosis factor-α, transforming growth factor-β, interleukin-6 and monocyte chemoattractant protein-1 [Citation44]. In addition to reducing fibrinolysis, Lp(a) affected platelet aggregation, with increases in expression of adhesion molecules, vascular remodelling through proliferative and migration changes in smooth muscle endothelial cells, oxidative adaptation and foam-cell formation [Citation45].
Lipoprotein(a) has prothrombotic properties through its interference with reactions in fibrinolysis regulation, due to its antifibrinolytic role [Citation46]. The thrombogenic properties of Lp(a) are a result of the homology between apo(a) and plasminogen. Lp(a) competes with plasminogen for binding sites on endothelial cells, which inhibits fibrinolysis and promotes intravascular thrombosis [Citation47]. Apo(a) affects plasminogen binding to endothelial cells, monocytes, and macrophages, which results in impairment of mural thrombus lysis. Lp(a) can also have antifibrinolytic effects due to the failure of plasminogen to bind to fibrin [Citation48]. Lp(a) inhibits the binding of t-PA to fibrin and inhibits plasminogen activation by t-PA, which is dependent on fibrin and fibrinogen particles. Lp(a) inhibits plasminogen activation by streptokinase, as it competes for plasminogen binding to monocytoid and epithelial cells. The plasma protein tetranectin binds reversibly to plasminogen to accelerate plasminogen activation by t-PA. Lp(a) binds to tetranectin with higher affinity than plasminogen [Citation33]. In vitro studies have shown that the apo(a) component of Lp(a) weakens fibrin clot lysis by interfering with t-PA-mediated plasminogen activation [Citation49]. Some studies have suggested that the mechanism by which apo(a) attenuates t-PA–mediated plasminogen activation is through direct competition of apo(a) with plasminogen for binding sites on fibrin or cell surfaces. Smaller apo(a) isoforms have higher affinities for fibrin when compared to larger isoforms, which suggests higher risk of thrombosis with smaller isoforms [Citation50]. Other studies have shown that apo(a) forms a complex with plasminogen, t-PA and fibrin, which leads to the formation of a quaternary complex that significantly reduces plasminogen activation [Citation51]. Increased Lp(a) levels promote plaque rupture and formation of thrombi in vessels. It is also possible that elevated Lp(a) levels promote thrombosis by promoting coagulation [Citation48]. Lp(a) has a prothrombotic effect on platelet activation via various agonists, such as platelet-activating factor and a thrombin-receptor-activating peptide. Lp(a) promotes coagulation by binding to and inhibiting tissue-factor pathway inhibitors [Citation52]. Apo(a) is localized with tissue-factor pathway inhibitor in the intima of human atherosclerotic regions [Citation53]. In the Copenhagen City Heart Study, a role for Lp(a) in coronary thrombosis was reported. There was evidence of a prothrombotic role for Lp(a) in 9330 individuals from a general population who were followed over a period of 10 years. Of these, 498 individuals developed acute MI, with acute MI incidence associated with higher Lp(a) levels [Citation54].
Clinical relevance
Risk stratification
Although LDL is considered a cause of atherosclerotic disease, studies have shown that even in patients with target LDL concentrations, Lp(a) carries an additional risk of CV events [Citation55]. In a substudy of the JUPITER trial, Lp(a) emerged as a significant determinant of residual risk for CV events, despite treatment with rosuvastatin [Citation56]. Furthermore, a recent analysis of two prospective cohorts (Copenhagen City Heart Study; Copenhagen General Population Study) reported that people with the highest levels of Lp(a) are at increased risk of CV events, regardless of their LDL concentrations [Citation57].
Lp(a)-associated risk for morbidity and mortality is mainly determined by the absolute Lp(a) concentration in the serum, whereby individuals with higher serum Lp(a) concentrations are at greater risk for MI, aortic valve stenosis and ischaemic stroke [Citation2, Citation58,Citation59]. Concentrations of Lp(a) above 30 mg/dL in the general population are associated with increased risk for MI, and therefore this value represents a cut-off for an abnormally elevated Lp(a) [Citation2, Citation60]. However, further large-scale observational and genetic studies have shown that clinically relevant Lp(a) concentrations might be higher. Among these studies, the ongoing observational cohort study known as the Copenhagen City Heart Study has shown higher risk for patients with higher Lp(a) concentrations, but without a threshold effect at 30 mg/dL [Citation54]. Although the robust and independent prognostic value of hyperlipoproteinemia(a) is well established, an impact of Lp(a) reduction with respect to CV events remains elusive. However, the very recent study by Madsen et al. has now provided an answer to this question. This study suggested that a reduction in Lp(a) by 50 mg/dL results in 20% lower risk of CVD [Citation61]. As we discuss below in terms of treatments, prospective, large-scale, intention-to-treat studies of Lp(a) therapies are warranted to explore the relationships between Lp(a) reduction and mortality outcomes.
Coronary artery disease
Higher plasma levels of Lp(a) have been linked to increased risk of CVD, and especially of MI, stroke, and aortic valve stenosis due to calcification [Citation46]. This risk is usually increased two-fold for patients who have mostly small apo(a) isoforms (which are smaller due to the lower levels of KIV subtype 2), as this leads to rapid production of, and thus higher levels of, Lp(a). Also, patients who have FH have greater risk of developing CVD [Citation62]. According to the 2019 European Society of Cardiology/European Atherosclerosis Society guidelines, Lp(a) measurements should be considered at least once in the lifetime of each adult, to identify those with very high inherited Lp(a) levels (>180 mg/dL; >430 nmol/L). These people might have a lifetime risk of atherosclerotic CVD that is equivalent to that associated with heterozygous FH [Citation10]. The 2016, the Canadian Cardiovascular Society guidelines for the management of dyslipidemia suggested that Lp(a) might aid risk assessment in subjects at intermediate Framingham risk or with a family history of premature CAD. Particular attention should be given to individuals with Lp(a) levels >30 mg/dL, for whom CVD risk is increased by approximately two-fold [Citation63]. The 2018 American College of Cardiology/American Heart Association guidelines on blood cholesterol defined Lp(a) ≥50 mg/dL, or ≥125 nmol/L, as a risk-enhancing factor; according to their guidelines, this is a relative indication for its measurement with a family history of premature CVD. HEART UK recommended that Lp(a) is only measured in specific cohorts, rather than in all adults, to identify those with a lower Lp(a) threshold than 430 nmol/L. They recommended the management of Lp(a)-associated risk in those with Lp(a) levels >90 nmol/L. They also recommend measuring Lp(a) levels in patients with personal or family history of premature atherosclerotic CVD (<60 years of age), in those with FH or other genetic dyslipidemias (e.g. familial combined hyperlipidaemia, familial dysbetalipoproteinemia, familial hypertriglyceridaemia), in those whose first-degree relative has Lp(a) levels >200 nmol/L, in those who have calcific aortic valve stenosis, and in those with borderline increased (but <15%) 10-year risk of CV events [Citation13]. A single measurement of serum Lp(a) appears sufficient for cardiovascular risk assessment [Citation13, Citation63], as for most patients repeat measurement is only indicated if a secondary cause is suspected (e.g. chronic kidney disease, nephrotic proteinuria, hypothyroidism, liver disease) or if therapeutic measures to lower levels have been introduced [Citation13]. Hyperlipoproteinemia(a) should be considered as a hereditary and quantitative risk factor that can be mitigated by controlling the CV risk factors, and in particular by reduction of Lp(a) levels by treatment [Citation64].
In a Mendelian randomization analysis from five studies, the data showed that the association of genetically predicted Lp(a) levels with the risk of CVD is most likely independent of changes in LDL cholesterol levels, due to genetic variants that link the relationship of statins, PCSK9 inhibitors, and ezetimibe to the risk of CVD. The clinical benefit of lowering Lp(a) levels is proportional to the absolute reduction in Lp(a) levels. An absolute reduction in Lp(a) levels of approximately 100 mg/dL should result in a clinically relevant reduction in the risk of CVD. Such a decrease in Lp(a) represents the same magnitude of CVD risk reduction achieved by lowering LDL cholesterol levels by 38.67 mg/dL [Citation65].
Degenerative aortic valve stenosis
Increased Lp(a) levels are not just a risk factor for CAD, but also for degenerative aortic valve stenosis (DAVS) [Citation59, Citation66,Citation67]. For a long time, DAVS was considered to be associated with age and calcium accumulation. However, more than a decade ago, it was shown unambiguously that this disease is influenced by classical atherosclerotic risk factors, such as smoking, arterial hypertension, diabetes mellitus, and lipid disorders [Citation68]. Among the lipids disorders, Lp(a) was shown to be a powerful predictor of DAVS. In 77,680 patients in the Copenhagen City Heart Study and the Copenhagen General Population Study who were followed for up to 20 years (mean, 5 years), 454 first time DAVS cases were reported. Elevated Lp(a) levels were associated with multivariate adjusted hazard ratio for DAVS [Citation59]. In patients with Lp(a) levels >95th percentile (>90 mg/dL), the risk for DAVS was 2.9-fold higher than in patients in the 22nd percentile (<5 mg/dL). Both homozygote and heterozygote carriers of rs10455872 were at increased risk for DAVS, while this risk was not seen for carriers of rs3798220. A trend towards increased risk was connected with lower number of KIV-2 repeats. In a study by Thanassoulis et al. in 6942 patients, they also showed that DAVS was increased in carriers of rs10455872 across all of the ethnic groups [Citation66]. In a multi-ethnic study of atherosclerosis, cut-off point for subclinical calcific aortic valve disease was shown to be lower in Caucasians (30 mg/dL) than in Black individuals (50 mg/dL) [Citation67]. This is in agreement with a study by Lee et al. [Citation28], who showed that Black individuals have higher Lp(a) levels than White individuals.
Treatment options
Given the unambiguous role of Lp(a) in the pathogenesis of atherosclerosis and the high CV events burden associated with hyperlipoproteinemia(a), there is an unmet need for specific Lp(a)-lowering therapies. Due to the lack of specific targets in Lp(a) metabolism, treatment remains challenging. There are two groups of drugs and interventions that can influence Lp(a). The first group includes the lipid-lowering therapies presented in , while in the second group, there are drugs that have no specific lipid-lowering effects, although they are known to also influence Lp(a) levels ().
Table 1. Overview of studies with lipid-lowering drugs and their effect on lipoprotein(a).
Table 2. Overview of studies with nonlipid-lowering drugs and their effect on lipoprotein(a).
Lipid-lowering therapies
Statins
Statins are the mainstay therapy in primary and secondary prevention of CV diseases, as they effective for the reduction of LDL cholesterol levels and have significant impact on CV mortality [Citation10]. However, their effects on the metabolism of Lp(a) are not completely understood, and there are conflicting opinions about the role of statins in the treatment of hyperlipoproteinemia(a). While some sub-analyses and meta-analyses of statin trials have suggested potential benefits of statin therapies for Lp(a) levels, this observation was not replicated in other studies [Citation56,Citation85–87]. A very recent individual patient meta-analysis that collected data from six randomized controlled trials showed important increases in Lp(a) levels following statin therapies [Citation75]. Among the heterogenous study population of 5256 subjects (after acute coronary syndrome, children with FH, patients with stable CAD, patients with moderate or severe aortic stenosis), for the different statins (atorvastatin, pravastatin, rosuvastatin, pitavastatin), the analysis revealed 8.5–19.6% increases in Lp(a) levels, compared to patients receiving placebo, who showed Lp(a) increases of 0.4–2.3%. Whether the observed increases in Lp(a) after initiation of statin therapy predicts adverse prognostic consequences remains a subject for further research. Nonetheless, elevated Lp(a) levels (>50 mg/dL) have been shown to be an independent predictor of future CV events despite therapies with statins and targeting of LDL cholesterol levels. This observation is in line with the general population, and further supports the important role of Lp(a) levels in the progression of atherosclerosis [Citation56,Citation76].
Ezetimibe
Ezetimibe is another potent LDL-lowering agent, and it has shown subtle, although significant, reductions in Lp(a) levels [Citation77]. However, Lp(a) reduction of 7% is probably too low to affect any outcomes, although ezetimibe might provide additional Lp(a) lowering effects on top of other Lp(a)-lowering therapies. Another lipid-lowering agent, lomitapide, appeared to reduce many lipoprotein fractions, including Lp(a) levels [Citation88]. Unfortunately, the high risk for nonCV side effects that is associated with lomitapide therapy resulted in its withdrawal from the market. This topic is reviewed in detail by van Capelleveen et al. [Citation89].
Fibrates
There are scarce data available on the impact of fibrates on Lp(a) levels. Moreover, the results of studies are conflicting [Citation90]. A meta-analysis that compared the effects of fibrates versus statins on plasma Lp(a) concentrations suggested that fibrates might neutralized the Lp(a)-raising effects of statins [Citation69].
Niacin
Niacin (i.e. vitamin B3) was for a long time the only approved lipid-lowering drug that showed potent reductions in Lp(a) levels [Citation10]. Niacin lowers Lp(a) by silencing apo(a) gene expression in hepatocytes, which encodes glycoprotein apo(a), a crucial element in the Lp(a) structure [Citation91]. The effects of niacin were dose-dependent and led to 25 and 38% decreases in Lp(a) levels with 2 and 4 g niacin daily, respectively [Citation92]. A similar efficacy of niacin was seen in a meta-analysis of extended-release niacin preparations [Citation93]. However, despite large reductions in Lp(a) levels, the treatment effects of niacin have not been mirrored in any overall reduction of CV events [Citation70, Citation72]. Indeed, a randomized double-blind trail of niacin in patients with low HDL cholesterol levels who were receiving intensive statin therapies (i.e. AIM-HIGH) found no differences in a comparison of simvastatin with placebo or with extended-release niacin in patients with established CVD, in terms of the endpoint of CV events [Citation73]. Also, addition of laropiprant to extended-release niacin did not improve the outcomes in the ‘Effects of extended-release niacin with laropiprant in high-risk patients’ (HPS2-THRIVE) study [Citation71]. Another limitation of the wider prescription of niacin is the high prevalence of side-effects. In particular, at the higher doses that are effective in reducing Lp(a) levels, niacin is poorly tolerated, and is often discontinued by patients.
PCSK9 inhibitors
PCSK9 inhibitors are the newest class of lipid-lowering agents that can effectively reduce Lp(a) levels, in addition to LDL cholesterol [Citation94]. Although the exact mechanisms of these effects remain poorly understood, the endpoint of reduction in Lp(a) levels was met in phase II trials, where both alirocumab and evolocumab promoted 25–30% reductions [Citation95,Citation96]. These effects were later confirmed in two large-scale phase III trials with PCSK9 inhibitors. The ‘Further cardiovascular outcomes research with PCSK9 inhibition in subjects with elevated risk’ (FOURIER) trial [Citation78] included 27,564 patients with stable atherosclerotic disease on high-intensity statin therapies, and investigated the effects of evolocumab and placebo on CV events (mean follow-up, 2.2 years). In contrast, the study population in the ‘Alirocumab and cardiovascular outcomes after acute coronary syndrome’ (ODYSSEY Outcome) trial [Citation80] included 18,924 patients in the more acute phase of atherosclerotic disease (i.e. recent prior MI), and investigated whether treatment with alirocumab improved CV outcomes (mean follow-up, 2.8 years). Importantly, in addition to significant reductions in Lp(a) levels, both drugs alirocumab and evolocumab showed trends for potential translation of this surrogate endpoint into improved outcomes [Citation80,Citation81]. A post hoc analysis of the FOURIER trial also revealed an overall 16% risk reduction for major adverse cardiovascular events in patients treated with evolocumab, independent of LDL cholesterol lowering [Citation81]. A secondary analysis of the ODYSSEY Outcome trial showed that changes in Lp(a) levels after alirocumab therapy reduced the risk of major adverse cardiovascular events by 0.6% for each 1 mg/dL reduction in Lp(a) levels, again independent of LDL cholesterol reduction [Citation79]. Both of these analyses indicated more pronounced and clinically meaningful prognostic benefits for patients with higher baseline Lp(a) levels. Although treatment of hyperlipoproteinemia(a) with PCSK9 inhibitors has not yet received any strong guideline recommendation, these drugs appear to be promising candidates for the treatment of the ‘residual’ risk carried by high Lp(a) levels [Citation10]. Encouragingly, and in contrast to niacin, PSCK9 inhibitors were well tolerated by these patients. Nevertheless, it needs to be underscored that current evidence for prognostic benefits of PCKS9 inhibitors has mainly been provided from secondary analyses, and not as the primary objectives of trials. Another gap in our knowledge is the role of PCSK9 inhibitors in primary prevention of patients with raised Lp(a) levels, as both trials assessed patients with established atherosclerotic disease. However, raised Lp(a) levels are associated with increased risk even in the absence of any evident atherosclerotic disease.
Antisense oligonucleotide therapies and RNA-based therapeutics
Lifestyle modifications are unlikely to have any effects on Lp(a) levels because of the primary genetic basis [Citation24]. On the other hand, a strong genetic background has become an interesting therapeutic target. Antisense oligonucleotide therapies are gene-based therapies that use small molecules that are structurally similar to short segments of DNA. These can interface with a specific region of a gene, thus enabling the transcription and translation process of, for instance, apo(a) mRNA. ISIS-APO(a)RX (also known as IONIS-APO(a)-RX) has already been validated in this setting. In a phase I, double-blind, place-controlled clinical trial, IONIS-APO(a)RX showed dose-dependent reductions in Lp(a) levels by up to 78% (at the highest dose of 300 mg) in otherwise healthy volunteers with Lp(a) levels >100 mg/L [Citation97]. In a phase 2 trial, the efficacy of IONIS-APO(a)RX was also confirmed in patients with higher Lp(a) levels [Citation98]. Furthermore, the structurally modified next generation IONIS-APO(a)-RX, known as IONIS-APO(a)-LRX, is more specifically targeted to hepatocytes. Due to this higher specificity, IONIS-APO(a)-LRX achieved the same magnitude of Lp(a) reduction at one-tenth of the dose [Citation98]. Moreover, at the highest administrated dose of 40 mg IONIS-APO(a)-LRX, the reduction in Lp(a) levels was particularly high, at 92%.
A phase 2 trial for AKCEA-APO(a)-LRx (new name for IONIS-APO(a)-LRX) was published very recently [Citation99], the results of which have fulfilled the promises from earlier studies. AKCEA-APO(a)-LRx has been shown to be a safe and feasible drug for patients with established CVD and elevated plasma Lp(a) levels (≥60 mg/dL; 150 nmol/dL) at screening. The reduction in Lp(a) levels was dose-dependent, as 35% for the lowest dose tested (20 mg, every months), and 80% at the highest dose (20 mg, every week). Importantly, no serious adverse events have been reported for these apo(a) antisense therapies, to date. This is in contrast to the use of mipomersen, which is an antisense therapy against apoB that has shown high rates of adverse effects on the liver [Citation100].
Another approach among the nucleic-acid-based therapies relies on small interfering RNAs (siRNAs). Inclisiran is a long-acting siRNA, and it is a representative of this drug class. By binding to PCSK9 mRNA, inclisiran promotes the degradation of this mRNA, which consequently inhibits the translation process for the PCSK9 protein [Citation101]. Although inclisiran was primary developed to target LDL and apoB, it also showed Lp(a)-lowering. In the phase 2 ORION-1 trial, inclisiran reduced Lp(a) by 25.6%, in addition to a 52.5% reduction in LDL [Citation102]. The same efficiency with respect to LDL was confirmed in phase 3 trials (ORION-10, ORION-11) [Citation103], although the effects on Lp(a) remain to be reported. Albeit acting by different mechanisms, both inclisrian and the PCSK9 inhibitors (e.g. evolocumab, arilocumab) affect PCSK9 metabolism. The same target is directly reflected in the comparable Lp(a) reductions obtained, although the long half-life of inclisiran (it was applied twice a year in the ORION-10, ORION-11 trials) might represent an important therapeutic advantage.
Lipoprotein apheresis
A more physical approach to treating hyperlipoproteinemia(a) is lipoprotein apheresis. This is a nonsurgical therapy that removes LDL cholesterol from the blood, and it is primarily indicated for patients with FH with extremely high LDL cholesterol levels that persist despite optimal hypolipemic therapy. The basis of lipoprotein apheresis is thus the elimination of lipoprotein particles, which include Lp(a) [Citation104]. Although this is an invasive procedure, lipoprotein apheresis is generally safe, with most complications related to the puncture site [Citation105]. Moreover, a recent observational multicenter study suggested that lipoprotein apheresis can provide prognostic benefits [Citation74]. An analysis of a large German registry from 2012 to 2016 reported an average of 68% decrease in Lp(a) and 78% reduction in coronary events after 1 year of adjuvant treatment with lipoprotein apheresis [Citation105]. It should be noted, however, that this evidence for prognostic benefit is based on nonrandomized registry data. Stronger evidence would be provided by a controlled multicenter intervention trial to explore the CV benefits of regular treatment with lipoprotein apheresis, compared to patients who receive only conventional lipid-lowering therapies.
Non-lipid lowering therapies and Lp(a)
Aspirin
Lipoprotein(a) contributes to atherosclerosis progression mainly through its pro-inflammatory and prothrombotic effects. Understanding this mechanistic link has provided a rationale for exploration of the potential role of low-dose aspirin as a treatment intervention in hyperlipoproteinemia(a) [Citation25,Citation82]. Although aspirin has shown to promote large reductions in Lp(a) levels, it has not been further tested in outcome trials [Citation83,Citation106]. Despite this lack of robust outcome data, aspirin is still one of the main drugs in preventive cardiology. However, recent trials on aspirin (i.e. ASPREE, ASCEND) have failed to confirm any overall beneficial role of aspirin in primary prevention settings [Citation107,Citation108]. Unfortunately, there remains a lack of evidence for populations with hyperlipoproteinemia(a). Given the thrombogenic nature of Lp(a), aspirin might have a place for people with high Lp(a) also in primary prevention settings.
Hormone-replacement therapy
As in the general population, raised Lp(a) levels are associated with increased CV risk also in women [Citation109]. Sex hormones are known to affect the metabolism of Lp(a), and thus they have become a subject of studies to explore their potential CV protective effects. The use of desogestrel-containing oral contraceptives has a marked lowering effect on lipoprotein levels, which is only seen for non-smoking women [Citation110]. A sub-analysis of the Heart and Oestrogen/Progestin Replacement Study (HERS) showed a mean reduction in Lp(a) levels of 15 mg/dL with oestrogen and progestin therapies. Among 2763 postmenopausal women with CAD, a greater drop was seen for those with high initial of Lp(a) levels, as opposed to those with normal Lp(a) levels [Citation84]. The favourable effects of hormone-replacement therapies on lipid profiles was also shown for women without established CV disease, with a possible impact on CV mortality over 10 years [Citation111]. Despite hormone-replacement therapy being associated with lowering of Lp(a) levels, convincing evidence of the impact of hormone-replacement therapy on CV outcomes is still missing. However, the evidence provided by some surrogate endpoints is promising (i.e. changes in carotid artery intima media thickness, coronary artery calcium) [Citation112,Citation113]. Accordingly, whether hormone-replacement therapy can provide benefits beyond symptomatic relief for high-risk postmenopausal women for reduction of CV mortality remains to be demonstrated definitively.
Conclusions
To summarize, although Lp(a) levels have been known to be a prognostic marker and risk factor for CVD for >50 years, it has only been in the past 10 years that this has gained additional clinical importance, with the emergence of new drugs such as the PCSK9 inhibitors and ISIS-APO(a)RX. Definition of the effective therapeutic strategies for hyperlipoproteinemia(a) remains a particular priority. Although many treatment approaches have been shown to effectively reduced Lp(a) levels, they have failed to improve patient outcome. The most recent results of trials with PCSK9 inhibitors and other up-coming approaches, especially the gene therapies, have revealed promising candidates to start into this new area of management of hyperlipoproteinemia(a). PCSK9 inhibitors can significantly reduce Lp(a) levels and decrease the incidence of acute coronary syndromes and CV death in patients with CAD. Subanalysis of the ODYSSEY study showed that lowering Lp(a) levels using PCSK9 inhibitors can reduce CV events, although only in patients with higher initial Lp(a) levels. Studies that determine the cut-off Lp(a) level for which therapy with PCSK9 inhibitors is optimal are strongly awaited. Furthermore, clinical trials that have investigated the effects of lowered Lp(a) levels only on CV morbidity and mortality are still missing, particularly as PCSK9 inhibitors promote large decreases in LDL cholesterol levels, which still remains the major culprit for the development and progression of atherosclerosis. To answer this question, studies using ISIS-APO(a)RX therapies in patients with LDL cholesterol levels <1.4 mmol/L and high Lp(a) levels are of the utmost importance. These drugs have been shown to be safe for the reduction of Lp(a) levels, although survival studies are definitely awaited. It would also be very interesting to evaluate the effects of combinations of PCSK9 inhibitors and ISIS-APO(a)RX therapy in high risk patients.
| Abbreviations | ||
| apo(a) | = | apolipoprotein(a) |
| apoB | = | apolipoproteinB100 |
| CAD | = | coronary artery disease |
| CV | = | cardiovascular |
| CVD | = | cardiovascular disease |
| DAVS | = | degenerative aortic valve stenosis |
| FH | = | familial hypercholesterolaemia |
| FOURIER | = | Further cardiovascular outcomes research with PCSK9 inhibition in subjects with elevated risk |
| KIV | = | kringle IV |
| LDL | = | low-density lipoprotein |
| Lp(a) | = | lipoprotein(a) |
| MI | = | myocardial infarction |
| ODYSSEY Outcome | = | Alirocumab and cardiovascular outcomes after acute coronary syndrome |
| PCSK9 | = | Proprotein convertase subtilisin/kexin type 9 |
| SNP | = | single nucleotide polymorphism |
| t-PA | = | tissue plasminogen activator |
Acknowledgments
Figures were created using Mind the Graph (https://mindthegraph.com/). The authors acknowledge Chris Berrie for scientific English editing of the manuscript.
Disclosure statement
The authors declare that they have no conflicts of interest regarding this review.
References
- Berg K. A new serum type system in man-the Lp system. Acta Pathol Microbiol Scand. 1963;59:369–382.
- Erqou S, Kaptoge S, Perry PL, et al.; Emerging Risk Factors Collaboration. Lipoprotein(a) concentration and the risk of coronary heart disease, stroke, and nonvascular mortality. JAMA. 2009;302(4):412–423.
- Rawther T, Tabet F. Biology, pathophysiology and current therapies that affect lipoprotein (a) levels. J Mol Cell Cardiol. 2019;131:1–11.
- Julius U, Tselmin S, Schatz U, et al. Lipoprotein(a) and proprotein convertase subtilisin/kexin type 9 inhibitors. Clin Res Cardiol Suppl. 2019;14(Suppl 1):45–50.
- Dangas G, Mehran R, Harpel PC, et al. Lipoprotein(a) and inflammation in human coronary atheroma: association with the severity of clinical presentation. J Am Coll Cardiol. 1998;32(7):2035–2042.
- Maranhão RC, Carvalho PO, Strunz CC, Pileggi F. Lipoprotein (a): structure, pathophysiology and clinical implications. Arq Bras Cardiol. 2014;103(1):76–84.
- Krempler F, Kostner G, Bolzano K, et al. Lipoprotein (a) is not a metabolic product of other lipoproteins containing apolipoprotein B. Biochim Biophys Acta. 1979;575(1):63–70.
- Reyes-Soffer G, Ginsberg HN, Ramakrishnan R. The metabolism of lipoprotein (a): an ever-evolving story. J Lipid Res. 2017;58(9):1756–1764.
- Ma L, Chan DC, Ooi EMM, et al. Fractional turnover of apolipoprotein(a) and apolipoprotein B-100 within plasma lipoprotein(a) particles in statin-treated patients with elevated and normal Lp(a) concentration. Metabolism. 2019;96:8–11.
- Mach F, Baigent C, Catapano AL, et al. ESC/EAS guidelines for the management of dyslipidaemias: lipid modification to reduce cardiovascular risk. Eur Heart J. 2020;41:111–188.
- Marcovina SM, Albers JJ. Lipoprotein (a) measurements for clinical application. J Lipid Res. 2016;57(4):526–537.
- Tsimikas S, Fazio S, Viney NJ, et al. Relationship of lipoprotein(a) molar concentrations and mass according to lipoprotein(a) thresholds and apolipoprotein(a) isoform size. J Clin Lipidol. 2018;12(5):1313–1323.
- Cegla J, Neely RDG, France M, et al. HEART UK consensus statement on lipoprotein(a): a call to action. Atherosclerosis. 2019;291:62–70.
- Wilson DP, Jacobson TA, Jones PH, et al. Use of Lipoprotein(a) in clinical practice: a biomarker whose time has come. A scientific statement from the National Lipid Association. J Clin Lipidol. 2019;13(3):374–392.
- Vuorio A, Watts GF, Schneider WJ, et al. Familial hypercholesterolemia and elevated lipoprotein(a): double heritable risk and new therapeutic opportunities. J Intern Med. 2020;287(1):2–18.
- Tsimikas S. A test in context: Lipoprotein(a): diagnosis, prognosis, controversies, and emerging therapies. J Am Coll Cardiol. 2017;69(6):692–711.
- Alonso R, Andres E, Mata N, et al. Lipoprotein(a) levels in familial hypercholesterolemia: an important predictor of cardiovascular disease independent of the type of LDL receptor mutation. J Am Coll Cardiol. 2014;63(19):1982–1989.
- Sebestjen M, Zegura B, Guzic-Salobir B, et al. Fibrinolytic parameters and insulin resistance in young survivors of myocardial infarction with heterozygous familial hypercholesterolemia. Wien Klin Wochenschr. 2001;113(3-4):113–118.
- Bartens W, Rader DJ, Talley G, et al. Decreased plasma levels of lipoprotein(a) in patients with hypertriglyceridemia. Atherosclerosis. 1994;108(2):149–147.
- Crawford DC, Peng Z, Cheng J-F, et al. LPA and PLG sequence variation and kringle IV-2 copy number in two populations. Hum Hered. 2008;66(4):199–209.
- Boerwinkle E, Leffert CC, Lin J, et al. Apolipoprotein(a) gene accounts for greater than 90% of the variation in plasma lipoprotein(a) concentrations. J Clin Invest. 1992;90(1):52–60.
- Berthold HK, Gouni-Berthold I. Hyperlipoproteinemia(a): clinical significance and treatment options. Atheroscler Suppl. 2013;14(1):1–5.
- Berglund L, Ramakrishnan R. Lipoprotein(a): an elusive cardiovascular risk factor. Arterioscler Thromb Vasc Biol. 2004;24(12):2219–2226.
- Clarke R, Peden JF, Hopewell JC, et al.; PROCARDIS Consortium. Genetic variants associated with Lp(a) lipoprotein level and coronary disease. N Engl J Med. 2009;361(26):2518–2528.
- Chasman DI, Shiffman D, Zee RYL, et al. Polymorphism in the apolipoprotein(a) gene, plasma lipoprotein(a), cardiovascular disease, and low-dose aspirin therapy. Atherosclerosis. 2009;203(2):371–376.
- Kathiresan S, Willer CJ, Peloso GM, et al. Common variants at 30 loci contribute to polygenic dyslipidemia. Nat Genet. 2009;41(1):56–65.
- Ober C, Nord AS, Thompson EE, et al. Genome-wide association study of plasma lipoprotein(a) levels identifies multiple genes on chromosome 6q. J Lipid Res. 2009;50(5):798–806.
- Lee S-R, Prasad A, Choi Y-S, et al. LPA gene, ethnicity, and cardiovascular events. Circulation. 2017;135(3):251–263.
- Saleheen D, Haycock PC, Zhao W, et al. Apolipoprotein(a) isoform size, lipoprotein(a) concentration, and coronary artery disease: a mendelian randomisation analysis. Lancet Diabetes Endocrinol. 2017;5(7):524–533.
- Cobbaert C, Kesteloot H. Serum lipoprotein(a) levels in racially different populations. Am J Epidemiol. 1992;136(4):441–449.
- Sandholzer C, Saha N, Kark JD, et al. Apo(a) isoforms predict risk for coronary heart disease. A study in six populations. Arterioscler Thromb. 1992;12(10):1214–1226.
- Jenner JL, Ordovas JM, Lamon-Fava S, et al. Effects of age, sex, and menopausal status on plasma lipoprotein(a) levels. The Framingham Offspring Study. Circulation. 1993;87(4):1135–1141.
- Singla S, Kaur K, Kaur G, et al. Lipoprotein (a) in type 2 diabetes mellitus: relation to LDL: HDL ratio and glycemic control. Int J Diab Dev Ctries. 2009;29(2):80–84.
- Allen S, Khan S, Tam S. p, et al. Expression of adhesion molecules by lp(a): a potential novel mechanism for its atherogenicity. Faseb J. 1998;12(15):1765–1776.
- Ornek E, Murat S, Duran M, et al. The relationship between lipoprotein(a) and coronary artery disease, as well as its variable nature following myocardial infarction. Clin Invest Med. 2011;34(1):E14–20.
- Argraves KM, Kozarsky KF, Fallon JT, et al. The atherogenic lipoprotein Lp(a) is internalized and degraded in a process mediated by the VLDL receptor. J Clin Invest. 1997;100(9):2170–2181.
- Wu HD, Berglund L, Dimayuga C, et al. High lipoprotein(a) levels and small apolipoprotein(a) sizes are associated with endothelial dysfunction in a multiethnic cohort. J Am Coll Cardiol. 2004;43(10):1828–1833.
- Scipione CA, Sayegh SE, Romagnuolo R, et al. Mechanistic insights into Lp(a)-induced IL-8 expression: a role for oxidized phospholipid modification of apo(a). J Lipid Res. 2015;56(12):2273–2285.
- Wang J, Hu B, Kong L, et al. Native, oxidized lipoprotein(a) and lipoprotein(a) immune complex in patients with active and inactive rheumatoid arthritis: plasma concentrations and relationship to inflammation. Clin Chim Acta. 2008;390(1-2):67–71.
- Borba EF, Santos RD, Bonfa E, et al. Lipoprotein(a) levels in systemic lupus erythematosus. J Rheumatol. 1994;21(2):220–223.
- Enkhmaa B, Anuurad E, Zhang W, et al. HIV disease activity as a modulator of lipoprotein(a) and allele-specific apolipoprotein(a) levels. Arterioscler Thromb Vasc Biol. 2013;33(2):387–392.
- Qi Q, Qi L. Lipoprotein(a) and cardiovascular disease in diabetic patients. Clin Lipidol. 2012;7(4):397–407.
- Qi Q, Workalemahu T, Zhang C, et al. Genetic variants, plasma lipoprotein(a) levels, and risk of cardiovascular morbidity and mortality among two prospective cohorts of type 2 diabetes. Eur Heart J. 2012;33(3):325–334.
- Ramharack R, Barkalow D, Spahr MA. Dominant negative effect of TGF-beta1 and TNF-alpha on basal and IL-6-induced lipoprotein(a) and apolipoprotein(a) mRNA expression in primary monkey hepatocyte cultures. Arterioscler Thromb Vasc Biol. 1998;18(6):984–990.
- Riches K, Porter KE. Lipoprotein(a): cellular effects and molecular mechanisms. Cholesterol. 2012;2012:923289.
- Tada H, Takamura M, Kawashiri M-A. Lipoprotein(a) as an old and new causal risk factor of atherosclerotic cardiovascular disease. J Atheroscler Thromb. 2019;26(7):583–591.
- Hajjar KA, Gavish D, Breslow JL, et al. Lipoprotein(a) modulation of endothelial cell surface fibrinolysis and its potential role in atherosclerosis. Nature. 1989;339(6222):303–305.
- Ellis KL, Boffa MB, Sahebkar A, et al. The renaissance of lipoprotein(a): Brave new world for preventive cardiology? Prog Lipid Res. 2017;68:57–82.
- Sangrar W, Bajzar L, Nesheim ME, et al. Antifibrinolytic effect of recombinant apolipoprotein(a) in vitro is primarily due to attenuation of tPA-mediated Glu-plasminogen activation. Biochemistry. 1995;34(15):5151–5157.
- Xue S, Madison EL, Miles LA. The Kringle V-protease domain is a fibrinogen binding region within Apo(a). Thromb Haemost. 2001;86(5):1229–1237.
- Kang C, Dominguez M, Loyau S, et al. Lp(a) particles mold fibrin-binding properties of apo(a) in size-dependent manner: a study with different-length recombinant apo(a), native Lp(a), and monoclonal antibody. Arterioscler Thromb Vasc Biol. 2002;22(7):1232–1238.
- Tsironis LD, Mitsios JV, Milionis HJ, et al. Effect of lipoprotein (a) on platelet activation induced by platelet-activating factor: role of apolipoprotein (a) and endogenous PAF-acetylhydrolase. Cardiovasc Res. 2004;63(1):130–138.
- Green D, Foiles N, Chan C, et al. Elevated fibrinogen levels and subsequent subclinical atherosclerosis: the CARDIA Study. Atherosclerosis. 2009;202(2):623–631.
- Kamstrup PR, Benn M, Tybjaerg-Hansen A, et al. Extreme lipoprotein(a) levels and risk of myocardial infarction in the general population: the Copenhagen city heart study. Circulation. 2008;117(2):176–184.
- Borén J, Chapman MJ, Krauss RM, Packard CJ, et al. Low-density lipoproteins cause atherosclerotic cardiovascular disease: pathophysiological, genetic, and therapeutic insights: a consensus statement from the European atherosclerosis society consensus panel. Eur Heart J. 2020:ehz962.
- Khera AV, Everett BM, Caulfield MP, et al. Lipoprotein(a) concentrations, rosuvastatin therapy, and residual vascular risk: an analysis from the JUPITER Trial (Justification for the Use of Statins in Prevention: an Intervention Trial Evaluating Rosuvastatin). Circulation. 2014;129(6):635–642.
- Langsted A, Kamstrup PR, Nordestgaard BG. High lipoprotein(a) and high risk of mortality. Eur Heart J. 2019;40(33):2760–2770.
- Kamstrup PR, Tybjaerg-Hansen A, Steffensen R, et al. Genetically elevated lipoprotein(a) and increased risk of myocardial infarction. JAMA. 2009;301(22):2331–2339.
- Kamstrup PR, Tybjaerg-Hansen A, Nordestgaard BG. Elevated lipoprotein(a) and risk of aortic valve stenosis in the general population. J Am Coll Cardiol. 2014;63(5):470–477.
- Kostner GM, Avogaro P, Cazzolato GME, et al. Lipoprotein Lp(a) and the risk for myocardial infarction. Atherosclerosis. 1981;38(1-2):51–61.
- Madsen CM, Kamstrup PR, Langsted A, et al. Lipoprotein(a)-lowering by 50 mg/dL (105 nmol/L) may be needed to reduce cardiovascular disease 20% in secondary prevention: a population-based study. ATVB. 2020;40(1):255–266.
- Gencer B, Kronenberg F, Stroes ES, et al. Lipoprotein(a): the revenant. Eur Heart J. 2017;38(20):1553–1560.
- Anderson TJ, Grégoire J, Pearson GJ, et al. Canadian cardiovascular society guidelines for the management of dyslipidemia for the prevention of cardiovascular disease in the adult. Can J Cardiol. 2016;32(11):1263–1282.
- Ellis KL, Chakraborty A, Moses EK, et al. To test, or not to test: that is the question for the future of lipoprotein(a). Expert Rev Cardiovasc Ther. 2019;17(4):241–250.
- Burgess S, Ference BA, Staley JR, et al.; European Prospective Investigation Into Cancer and Nutrition–Cardiovascular Disease (EPIC-CVD) Consortium. Association of LPA variants with risk of coronary disease and the implications for lipoprotein(a)-lowering therapies: a Mendelian randomization analysis. JAMA Cardiol. 2018;3(7):619–627.
- Thanassoulis G, Campbell CY, Owens DS, et al. Genetic associations with valvular calcification and aortic stenosis. N Engl J Med. 2013;368(6):503–512.
- Cao J, Steffen BT, Budoff M, et al. Lipoprotein(a) levels are associated with subclinical calcific aortic valve disease in white and black individuals: the multi-ethnic study of atherosclerosis. Arterioscler Thromb Vasc Biol. 2016;36(5):1003–1009.
- Rajamannan NM, Evans FJ, Aikawa E, et al. Calcific aortic valve disease: not simply a degenerative process: a review and agenda for research from the National Heart and Lung and Blood Institute Aortic Stenosis Working Group. Executive summary: Calcific aortic valve disease-2011 update. Circulation. 2011;124(16):1783–1791.
- Sahebkar A, Simental-Mendía LE, Watts GF, et al.; Lipid and Blood Pressure Meta-Analysis Collaboration (LBPMC) group. Comparison of the effects of fibrates versus statins on plasma lipoprotein(a) concentrations: a systematic review and meta-analysis of head-to-head randomized controlled trials. BMC Med. 2017;15(1):22.
- Parish S, Hopewell JC, Hill MR, et al. Impact of apolipoprotein(a) isoform size on lipoprotein(a) lowering in the HPS2-THRIVE Study. Circ Genomic Precis Med. 2018;11(2):e001696.
- Landray MJ, Haynes R, Hopewell JC, et al. HPS2-THRIVE collaborative group. Effects of extended-release niacin with laropiprant in high-risk patients. N Engl J Med. 2014;371(3):203–212.
- Albers JJ, Slee A, O'Brien KD, et al. Relationship of apolipoproteins A-1 and B, and lipoprotein(a) to cardiovascular outcomes: the AIM-HIGH trial (Atherothrombosis Intervention in Metabolic Syndrome with Low HDL/High Triglyceride and Impact on Global Health Outcomes). J Am Coll Cardiol. 2013;62(17):1575–1579.
- The AIM-HIGH Investigators. Niacin in patients with low HDL cholesterol levels receiving intensive statin therapy. N Engl J Med. 2011;365(24):2255–2267.
- Leebmann J, Roeseler E, Julius U, et al. Lipoprotein apheresis in patients with maximally tolerated lipid-lowering therapy, lipoprotein(a)-hyperlipoproteinemia, and progressive cardiovascular disease: prospective observational multicenter study. Circulation. 2013;128(24):2567–2576.
- Tsimikas S, Gordts P, Nora C, et al. Statin therapy increases lipoprotein(a) levels. Eur Heart J. 2019;ehz310.
- Willeit P, Ridker PM, Nestel PJ, et al. Baseline and on-statin treatment lipoprotein(a) levels for prediction of cardiovascular events: individual patient-data meta-analysis of statin outcome trials. Lancet Lond Engl. 2018;392(10155):1311–1320.
- Awad K, Mikhailidis DP, Katsiki N, et al.; Lipid and Blood Pressure Meta-Analysis Collaboration (LBPMC) Group. On behalf of Lipid and Blood Pressure Meta-Analysis Collaboration (LBPMC) group. Effect of ezetimibe monotherapy on plasma lipoprotein(a) concentrations in patients with primary hypercholesterolemia: a systematic review and meta-analysis of randomized controlled trials. Drugs. 2018;78(4):453–462.
- Schwartz GG, Steg PG, Szarek M, et al. Alirocumab and cardiovascular outcomes after acute coronary syndrome. N Engl J Med. 2018;379(22):2097–2107.
- Bittner VA, Szarek M, Aylward PE, et al.; ODYSSEY OUTCOMES Committees and Investigators. Effect of alirocumab on lipoprotein(a) and cardiovascular risk after Acute Coronary Syndrome. J Am Coll Cardiol. 2020;75(2):133–144.
- Sabatine MS, Giugliano RP, Keech AC, et al. Evolocumab and clinical outcomes in patients with cardiovascular disease. N Engl J Med. 2017;376(18):1713–1722.
- O’Donoghue ML, Fazio S, Giugliano RP, et al. Lipoprotein(a), PCSK9 inhibition, and cardiovascular risk: Insights from the FOURIER trial. Circulation. 2019;139(12):1483–1492.
- Shiffman D, Chasman DI, Ballantyne CM, et al. Coronary heart disease risk, aspirin use, and apolipoprotein(a) 4399Met allele in the Atherosclerosis Risk in Communities (ARIC) study. Thromb Haemost. 2009;102(1):179–180.
- Akaike M, Azuma H, Kagawa A, et al. Effect of aspirin treatment on serum concentrations of lipoprotein(a) in patients with atherosclerotic diseases. Clin Chem. 2002;48(9):1454–1459.
- Shlipak MG. Estrogen and progestin, lipoprotein(a), and the risk of recurrent coronary heart disease events after menopause. JAMA. 2000;283(14):1845.
- van Wissen S, Smilde TJ, Trip MD, et al. Long term statin treatment reduces lipoprotein(a) concentrations in heterozygous familial hypercholesterolaemia. Heart Br Card Soc. 2003;89(8):893–896.
- Yeang C, Hung M-Y, Byun Y-S, et al. Effect of therapeutic interventions on oxidized phospholipids on apolipoprotein B100 and lipoprotein(a). J Clin Lipidol. 2016;10(3):594–603.
- Takagi H, Umemoto T. Atorvastatin decreases lipoprotein(a): a meta-analysis of randomized trials. Int J Cardiol. 2012;154(2):183–186.
- Rader DJ, Kastelein J. Lomitapide and mipomersen: two first-in-class drugs for reducing low-density lipoprotein cholesterol in patients with homozygous familial hypercholesterolemia. Circulation. 2014;129(9):1022–1032.
- van Capelleveen JC, van der Valk FM, Stroes E. Current therapies for lowering lipoprotein (a). J Lipid Res. 2016;57(9):1612–1618.
- Tziomalos K, Athyros VG, Wierzbicki AS, et al. Lipoprotein a: where are we now? Curr Opin Cardiol. 2009;24(4):351–357.
- Chennamsetty I, Kostner KM, Claudel T, et al. Nicotinic acid inhibits hepatic APOA gene expression: studies in humans and in transgenic mice. J Lipid Res. 2012;53(11):2405–2412.
- Carlson LA, Hamsten A, Asplund A. Pronounced lowering of serum levels of lipoprotein Lp(a) in hyperlipidaemic subjects treated with nicotinic acid. J Intern Med. 1989;226(4):271–276.
- Sahebkar A, Reiner Ž, Simental-Mendía LE, et al. Effect of extended-release niacin on plasma lipoprotein(a) levels: A systematic review and meta-analysis of randomized placebo-controlled trials. Metabolism. 2016;65(11):1664–1678.
- Cao Y-X, Liu H-H, Li S, et al. A meta-analysis of the effect of PCSK9-monoclonal antibodies on circulating lipoprotein (a) Levels. Am J Cardiovasc Drugs. 2019;19(1):87–97.
- Gaudet D, Kereiakes DJ, McKenney JM, Roth EM, et al. Effect of alirocumab, a monoclonal proprotein convertase subtilisin/kexin 9 antibody, on lipoprotein(a) concentrations (a pooled analysis of 150 mg every two weeks dosing from phase 2 trials). Am J Cardiol. 2014;114(5):711–715.
- Raal FJ, Giugliano RP, Sabatine MS, et al. Reduction in lipoprotein(a) with PCSK9 monoclonal antibody evolocumab (AMG 145). J Am Coll Cardiol. 2014;63(13):1278–1288.
- Tsimikas S, Viney NJ, Hughes SG, et al. Antisense therapy targeting apolipoprotein(a): a randomised, double-blind, placebo-controlled phase 1 study. The Lancet. 2015;386(10002):1472–1483.
- Viney NJ, van Capelleveen JC, Geary RS, et al. Antisense oligonucleotides targeting apolipoprotein(a) in people with raised lipoprotein(a): two randomised, double-blind, placebo-controlled, dose-ranging trials. The Lancet. 2016;388(10057):2239–2253.
- Tsimikas S, Karwatowska-Prokopczuk E, Gouni-Berthold I, et al. Lipoprotein(a) reduction in persons with cardiovascular disease. N Engl J Med. 2020;382(3):244–255.
- Raal FJ, Santos RD, Blom DJ, et al. Mipomersen, an apolipoprotein B synthesis inhibitor, for lowering of LDL cholesterol concentrations in patients with homozygous familial hypercholesterolaemia: a randomised, double-blind, placebo-controlled trial. The Lancet. 2010;375(9719):998–1006.
- Gaudet D, Alexander VJ, Baker BF, et al. Antisense inhibition of apolipoprotein C-III in patients with hypertriglyceridemia. N Engl J Med. 2015;373(5):438–447.
- Ray KK, Landmesser U, Leiter LA, et al. Inclisiran in patients at high cardiovascular risk with elevated LDL cholesterol. N Engl J Med. 2017;376(15):1430–1440.
- Ray KK, Wright RS, Kallend D, et al. Two phase 3 trials of inclisiran in patients with elevated LDL cholesterol. N Engl J Med. 2020;382(16):1507–1519.
- Thompson G, Parhofer KG. Current role of lipoprotein apheresis. Curr Atheroscler Rep. 2019;21(7):26.
- Schettler VJJ, Neumann CL, Peter C, Scientific Board of GLAR for, et al. the German Lipoprotein Apheresis Registry (GLAR) - almost 5 years on. Clin Res Cardiol Suppl. 2017;12(Suppl 1):44–49.
- Ranga GS, Kalra OP, Tandon H, et al. Effect of aspirin on lipoprotein(a) in patients with ischemic stroke. J Stroke Cerebrovasc Dis. 2007;16(5):220–224.
- McNeil JJ, Nelson MR, Woods RL, et al.; ASPREE Investigator Group. Effect of aspirin on all-cause mortality in the healthy elderly. N Engl J Med. 2018;379(16):1519–1528.
- The ASCEND Study Collaborative Group. Effects of aspirin for primary prevention in persons with diabetes mellitus. N Engl J Med. 2018;379(16):1529–1539.
- Costello BT, Silverman ER, Doukky R, et al. Lipoprotein(a) and increased cardiovascular risk in women. Clin Cardiol. 2016;39(2):96–102.
- Porkka KV, Erkkola R, Taimela S, et al. Influence of oral contraceptive use on lipoprotein (a) and other coronary heart disease risk factors. Ann Med. 1995;27(2):193–198.
- Suk Danik J, Rifai N, Buring JE, et al. Lipoprotein(a), hormone replacement therapy and risk of future cardiovascular events. J Am Coll Cardiol. 2008;52(2):124–131.
- Harman SM, Black DM, Naftolin F, et al. Arterial imaging outcomes and cardiovascular risk factors in recently menopausal women: A randomized trial. Ann Intern Med. 2014;161(4):249–260.
- Hodis HN, Mack WJ, Henderson VW, et al. Vascular effects of early versus late postmenopausal treatment with estradiol. N Engl J Med. 2016;374(13):1221–1231.