
Abstract
Statins are the first-line choice in Lipid-lowering therapy to reduce cardiovascular risk. In a continuous attempt to optimise treatment success, there is a need for additional research on genes and related molecular pathways that can determine the efficacy and toxicity of lipid-lowering drugs. Several variations within genes associated with lipid metabolism, including those involved in uptake, distribution and metabolism of statins have been reported. The purpose of this study was to evaluate the effect of genetic variations in the key genes responsible for statins’ metabolism and their role in personalised medicine and pharmacogenetic testing (PGx) in patients treated with such drugs. Genetic assessment for specific known SNPs within the most known genes such as ABCG2, SLCO1B1, CYP3A4, and HMGCR, appears likely to predict the efficacy of statin therapy and prevent their side effects but does not necessarily reduce the risk of cardiovascular events.
Hypercholesterolaemia patients show different response to statin therapy.
Several variations within genes associated with statin metabolism have been investigated.
Genetic assessment for specific known SNPs within the most known genes may improve the efficacy of statins treatment and prevent their side effects.
Key Messages
Introduction
In recent years, we have seen significant improvements in lipid-lowering therapy, with several new classes of drugs currently available or being tested in late-phase clinical trials. However, statins are still the most widely used lipid-lowering agents to reduce cardiovascular risk [Citation1]. Statins are prescribed to lower serum LDL-C by controlling the rate-limiting enzyme in cholesterol synthesis i.e. 3-hydroxy-3-methylglutaryl coenzyme A reductase (HMGCR). This process can reduce the production of hepatic cholesterol, upregulate LDLR and increase LDL uptake [Citation2]. The goal of lowering lipids is to reduce LDL-C by 100 mg/dL in high-risk patients and an optional target of less than 40 mg/dL in patients at highest risk [Citation3–5].
Based on current evidence, European Society of Cardiology and the European Atherosclerosis Association (ECS/EAS) recommended the initiation of high-intensity statin therapy during the first two weeks of hospitalisation for the index acute coronary syndrome (ACS) [Citation6,Citation7]. Moreover, pre-treatment (or loading dose for patients already on a statin) with a high-intensity statin should be considered in ACS patients with planned invasive management [Citation8]. Several clinical trials have revealed the effective role of statins in reducing cardiovascular events following lowering LDL-C levels [Citation9,Citation10]. Some medications reduce the effectiveness of statins by reducing bioavailability or increasing the metabolism of statins (e.g. rifampicin) [Citation11]. Moreover, some clinical complications affecting the metabolism of cholesterol, such as hypothyroidism, should be considered [Citation12,Citation13]. Besides these factors, several variations within genes associated with lipid metabolism such as genes involved in uptake, distribution and metabolism can exert their influence [Citation14].
According to the previous reports, one of the side effects of statin treatment is myopathy that may result in treatment discontinuation [Citation15]. There are several risk factors for statin-induced myopathy that are related to patients’ characteristics such as age and genetics, and also drug-related transporter systems [Citation16].
Statin therapy is limited in case of skeletal muscle toxicity which can be correlated with elevated systemic drug exposure. Up to 10% of statin-treated patients will experience muscle pain or weakness [Citation17,Citation18] and life-threatening rhabdomyolysis [Citation19], which tend to occur several months to years after statin initiation.
In addition, LDL-C remains above the target still in a considerable proportion of the treated patients (>40%) [Citation20]. Barriers to attain LDL-C goals include failure to initiate therapy, non-adherence, side effects, inappropriate drug/dose selection, and inadequate dose titration. Although several algorithms for dose selection have been introduced to minimise dose titration, they are not widely used, resulting in over 20% of patients with LDL-C still remaining above the target value [Citation21]. Some studies have shown that statin therapy may change the course of clinical events without notable decrease in LDL-C [Citation22,Citation23]. In a study it has been showed significantly greater benefit from intensive statin therapy in carriers of rs20455 (Trp719Arg) than non-carriers which appears to be due to a mechanism distinct from lipid or CRP lowering [Citation22]. This highlights the importance of pharmacogenomics and pharmacogenetics in predicting the therapeutic outcome. Additional research on genes, related molecular pathways, and their impact on lipid-lowering drugs would pave the road for more effective treatments [Citation24]. Therefore, the purpose of this study was to evaluate the effect of genetic variation in the key genes responsible for statins metabolism () and their role in personalised medicine and pharmacogenetic testing (PGx) in patients treated with these drugs.
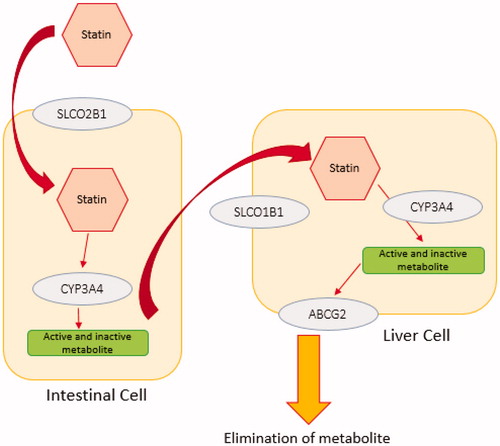
Figure 1. The simple schematic pathway for statin transporters. Statins undergo passive intestinal absorption and subsequently are taken up from the blood stream into the liver by members of solute carrier transporter family (SLCO1B1, SLCO1B3, SLCO2B1). Statins are metabolised by phase I and II enzymes and eliminated via efflux transporters mediated biliary excretion. Different enzymes are involved in phase I statin metabolism such as CYP3A4. Elimination of statins is carried out by members of the efflux family transporters ABCB1, ABCC2, ABCG2 or ABCB11. Statin metabolism and elimination take place primarily in the liver and to a lesser extent in the kidney.
Genetic variants and statin efficacy
Although many of the new guidelines for cardiovascular treatments no longer specify lipid targets, many clinicians have used lipid-lowering responses to monitor treatment efficacy and therefore optimise the statin dose. In addition to various constitutional or clinical factors (e.g. race, gender, ethnicity, and side effects) that affect statins function, genetic background can also play an important role in lipid reduction success. Increasing the dose in patients who do not show a significant decrease in lipids may also increase the risk of statin toxicity [Citation24].
Hundreds of candidate gene studies and several genome-wide association studies (GWASs) have focussed on the influence of genetic variants on statin pharmacokinetics (e.g. drug and metabolite levels in blood, area under the time–concentration curve [AUC]) and statin pharmacodynamics (lipid lowering, incidence of adverse events, incidence of cardiovascular events). Discussing relevant clinical research, this contemporary review focuses on polymorphisms in several key genes that affect statin pharmacokinetics (e.g. transporters and metabolising enzymes), statin efficacy (e.g. drug targets and pathways), and end-organ toxicity (e.g. myopathy pathways) [Citation17,Citation25]. Some of key genes that affect statin pharmacokinetics include ABCG2, CYP3A4, HMGCR and SLCO1B1 are discussed in this review.
ABCG2
ATP-binding cassette G2 protein (ABCG2, also known as breast cancer resistance protein) is an efflux transporter which is expressed in a number of normal tissues such as brain, heart, colon, small intestine, kidney, liver, placenta, mammary gland and stem cells as well as in cancer cells [Citation26]. ABCG2 is a mediator of the cellular efflux of a wide range of xenobiotics including antibiotics, chemotherapeutic agent, and dietary toxins as well as endogenous compounds such as oestrogens and oestrogen conjugates. It seems that ABCG2 protects normal cells against cytotoxic materials; however, it is also a tumour resistance inducer to several anticancer drugs [Citation27]. ABCG2 gene encodes the efflux transporter BCRP (Breast Cancer Resistance Protein) which is expressed on the canicular membrane of hepatocytes and in enterocytes constitutively. Rosuvastatin is actively pumped from the portal blood into the enterocytes and is thus resulting in faecal excretion of the drug. Similarly, changes in BCRP activity affects the amount of rosuvastatin circulating in the blood. It has been shown that ABCG2c.421C>A genetic variant leads to increased plasma concentrations of simvastatin [Citation28] which is associated with increased risk of simvastatin induced myopathy (SIM) [Citation29].
Absorption of ABCG2 substrates from the gut, their entry through the blood–brain barrier and through the placenta into the foetus is limited by the ABCG2 action. Moreover, by increasing the excretion of its substrates into the bile and urine, systemic exposure to many drugs is reduced potentially [Citation30]. Rosuvastatin is a known substrate for ABCG2. One of the best-characterized sequence variations in the ABCG2 gene is c.421C>A SNP (rs2231142), resulting in an amino acid change p. Gln141Lys. In vitro studies showed that the c.421A variant has been associated with reduced ABCG2 transport activity with respect to estrone-sulfate, methotrexate, mitoxantrone, and topotecan [Citation31]. Some studies have suggested that this SNPresults in reduced expression ofABCG2 [Citation32,Citation33]. Although other studies have suggested that it decreases the ATPase activity of ABCG2 protein [Citation30,Citation34] while the p.Gln141Lys variant is located within the cell apart from the ATP-binding region. It has been found that in humans in vivo, the c.421A allele is associated with an increase in plasma concentrations of topotecan, diflomotecan, and sulfasalazine, as compared with the c.421C allele [Citation35,Citation36]. The ABCG2 c.421C>A SNP affects the pharmacokinetics of atorvastatin and rosuvastatin significantly, thus this indicates that ABCG2 plays an important role in limiting the absorption of these statins in the gut. This effect was shown for rosuvastatin than in atorvastatin, suggesting different impact of rs2231142 on the pharmacokinetics of different statins. ABCG2 genotyping, along with other genetic markers such as the SLCO1B1 c.521T>C SNP, might be used to personalise selection of a statin and related dose [Citation28].
CYP3A4
It has been known that hepatic isoenzymeCYP3A4 is metabolising simvastatin, and to a lesser extent, atorvastatin [Citation37]. Inhibitors or inducers of this enzyme affect the plasma concentration of these statins, then results either in an increased risk of side effects such as myopathy and rhabdomyolysis or a reduction in the efficacy of treatment [Citation38]. The primary metabolic pathway for statins such as lovastatin, simvastatin and atorvastatin is CYP3A4. It has been shown that CYP3A4 activity can be varied up to 10-fold among groups of affected individuals which may result from variations in genes encoding for these enzymes [Citation39]. A study reported association of CYP3A4 promoter variant (rs2740574) with higher LDL-C levels after treatment in 340 hypercholesterolaemia patients treated with atorvastatin 10 mg/day, though carriers of the missense variant (rs4986910) revealed an enhanced efficacy response and lower LDL-C levels in comparison with the wild-type allele [Citation39]. rs2740574- the promoter variant- alters efficiency of transcription with high CYP3A4 activity resulting in rapid drug metabolism and subsequent lower plasma concentration of atorvastatin [Citation40]. It is reported that individuals with wild‐type genotype for rs2740574 variant, had a significantly greater reduction in LDL-C as compared to the variant allele (G allele). Though, they found no association between rs4986910 variant and response to atorvastatin [Citation41]. Moreover, another study on Indian population demonstrated no significant association between rs2740574 and lower LDL‐C levels in response to atorvastatin [Citation42]. Despite the significant pharmacokinetic interactions between inhibitors and inducers of CYP3A4 and lovastatin/simvastatin, their clinical relevance appears to be limited, especially at lower statin doses. However, combination of statins with fibrates, mainly gemfibrozil considerably increases the potential for muscular toxicity [Citation43].
HMGCR
As above described, statins inhibit the rate-limiting enzyme in cholesterol synthesis – namely, HMGCR [Citation2]. There are wide inter-individual differences in response to statin therapy and serum lipid levels [Citation44]. Despite the undeniable environmental impact, variations observed in serum lipid levels in different individuals are primarily described by genetic variability [Citation45,Citation46]. Although recent evidence suggests that HMGCR genotypes elucidated less than 2% of the variance in statin response, alternative splicing of HMGCRcan explain6 to 15% of its variance [Citation47,Citation48]. Alternative splicing of HMGCR pre-mRNA on exon 13, produces two transcripts as full-length (FL) HMGCR and Δ13 HMGCR which is related to rs3846662 [Citation49]. Exon 13 encodes a part of the catalytic/statin-binding domain of the HMGCR [Citation50,Citation51]. It is likely that rs3846662 modifies the binding motif of heterozygous nuclear ribonucleoprotein A1 (HNRNPA1) which regulates alternative splicing of HMGCR [Citation52]. According to these findings, it is suggested that an increase in the amount of Δ13 HMGCR mRNA in individuals carrying the rs3846662 A allele [Citation53], results in probable lower activity in HMGCR [Citation53], as well as lower levels of baseline LDL-C and reduced sensitivity and response to the statin inhibition [Citation48,Citation54]. A cohort of familial hypercholesterolaemia patients indicated that alternative splicing of HMGCR would explain 22 to 55% of the variance in statin response. A Study showed that rs3846662 and the alternative splicing of HMGCR mRNA have a significant effect on women’s response to statin therapy [Citation55].
LDLR
The presence and type of LDLR mutations influence response to lipid-lowering therapy. Numerous randomised trials have demonstrated that therapies that lower LDL-C levels by reducing LDL particles through upregulation of the LDL receptor (LDLR) reduce the risk of cardiovascular events [Citation56,Citation57]. There is recent evidence that the 3′-untranslated region [3-UTR] of LDLR is required for berberine-mediated LDLC reduction through a mechanism that increases LDLR mRNA stability. Single-nucleotide polymorphisms (SNPs) in the 3-UTR have been associated with in vivo LDLC levels, as well as in vitro LDLR mRNA stability [Citation58], and variation in this region may also influence statin-mediated lipid reduction [Citation59]. It has been identified that some variants within the LDLR gene areassociated with attenuated lipid-lowering response to simvastatin treatment. Response was further reduced in individuals with both LDLR and previously described HMGCR haplotypes [Citation60]. Numerous variants through the LPL pathway and current therapies that lower LDL-C levels through the LDLR pathway were also associated with similar lower CHD risk per unit lower plasma ApoB levels [Citation61].
SLCO1B1
The SLCO1B1 gene encodes a protein called organic anion transporting polypeptide 1B1, or OATP1B1 which is found in liver cells. OATP1B1 transports bilirubin, certain hormones, toxins, and drugs into the liver for removal from the body [Citation62]. Several drugs are normally transported by the OATP1B1 protein including statins, heart disease medications, certain antibiotics and some cancer therapy drugs [Citation63,Citation64]. Among statins, rosuvastatin is an inhibitor of 3-hydroxymethylglutaryl co-enzyme A reductase which Inhibits 3-hydroxymethylglutaryl co-enzyme A reductase, and therefore leads to reduced production of mevalonic acid by hepatocytes. This results in reduction of total circulating levels of cholesterol and LDL-C in the blood. One of the most commonly used drugs in the treatment of hypercholesterolaemia, hypertriglyceridaemia, hyperlipidaemia, mixed dyslipidemia and homozygous familial hypercholesterolaemia is rosuvastatin [Citation65]. Additionally, rosuvastatin and pitavastatin have been proposed as probe substrates for OATP1B. Prueksaritanont et al. showed relative sensitivity and selectivity to OATP1B inhibitors. They recommended pitavastatin over rosuvastatin as a more sensitive and selective clinical OATP1B probe [Citation66].
The most comprehensively studied example is the effect of the missense variant rs4149056 in SLCO1B1 (SLCO1B1*5 and part of SLCO1B1*15) that results in elevated statin plasma concentrations due to impaired hepatic clearance [Citation67,Citation68] and, in the case of simvastatin use, this increased exposure has convincingly been associated with increased risks of developing myopathy [Citation69]. Based on these and other similar reports, important informationon SLCO allele frequency with putative clinical relevancehave been provided [Citation70–73]. The pharmacogenetics of rosuvastatin has been influenced by SLCO1B1c.521T>C (rs4149056, p. Val174Ala) and ABCG2c.421C>A (rs2231142, p. Gln141Lys) genetic variants. OATP1B1- encoded by SLCO1B1- is responsible for the uptake of rosuvastatin by hepatocytes and from the intestinal lumen by enterocytes. Consequently, each polymorphism that alters OATP1B1 activity affect plasma levels of rosuvastatin. The SLCO1B1 c.521T>C SNP has been shown to reduce OATP1B1 transporter activity [Citation74] and therefore, elevatesrosuvastatin concentrations/exposure in plasma which leads to an increased risk of statin-inducedmyopathy [Citation67].
Statins structure and types
Statins are made in two ways, either of fungal-derived or synthetically products. Fungal-derived statins includes lovastatin, pravastatin, and simvastatin andsynthetic compounds consists of atorvastatin, cerivastatin, fluvastatin, pravastatin, pitavastatin, and rosuvastatin [Citation75,Citation76]. There are functional differences between natural and synthetic statins that depend on their ability to interact and inhibit HMG-CoA reductase and their hydrophobicity. Due to the structural properties of statins, synthetic forms are more likely to interact with HMG-CoA reductase. Atorvastatin and rosuvastatin are more likely to interact with hydrogen. In addition, rosuvastatin also has a polar interaction between the methane sulphonamide group and the HMG-CoA reductase enzyme. Therefore, rosuvastatin has the greatest effect on reducing HMG-CoA reductase activity by up to 50% [Citation76,Citation77]. Due to the polar hydroxyl group and methane sulphonamide, pravastatin and rosuvastatin are hydrophobic. Lovastatin, fluvastatin, simvastatin and atorvastatin are relatively lipophilic compounds. Lipophilic statins (except pitavastatin and cerivastatin) are less bioavailable compounds because of their first-pass effect on the liver surface [Citation77,Citation78]. Side effects, such as muscle toxicity, have been reported due to the penetration of lipophilic statins into the extrahepatic tissue. But hydrophilic statins are excluded from the extracellular tissue because of the need for active transport mechanisms in the liver. However, there is a balance between the favourable and side effects of lipophilic and hydrophobic statins [Citation77].
As mentioned previously, a common nonsynonymous variant in the drug transporter SLCO1B1 (rs4149056) has beenassociated with a 4-fold increase in the risk of myopathy among patients with high dose simvastatin usage at genome-wide levels of statistical significance [Citation69]. This finding has been widely proved in several studies [Citation63,Citation79,Citation80]. Attempts to find additional common variants related to statin-related muscle injury have failed to yield replicable results [Citation81,Citation82]. A recent study conducted whole-exome sequencing on 88 Czech patients with mild statin-associated muscle toxicity, demonstrated an increased burden of rare variants in 24 genes [Citation83].
Individual polymorphic forms of the same drug substance may differ in their physical properties such as chemical reactivity, solubility and dissolution rate, stability, melting and sublimation temperature, density, hardness, adsorption, hygroscopicity and refractive index. It is particularly crucial for statins, which arepoorly soluble and have low bioavailability. The low total bioavailability of statins creates the need for new polymorphic forms that will increase the therapeutic effect and reduce the dose of the drug taken by the patient. Based on the available scientific reports, it can be concluded that amorphous forms of statins create the possibility of increasing the solubility and bioavailability of this group of drugs, which in turn is an opportunity to increase theireffectiveness in the treatment of cardiovascular diseases [Citation84].
Statins and personalised medicine
Personalised medicine in cardiovascular diseases hasadvancedrapidly, with PGx applications accessible for both researchers and clinicians. This may help selection, dosing and monitoring of drugs [Citation85,Citation86]. It seems that the variations in genes involved in statins metabolic pathways may influence the therapeutic effectiveness of these drugs (). Genetic assessment for specific known SNPs appears likely to improve the efficacy of statins treatment and prevent their side effects but does not necessarily predict a reduction in the risk of cardiovascular events by statins [Citation94]. In addition, PGx test can be performed as a valuable tool for clinicians to monitor the treatment process of patients with statin therapy. Catalan and LeLorier [Citation95] showed that only 33% of patients still adhere to statin therapy after 1 year. Another study by Dorais et al. [Citation96], showed that among the 19,727 statin-treated patients, 53.3% stopped taking the drug after 1 year.
Table 1. The most known genetic variants in statins pharmacogenetics reported on ClinVar/PharmKG.
It is likely that pharmacogenetic testing will become a routine part of clinical practice in the near future. Notably, the rapid progress in technology and increased application of NGS techniques have allowed a paradigm shift from candidate variant researches towards comprehensive analyses of genomic makeup. Personalised medicine is still in its infancy; however, there are challenges such as ethical, behavioural, methodological, technical and financial issues in providing an available service for all patients around the world based on causal variants at associated loci.
According to the results of clinical trials as well assystematic reviews and well-powered GWAS, variants in several genes such as APOE, LPA, ABCG2, SLCO1B1 have been confirmed to affect LDL-C response to statins and are likely to have a limited effect on clinical benefit. Although there are various reports of association of different SNPs affecting only clinical benefit of statin therapy, such claims mostly rely on questionable evidence. Genetic variation, without a doubt, can affect statin response; however, due to the small effect sizes of these variants, using such information in clinical decision-making should be made with caution.The consideration of interaction between gene and environment and the application of “omics” data in PGx studies will assist the reliable outcomes and new strategies of pharmacogenomics research and development.
Disclosure statement
The authors declare that they have no competing interests.
References
- Alfonsi JE, Hegele RA, Gryn SE. Pharmacogenetics of lipid-lowering agents: precision or indecision medicine? Curr Atheroscler Rep. 2016;18(5):24.
- Goldstein JL, Brown MS. Regulation of the mevalonate pathway. Nature. 1990;343(6257):425–430.
- Grundy SM, Cleeman JI, Merz CNB, et al. Implications of recent clinical trials for the national cholesterol education program adult treatment panel III guidelines. J Am Coll Cardiol. 2004;44(3):720–732.
- Expert Panel on Detection E. Executive summary of the third report of the National Cholesterol Education Program (NCEP) expert panel on detection, evaluation, and treatment of high blood cholesterol in adults (Adult Treatment Panel III). JAMA. 2001;285(19):2486.
- Mach F, Baigent C, Catapano AL, et al.; ESC Scientific Document Group. 2019 ESC/EAS Guidelines for the management of dyslipidaemias: lipid modification to reduce cardiovascular risk: The Task Force for the management of dyslipidaemias of the European Society of Cardiology (ESC) and European Atherosclerosis Society (EAS). Eur Heart J. 2020;41(1):111–188.
- de Lemos JA, Blazing MA, Wiviott SD, et al.; Investigators. Early intensive vs a delayed conservative simvastatin strategy in patients with acute coronary syndromes: phase Z of the A to Z trial. JAMA. 2004;292(11):1307–1316.
- Schwartz G. Myocardial Ischemia Reduction with Aggressive Cholesterol Lowering (MIRACL) Study Investigators. Effects of atorvastatin on early recurrent ischemic events in acute coronary syundromes; the MIRACL study; a randomized controlled trial. JAMA. 2001;285(13):1711–1718.
- Pourhosseini H, Lashkari R, Aminorroaya A, et al. Effects of high dose atorvastatin before elective percutaneous coronary intervention on highly sensitive troponin T and one year major cardiovascular events; a randomized clinical trial. Int J Cardiol Heart Vasc. 2019;22:96–101.
- Mangravite L, Thorn C, Krauss R. Clinical implications of pharmacogenomics of statin treatment. Pharmacogenomics J. 2006;6(6):360–374.
- Simon JA, Lin F, Hulley SB, et al. Phenotypic predictors of response to simvastatin therapy among African-Americans and Caucasians: the Cholesterol and Pharmacogenetics (CAP) Study. The Am J Cardiol. 2006;97(6):843–850.
- Sahebkar A, Serban M-C, Gluba-Brzozka A, et al. Lipid-modifying effects of nutraceuticals: an evidence-based approach. Nutrition. 2016;32(11-12):1179–1192.
- Catapano AL, Graham I, De Backer G, et al.; Authors/Task Force Members. 2016 ESC/EAS guidelines for the management of dyslipidaemias: the task force for the management of dyslipidaemias of the European Society of Cardiology (ESC) and European Atherosclerosis Society (EAS) developed with the special contribution of the European Assocciation for Cardiovascular Prevention & Rehabilitation (EACPR)). Atherosclerosis. 2016;253:281–344.
- Bates T, Connaughton V, Watts G. Non-adherence to statin therapy: a major challenge for preventive cardiology. Expert Opin Pharmacother. 2009;10(18):2973–2985.
- Schmitz G, Langmann T. Pharmacogenomics of cholesterol-lowering therapy. Vascul Pharmacol. 2006;44(2):75–89.
- Mitchell D, Guertin JR, Iliza AC, et al. Economic evaluation of a pharmacogenomics test for statin-induced myopathy in cardiovascular high-risk patients initiating a statin. Mol Diagn Ther. 2017;21(1):95–105.
- Abd TT, Jacobson TA. Statin-induced myopathy: a review and update. Expert Opin Drug Saf. 2011;10(3):373–387.
- Bruckert E, Hayem G, Dejager S, et al. Mild to moderate muscular symptoms with high-dosage statin therapy in hyperlipidemic patients—the PRIMO study. Cardiovasc Drugs Ther. 2005;19(6):403–414.
- Joy TR, Hegele RA. Narrative review: statin-related myopathy. Ann Intern Med. 2009;150(12):858–868.
- Nichols GA, Koro CE. Does statin therapy initiation increase the risk for myopathy? An observational study of 32,225 diabetic and nondiabetic patients. Clin Ther. 2007;29(8):1761–1770.
- Yan AT, Yan RT, Tan M, et al. Contemporary management of dyslipidemia in high-risk patients: targets still not met. Am J Med. 2006;119(8):676–683.
- Martineau P, Gaw A, De Teresa E, et al. Effect of individualizing starting doses of a statin according to baseline LDL-cholesterol levels on achieving cholesterol targets: the achieve cholesterol targets fast with atorvastatin stratified titration (ACTFAST) study. Atherosclerosis. 2007;191(1):135–146.
- Iakoubova OA, Sabatine MS, Rowland CM, et al. Polymorphism in KIF6 gene and benefit from statins after acute coronary syndromes: results from the PROVE IT-TIMI 22 study. J Am Coll Cardiol. 2008;51(4):449–455.
- Assimes TL, Hólm H, Kathiresan S, et al. Lack of association between the trp719Arg polymorphism in kinesin-like protein-6 and coronary artery disease in 19 case-control studies. J Am Coll Cardiol. 2011;56(4):155–263.
- Kitzmiller JP, Mikulik EB, Dauki AM, et al. Pharmacogenomics of statins: understanding susceptibility to adverse effects. Pharmgenomics Pers Med. 2016;9:97–106.
- de Denus S, Spinler SA, Miller K, Peterson AM. Statins and liver toxicity: a meta-analysis. Pharmacotherapy J Hum Pharmacol Drug Therapy. 2004;24(5):584–591.
- Gradhand U, Kim RB. Pharmacogenomics of MRP transporters (ABCC1-5) and BCRP (ABCG2). Drug Metab Rev. 2008;40(2):317–354.
- Sparreboom A, Loos WJ, Burger H, et al. Effect of ABCG2 genotype on the oral bioavailability of topotecan. Cancer Biol Ther. 2005;4(6):650–653.
- Keskitalo J, Zolk O, Fromm M, et al. ABCG2 polymorphism markedly affects the pharmacokinetics of atorvastatin and rosuvastatin. Clin Pharmacol Ther. 2009;86(2):197–203.
- DeGorter MK, Tirona RG, Schwarz UI, et al. Clinical and pharmacogenetic predictors of circulating atorvastatin and rosuvastatin concentrations in routine clinical care. Circ Cardiovasc Genet. 2013;6(4):400–408.
- Morisaki K, Robey RW, Özvegy-Laczka C, et al. Single nucleotide polymorphisms modify the transporter activity of ABCG2. Cancer Chemother Pharmacol. 2005;56(2):161–172.
- Kondo C, Suzuki H, Itoda M, et al. Functional analysis of SNPs variants of BCRP/ABCG2. Pharm Res. 2004;21(10):1895–1903.
- Imai Y, Nakane M, Kage K, et al. C421A polymorphism in the human breast cancer resistance protein gene is associated with low expression of Q141K protein and low-level drug resistance 1 supported in part by grants from the ministry of education, culture, sports, science and technology, the ministry of health, labour and welfare, Japan, and the virtual research institute of aging of Nippon Boehringer Ingelheim. Mol Cancer Therapeutics. 2002;1(8):611–616.
- Kobayashi D, Ieiri I, Hirota T, et al. Functional assessment of ABCG2 (BCRP) gene polymorphisms to protein expression in human placenta. Drug Metab Dispos. 2005;33(1):94–101.
- Mizuarai S, Aozasa N, Kotani H. Single nucleotide polymorphisms result in impaired membrane localization and reduced atpase activity in multidrug transporter ABCG2. Int J Cancer. 2004;109(2):238–246.
- Sparreboom A, Gelderblom H, Marsh S, et al. Diflomotecan pharmacokinetics in relation to ABCG2 421C>A genotype. Clin Pharmacol Ther. 2004;76(1):38–44.
- Yamasaki Y, Ieiri I, Kusuhara H, et al. Pharmacogenetic characterization of sulfasalazine disposition based on NAT2 and ABCG2 (BCRP) gene polymorphisms in humans. Clin Pharmacol Ther. 2008;84(1):95–103.
- Baxter K, Preston C. Stockley’s drug interactions. London: Pharmaceutical Press
- Voora D, Shah SH, Reed CR, et al. Pharmacogenetic predictors of statin-mediated low-density lipoprotein cholesterol reduction and dose response. Circ Cardiovasc Genet. 2008;1(2):100–106.
- Kajinami K, Takekoshi N, Brousseau ME, et al. Pharmacogenetics of HMG-CoA reductase inhibitors: exploring the potential for genotype-based individualization of coronary heart disease management. Atherosclerosis. 2004;177(2):219–234.
- Kajinami K, Brousseau ME, Ordovas JM, et al. CYP3A4 genotypes and plasma lipoprotein levels before and after treatment with atorvastatin in primary hypercholesterolemia. Am J Cardiol. 2004;93(1):104–107.
- Kadam P, Ashavaid TF, Ponde CK, et al. Genetic determinants of lipid-lowering response to atorvastatin therapy in an Indian population. J Clin Pharm Ther. 2016;41(3):329–333.
- Poduri A, Khullar M, Bahl A, et al. Common variants of HMGCR, CETP, APOAI, ABCB1, CYP3A4, and CYP7A1 genes as predictors of lipid-lowering response to atorvastatin therapy. DNA and Cell Biology. 2010;29(10):629–637.
- Tirkkonen T, Ryynänen A, Vahlberg T, et al. Frequency and clinical relevance of drug interactions with lovastatin and simvastatin: an observational database study. Drug Saf. 2008;31(3):231–240.
- Jermendy G, Horváth T, Littvay L, et al. Effect of genetic and environmental influences on cardiometabolic risk factors: a twin study. Cardiovasc Diabetol. 2011;10(1):96.
- Elder SJ, Lichtenstein AH, Pittas AG, et al. Genetic and environmental influences on factors associated with cardiovascular disease and the metabolic syndrome. J Lipid Res. 2009;50(9):1917–1926.
- Li S, Duan H, Pang Z, et al. Heritability of eleven metabolic phenotypes in Danish and Chinese twins: A cross-population comparison. Obesity (Silver Spring)). 2013;21(9):1908–1914.
- Medina MW, Krauss RM. Alternative splicing in the regulation of cholesterol homeostasis. Curr Opin Lipidol. 2013;24(2):147–152.
- Medina MW, Krauss RM. The role of HMGCR alternative splicing in statin efficacy. Trends Cardiovasc Med. 2009;19(5):173–177.
- Johnson JM, Castle J, Garrett-Engele P, et al. Genome-wide survey of human alternative pre-mRNA splicing with exon junction microarrays. Science. 2003;302(5653):2141–2144.
- Istvan ES, Palnitkar M, Buchanan SK, et al. Crystal structure of the catalytic portion of human HMG-CoA reductase: insights into regulation of activity and catalysis. Embo J. 2000;19(5):819–830.
- Istvan ES, Deisenhofer J. Structural mechanism for statin inhibition of HMG-CoA reductase. Science. 2001;292(5519):1160–1164.
- Yu C-Y, Theusch E, Lo K, et al. HNRNPA1 regulates HMGCR alternative splicing and modulates cellular cholesterol metabolism. Hum Mol Genet. 2014;23(2):319–332.
- Burkhardt R, Kenny EE, Lowe JK, et al. Common SNPs in HMGCR in micronesians and whites associated with LDL-cholesterol levels affect alternative splicing of exon13. Arterioscler Thromb Vasc Biol. 2008;28(11):2078–2084.
- Chung JY, Cho SK, Oh ES, et al. Effect of HMGCR variant alleles on low-density lipoprotein cholesterol—lowering response to atorvastatin in healthy Korean subjects. J Clin Pharmacol. 2012;52(3):339–346.
- Leduc V, Bourque L, Poirier J, et al. Role of rs3846662 and HMGCR alternative splicing in statin efficacy and baseline lipid levels in familial hypercholesterolemia. Pharmacogenet Genomics. 2016;26(1):1.
- Catapano AL, Graham I, De Backer G, et al.; ESC Scientific Document Group. 2016 ESC/EAS guidelines for the management of dyslipidaemias. Eur Heart J. 2016;37(39):2999–3058.
- Silverman MG, Ference BA, Im K, et al. Association between lowering LDL-C and cardiovascular risk reduction among different therapeutic interventions: a systematic review and meta-analysis. Jama. 2016;316(12):1289–1297.
- Muallem H, North KE, Kakoki M, et al. Quantitative effects of common genetic variations in the 3′ UTR of the human LDL-receptor gene and their associations with plasma lipid levels in the Atherosclerosis Risk in Communities study. Hum Genet. 2007;121(3-4):421–431.
- Polisecki E, Muallem H, Maeda N, et al.; Prospective Study of Pravastatin in the Elderly at Risk (PROSPER) Investigators. Genetic variation at the LDL receptor and HMG-CoA reductase gene loci, lipid levels, statin response, and cardiovascular disease incidence in PROSPER. Atherosclerosis. 2008;200(1):109–114.
- Mangravite LM, Medina MW, Cui J, et al. Combined influence of LDLR and HMGCR sequence variation on lipid-lowering response to simvastatin. Arterioscler Thromb Vasc Biol. 2010;30(7):1485–1492.
- Ference BA, Kastelein JJ, Ray KK, et al. Association of triglyceride-lowering LPL variants and LDL-C-lowering LDLR variants with risk of coronary heart disease. JAMA. 2019;321(4):364–373.
- Kniffin CL. Solute carrier organic anion transporter family, Member 1B1: OMIM; 2015.
- Voora D, Shah SH, Spasojevic I, et al. The SLCO1B1* 5genetic variant is associated with statin-induced side effects. J Am Coll Cardiol. 2009;54(17):1609–1616.
- Group SC. SLCO1B1 variants and statin-induced myopathy—a genomewide study. New Eng J Med. 2008;359(8):789–799.
- Olsson AG, Istad H, Luurila O, et al. Effects of rosuvastatin and atorvastatin compared over 52 weeks of treatment in patients with hypercholesterolemia. American Heart Journal. 2002;144(6):1044–1051.
- Prueksaritanont T, Chu X, Evers R, et al. Pitavastatin is a more sensitive and selective organic anion-transporting polypeptide 1B clinical probe than rosuvastatin. Br J Clin Pharmacol. 2014;78(3):587–598.
- Tornio A, Vakkilainen J, Neuvonen M, et al. SLCO1B1 polymorphism markedly affects the pharmacokinetics of lovastatin acid. Pharmacogenet Genomics. 2015;25(8):382–387.
- Pasanen MK, Fredrikson H, Neuvonen PJ, et al. Different effects of SLCO1B1 polymorphism on the pharmacokinetics of atorvastatin and rosuvastatin. Clin Pharmacol Ther. 2007;82(6):726–733.
- Link E, Parish S, Armitage J, Bowman L, et al.; The Study of the Effectiveness of Additional Reductions in Cholesterol and Homocysteine (SEARCH) Collaborative Group. SLCO1B1 variants and statin‐induced myopathy‐a genomewide study. N Engl J Med. 2008;359(8):789–799.
- Kozyra M, Ingelman-Sundberg M, Lauschke VM. Rare genetic variants in cellular transporters, metabolic enzymes, and nuclear receptors can be important determinants of interindividual differences in drug response. Genet Med. 2017;19(1):20–29.
- Bush WS, Crosslin DR, Owusu-Obeng A, et al. Genetic variation among 82 pharmacogenes: the PGRNseq data from the eMERGE network. Clin Pharmacol Ther. 2016;100(2):160–169.
- Ingelman-Sundberg M, Mkrtchian S, Zhou Y, et al. Integrating rare genetic variants into pharmacogenetic drug response predictions. Hum Genomics. 2018;12(1):26.
- Fujikura K, Ingelman-Sundberg M, Lauschke VM. Genetic variation in the human cytochrome P450 supergene family. Pharmacogenet Genomics. 2015;25(12):584–594.
- Tirona RG, Leake BF, Merino G, et al. Polymorphisms in OATP-C: identification of multiple allelic variants associated with altered transport activity among European- and African-Americans. J Biol Chem. 2001;276(38):35669–35675.
- Schachter M. Chemical, pharmacokinetic and pharmacodynamic properties of statins: an update. Fundam Clin Pharmacol. 2005;19(1):117–125.
- Davidson MH. Rosuvastatin: a highly efficacious statin for the treatment of dyslipidaemia. Expert Opin Investig Drugs. 2002;11(1):125–141.
- Gazzerro P, Proto MC, Gangemi G, et al. Pharmacological actions of statins: a critical appraisal in the management of cancer. Pharmacol Rev. 2012;64(1):102–146.
- Garcia M, Reinoso R, Sanchez Navarro A, et al. Clinical pharmacokinetics of statins. Methods Find Exp Clin Pharmacol. 2003;25(6):457–481.
- Marciante KD, Durda JP, Heckbert SR, et al. Cerivastatin, genetic variants, and the risk of rhabdomyolysis. Pharmacogenet Genomics. 2011;21(5):280.
- Carr D, O'meara H, Jorgensen A, et al. SLCO1B1 genetic variant associated with statin-induced myopathy: a proof-of-concept study using the clinical practice research datalink. Clin Pharmacol Ther. 2013;94(6):695–701.
- Mangravite LM, Engelhardt BE, Medina MW, et al. A statin-dependent QTL for GATM expression is associated with statin-induced myopathy. Nature. 2013;502(7471):377–380.
- Carr D, Alfirevic A, Johnson R, et al. GATM gene variants and statin myopathy risk. Nature. 2014;513(7518):E1.
- Neřoldová M, Stránecký V, Hodaňová K, et al. Rare variants in known and novel candidate genes predisposing to statin-associated myopathy. Pharmacogenomics. 2016;17(13):1405–1414.
- Karaźniewicz-Łada M, Bąba K, Dolatowski F, et al. The polymorphism of statins and its effect on their physicochemical properties. Polim Med. 2018;48(2):77–82.
- O'Donnell CJ, Nabel EG. Genomics of cardiovascular disease. N Engl J Med. 2011;365(22):2098–2109.
- Wang L, McLeod HL, Weinshilboum RM. Genomics and drug response. N Engl J Med. 2011;364(12):1144–1153.
- Niemi M, Schaeffeler E, Lang T, et al. High plasma pravastatin concentrations are associated with single nucleotide polymorphisms and haplotypes of organic anion transporting polypeptide-C (OATP-C, SLCO1B1). Pharmacogenet Genomics. 2004;14(7):429–440.
- Niemi M, Neuvonen PJ, Hofmann U, et al. Acute effects of pravastatin on cholesterol synthesis are associated with SLCO1B1 (encoding OATP1B1) haplotype *17. Pharmacogenet Genomics. 2005;15(5):303–309.
- Oh ES, Kim CO, Cho SK, Park MS, et al. Impact of ABCC2, ABCG2 and SLCO1B1 polymorphisms on the pharmacokinetics of pitavastatin in humans. Drug Metab Pharmacokinet. 2013;28(3):196–202.
- Tomlinson B, Hu M, Lee V, et al. ABCG2 polymorphism is associated with the low-density lipoprotein cholesterol response to rosuvastatin . Clin Pharmacol Ther. 2010;87(5):558–562.
- Hu M, Lui SS, Mak VW, et al. Pharmacogenetic analysis of lipid responses to rosuvastatin in Chinese patients. Pharmacogenet Genomics. 2010;20(10):634–637.
- Medina MW, Sangkuhl K, Klein TE, et al. PharmGKB: very important pharmacogene-HMGCR. Pharmacogenet Genomics. 2011;21(2):98–101.
- Kathiresan S, Melander O, Anevski D, et al. Polymorphisms associated with cholesterol and risk of cardiovascular events. N Engl J Med. 2008;358(12):1240–1249.
- Hu M, To KK, Mak VW, et al. The ABCG2 transporter and its relations with the pharmacokinetics, drug interaction and lipid-lowering effects of statins. Expert Opin Drug Metab Toxicol. 2011;7(1):49–62.
- Catalan VS, LeLorier J. Predictors of long-term persistence on statins in a subsidized clinical population. Value Health. 2000;3(6):417–426.
- Dorais M, Chirovsky D, Ambegaonkar B, et al. Utilization patterns of extended-release niacin in Canada: analysis of an administrative claims database. Can J Cardiol. 2010;26(7):e229–e35.