
Abstract
Objectives
The aim of this study is to share our experience in the prenatal diagnosis of omphalocele by karyotyping, chromosomal microarray analysis (CMA) and whole exome sequencing (WES).
Methods
In this retrospective study, 81 cases of omphalocele were identified from 2015 to 2020. Associated anomalies and prenatal diagnosis based on karyotyping, CMA and WES were analysed.
Results
Fifty-eight (71.6%) of the 81 foetuses had other ultrasound anomalies. Giant omphalocele was present in 11 cases (13.6%) and small omphalocele was present in 70 cases (86.4%). Chromosomal abnormalities were found in 24 foetuses (29.6%, 24/81), the most common of which were trisomy 18 (58.8%, 11/24) and trisomy 13 (29.2%, 7/24). Compared to isolated omphalocele, non-isolated omphalocele was accompanied by an increased prevalence of chromosomal abnormalities (4.3% (1/23) vs. 39.7% (23/58), χ2 = 8.226, p = .004). All chromosomal abnormalities were found in small omphalocele. Aside from aneuploidy, CMA showed one pathogenic copy number variants (CNVs) for a detection rate of 1.2%, one variants of unknown significance (VOUS) and one instance of loss of heterozygosity (LOH). WES was performed on 3 non-isolated cases, and one was found to have pathogenic variants.
Conclusions
The most common genetic cause of omphalocele is aneuploidy and the prevalence of chromosomal abnormalities is increased with non-isolated and small omphalocele. CMA and WES can be useful for providing further genetic information to assist in prenatal counselling and pregnancy management.
Introduction
Omphalocele is a congenital defect in the abdominal wall characterized by absent abdominal muscles, fascia and skin and is one of the most common types of abdominal wall defects. Omphalocele is easily recognizable on ultrasound examination. Approximately, 50% of cases are associated with multiple malformation syndromes, including cardiac, gastrointestinal, genitourinary, musculoskeletal and central nervous system abnormalities [Citation1]. It has been reported that chromosomal abnormalities are present in 30–70% of cases where omphalocele is accompanied by other malformation syndromes. The most frequent chromosomal anomalies are trisomies 18 and 13, pentalogy of Cantrell and Beckwith–Wiedemann syndrome (BWS), and autosomal dominant and X-linked inheritance [Citation2,Citation3]. Therefore, understanding the genetic abnormalities underlying omphalocele is important for the diagnosis of this disease, which will aid in parental counselling and assist pregnancy management.
G-banding karyotyping has been the predominant strategy applied for detecting chromosomal abnormalities in foetuses with omphalocele in clinical practice over the past few decades. However, it has some limited resolutions. It is time-consuming and low-resolution. Chromosomal microarray analysis (CMA) has been recommended as the first-tier test for foetal structural abnormalities in identification of microscopic or submicroscopic copy number variants (CNVs). However, CMA cannot detect single-nucleotide variants (SNVs) and small insertions/deletions. With advances in genetic technology, whole-exome sequencing (WES) has been recently introduced. According to the PAGE cohort study, the yield of WES was 8.5% in foetuses with structural defects after the exclusion of aneuploidies and CNVs [Citation4]. However, there are very few case reports of prenatal diagnosis of omphalocele detected by CMA or WES in the literature. We designed a retrospective study of foetuses with omphalocele undergoing karyotyping, CMA and WES at our institution over five years to evaluate their feasibility in prenatal diagnosis for foetuses with omphalocele.
Materials and methods
This retrospective study was conducted at Guangdong women and children hospital between 2015 and 2020. The study was approved by our institutional review board and clinical research ethics committee. Foetuses were included if they had a prenatal ultrasound report showing omphalocele.
Giant omphalocele was defined by abdominal wall defects larger than 5 cm in diameter. Small omphalocele was defined by abdominal wall defects smaller than 5 cm in diameter. Isolated omphalocele was defined as sole omphalocele. Non-isolated omphalocele was defined as omphalocele in combination with another soft marker or a structural abnormality.
After diagnosis, all parents were given comprehensive counselling. Karyotyping and CMA analysis were presented to all foetuses. When no abnormalities were detected by karyotyping and CMA, WES was offered as an option.
Metaphase chromosome G-banding karyotyping was performed at a level of 320–400 bands. CMA was performed using a high-resolution genotyping single nucleotide polymorphism microarray, Affymetrix CytoScan 750 K Array (Affymetrix, Santa Clara, CA). CNVs were identified based on records associated with the human reference genome 37(NCBI37hg19) of the National Centre for Biotechnology Information.
Subsequently, genomic DNA samples from parent–foetus trios were subjected to WES using an Illumina Nextera Rapid Capture Exome Kit for library preparation. Sequencing was performed with the HiSeq 2500 platform (Illumina, San Diego, CA). Only variants related to the phenotype were reported. Variants were classified according to the American College of Medical Genetics and Genomics (ACMG) guidelines by the laboratory.
Statistical analysis was performed with the SPSS version 24.0 (IBM, Armonk, NY). Quantitative variables are expressed as the mean ± standard deviation, and categorical variables are expressed as the frequency and percentage. Differences between categorical variables were analysed using Chi-square test or Fisher’s exact test. p < .05 was considered statistically significant.
Results
There were 81 foetuses prenatally diagnosed with omphalocele during the study period. The median maternal age at the time of sampling was 30.9 ± 5.4 years and the gestational age at the time of invasive prenatal diagnosis was 18.0 ± 5.7 weeks. The methods of invasive test were chorionic villus sampling in 31 cases (38.3%, 31/81), amniocentesis sampling in 41 cases (50.6%, 41/81) and cordocentesis sampling in 9 cases (11.1%, 9/81).
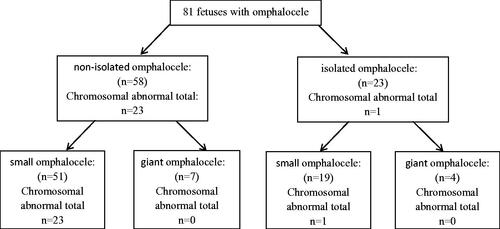
Among these foetuses, there were 23 cases (28.4%, 23/81) of isolated omphalocele and 58 cases (71.6%, 58/81) with other ultrasound anomalies. There were 11 cases of giant omphalocele (13.6%, 11/81) and 70 cases of small omphalocele (86.4%, 70/81).
Prenatal diagnostic results of karyotyping
Karyotyping results showed that chromosomal abnormalities were found in 24 foetuses, including 23 aneuploidies and 1 unbalanced translocation (). Overall, the prevalence of chromosomal abnormalities was 29.6% (24/81). The most common chromosomal abnormalities were trisomy 18 (58.8%, 11/24) and trisomy 13 (29.2%, 7/24), followed by trisomy 21(16.6%, 4/24), Klinefelter’s syndrome (4.2%, 1/24) and unbalanced translocation (4.2%, 1/24).
Table 1. Abnormal karyotype in foetuses with omphalocele.
In isolated omphalocele, chromosomal abnormalities were found in 1 foetus (trisomy 21) at 12 weeks. In non-isolated omphalocele, chromosomal abnormalities were found in 23 foetuses. Compared to isolated omphalocele, the prevalence of chromosomal abnormalities in non-isolated omphalocele was increased (4.3 vs. 39.7%, χ2=8.226, p = .004, Chi-square test for continuous calibration). In giant omphalocele, no chromosomal abnormalities were found, and all were found in small omphalocele ().
Figure 1. Prenatal diagnosis for isolated and non-isolated omphalocele.
Prenatal diagnostic results of CMA
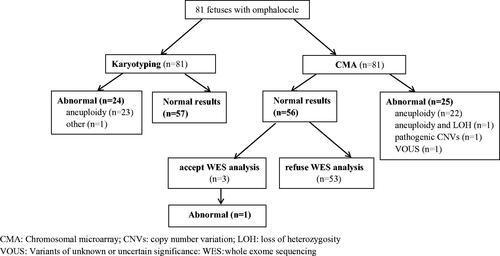
Of the 81 cases in which CMA was performed, aneuploidy was detected in 23 cases. In addition, there was 1 case of pathogenic CNVs, 1 case of VOUS and 1 case of LOH. CMA results (except aneuploidy) are outlined in . The total yield of pathogenic CNVs in CMA testing was 1.2% (1/81).
Table 2. Abnormal CMA (except aneuploidy) in foetuses with omphalocele.
In Case 2, omphalocele and placentomegaly were prenatally diagnosed by ultrasound. CMA showed a microdeletion in 11p15.5, which may cause DNA methylation aberrance leading to Beckwith–Wiedemann syndrome. Methylation-sensitive multiplex ligation probe analysis (MS-MLPA) revealed a loss of methylation at imprinting control region 2 and a 50% reduction of copy numbers of KCNQ1 gene. We finally confirmed the diagnosis of Beckwith–Wiedemann syndrome [Citation5].
In Case 3, the foetus was diagnosed with omphalocele, cystic hygroma and increased nuchal translucency and had already been diagnosed with trisomy 21 based on the prenatal karyotype. LOH at chromosome 14 suggests the existence of uniparental disomy (UPD) (14) and may be associated with Temple syndrome or Kagami–Ogata syndrome. Unfortunately, further analysis could not be performed because the pregnancy was terminated.
Prenatal diagnostic results of WES
After the exclusion of aneuploidies and pathogenic CNVs, there were 56 cases. Three non-isolated omphalocele cases accepted WES, and one had pathogenic variants ().
Figure 2. Prenatal diagnosis for foetuses with omphalocele according to the type of genetic analysis. CMA: chromosomal microarray; CNVs: copy number variation; LOH: loss of heterozygosity; VOUS: Variants of unknown or uncertain significance: WES: whole-exome sequencing.
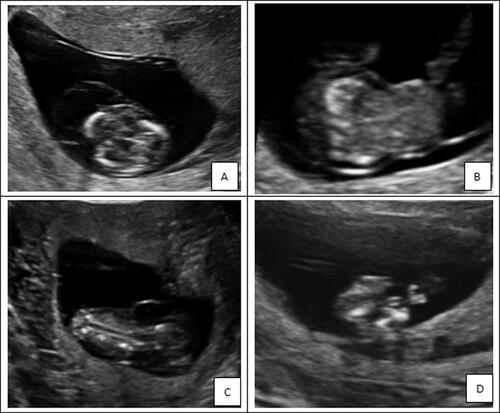
In this case, omphalocele, cystic hygroma, spinal malformation and short limbs were prenatally diagnosed by ultrasound at 11+3 weeks (). Moreover, the woman had two similar pregnancies before. WES showed a heterozygous point variation of the SLC26A2 gene: c.1020_1022delTGT (p.Val341del) from the father and the heterozygosity exon 2–3 deletion from the mother, forming a compound heterozygosity. Mutations in SLC26A2 gene result in a spectrum of autosomal recessive chondrodysplasias that range from the mildest recessive form of multiple epiphysial dysplasia (rMED) through the most common diastrophic dysplasia (DTD) to lethal atelosteogenesis type II and achondrogenesis IB.
Figure 3. The ultrasound examination of the case with diastrophic dysplasia. The ultrasound examination showed that cystic hygroma (A), omphalocele (B), spinal malformation (C) and short limbs (D).
DTD (OMIM 222600) is a rare osteochondrodysplasia characterized by short-limbed short stature and joint dysplasia. DTD is caused by mutations in SLC26A2. The variant c.1020_1022delTGT (p.Val341del) has been previously reported and classified as pathogenic. The deletion of exon 2–3 has not been reported, but was predicted to be a disease-causing variant by in-silico prediction tools and classified as likely-pathogenic. Finally, the foetus was diagnosed with DTD by combined prenatal ultrasonography and whole exome sequencing (WES).
Discussion
In the literature, Omphalocele is complicated with other organ anomalies and chromosomal abnormalities in 30–70% of cases. The most frequent chromosomal anomalies of omphalocele are trisomies 18 and 13, and small omphaloceles are more likely to be associated with abnormal karyotypes [Citation6–8]. In our study, karyotyping showed that the prevalence of chromosomal abnormalities was 29.6%, and trisomy 18 and trisomy 13 accounted for 58.8 and 29.2%, respectively. Our study also demonstrated that 71.6% of cases had other ultrasound anomalies. For isolated cases, the prevalence of chromosomal abnormalities was 4.3%, while 39.7% in non-isolated omphalocele cases. Moreover, all chromosomal abnormalities were found in small omphalocele. These results were consistent with previous research and further support the idea that the prevalence of chromosomal abnormalities was increased with non-isolated and small omphalocele.
Khalil et al. [Citation9] recommend delaying foetal karyotyping until after the early second-trimester scan in cases of omphalocele with normal nuchal translucency. In their cohort, they observed that many of the omphalocele cases had resolved by 16 weeks of gestation. However, as reported in our study, one foetus with isolated omphalocele was diagnosed with trisomy 21 at 12 weeks. Therefore, isolated omphalocele may be associated with chromosomal abnormalities even in the first-trimester, so we suggest that prenatal diagnosis should be presented to all foetuses with omphalocele.
It has been shown that CMA has the ability to detect clinically significant CNVs in 6–8% of pregnancies with ultrasound abnormalities [Citation10]. However, previous studies looking at prenatal CMA do not specifically report cases of omphalocele, therefore, the exact utility of CMA in cases of omphalocele remains unclear. In our study, there was 1 case (1.2%) of pathogenic CNVs, and the most likely explanation for the low yield of CMA in cases of omphalocele is that most common genetic cause of omphalocele is due to aneuploidy, rather than CNVs. Jasmin Beygo et al. [Citation11] reported that a maternal deletion upstream of the imprint control region 2 in 11p15 causes loss of methylation and familial Beckwith–Wiedemann syndrome. In our study, CMA found an 11p15.5 microdeletion in a foetus affected by omphalocele and placentomegaly. As the deletion also includes the promoter and 5′ part of the KCNQ1 gene, we considered it may cause BWS. To verify the initial clinical diagnosis of BWS, molecular genetic analysis was performed using MS-MLPA of chromosome 11p15 and finally confirmed the diagnosis [Citation5]. We also found a LOH at chromosome 14 in a foetus. UPD (14) may be associated with Temple syndrome or Kagami–Ogata syndrome. Temple syndrome and Kagami–Ogata syndrome are both rare imprinting disorders. These results demonstrate that CMA is a valuable tool for identifying unbalanced submicroscopic chromosomal abnormalities and LOH in the prenatal diagnosis of omphalocele.
BWS is the most common paediatric overgrowth syndrome, classically characterized by omphalocele, macroglossia and overgrowth. Omphalocele can be associated with BWS. Omphaloceles have seen with BWS often contain only the small bowel, and additional findings in BWS include macrosomia, organomegaly, macroglossia, polyhydramnios, and placentomegaly [Citation12,Citation13]. Previous studies have reported BWS in association with prenatally diagnosed omphalocele in the range of 2–23% [Citation14,Citation15]. Abbasi N et al. [Citation16] conducted a retrospective review of 92 prenatally diagnosed omphalocele with molecular confirmation of BWS and showed that BWS was identified in 37 and 7% of isolated and non-isolated omphalocele, respectively, after exclusion of aneuploidy. However, we found only one case (1.2%, 1/81) of BWS in our study. Multiple possible explanations exist, and a potential reason for this discrepancy is that major underlying pathogenetic mechanisms of BWS include loss or gain of DNA methylation, paternal UPD, chromosomal rearrangements and gene sequencing. However, in our study, we did not provide MS-MLPA to all foetuses, which may lead to missed diagnosis in some cases. Another possible reason is that many of the features of BWS develop late in gestation or postnatally, and some foetuses with non-isolated omphalocele were terminated prior to expanded molecular analysis.
A wide spectrum of genetic disorders has been reported in association with omphalocele. These disorders, such as Donnai–Barrow syndrome, acrocallosal syndrome and hydrolethalus syndrome, are not detected by either CMA or karyotype and are likely to explain a significant portion of unexplained cases. It may be reasonable to consider exome sequencing as the next step when standard genetic tests do not yield a diagnosis. A recent report shows the prenatal use of WES in diagnosing Donnai–Barrow syndrome in the setting of omphalocele and associated anomalies [Citation17]. In our study, WES was performed to identify possible causal variants in three non-isolated omphalocele cases, and one pathogenic variant was successfully identified. Thus, WES should be a part of the diagnostic workup for any euploid foetus with non-isolated omphalocele, particularly if abnormal pregnancies have occurred repeatedly, as in our case.
Conclusion
In summary, our study demonstrates that the most common genetic cause of omphalocele is due to aneuploidy, and the prevalence of chromosomal abnormalities was increased with non-isolated and small omphalocele. CMA and WES can be useful in providing further genetic information to assist in prenatal counselling and pregnancy management. This study was limited by its small sample size and retrospective design taking place at a single subspecialty referral centre. Another limitation is that genetic testing, such as MS-MLPA for BWS is not offered for all foetuses. Further investigation through multicentre and prospective research is warranted to evaluate the prenatal diagnosis of omphalocele.
Ethical approval
The study protocol was approved by institutional review board and clinical research ethics committee of Guangdong Women and Children Hospital and conforms to the ethical guidelines of the 1975 Declaration of Helsinki modified in 2013. All participating subjects signed an informed consent before enrolment.
Author contributions
Shi Xiaomei: analysed and interpreted the data, and wrote the final version of article. Tang Hui and Yang Xiue: data collection and manuscript editing. Lu Jian and Ding Hongke: karyotyping, CMA and WES analysis. Wu Jing: project development and manuscript writing.
Acknowledgements
The authors thank all the parents who pleasantly agreed to use the prenatal diagnosis results and ultrasonography images of the foetus.
Disclosure statement
The authors have no conflicts to declare.
Data availability statement
All data included in this study are available upon request by contact with the corresponding author.
Additional information
Funding
References
- Calvert N, Damiani S, Sunario J, et al. The outcomes of pregnancies following a prenatal diagnosis of fetal exomphalos in Western Australia. Aust N Z J Obstet Gynaecol. 2009;49(4):371–375.
- Verla MA, Style CC, Olutoye OO. Prenatal diagnosis and management of omphalocele. Semin Pediatr Surg. 2019;28(2):84–88.
- Adams AD, Stover S, Rac MW. Omphalocele-What should we tell the prospective parents? Prenat Diagn. 2021;41(4):486–496.
- Lord J, McMullan DJ, Eberhardt RY, et al. Prenatal exome sequencing analysis in fetal structural anomalies detected by ultrasonography (PAGE): a cohort study. Lancet. 2019;393(10173):747–757.
- Tong H, Lu J, Liu L, et al. Prenatal diagnosis of suspected recurrent Beckwith-Wiedeman syndrome:a case report and literature review. Chin J Perinat Med. 2021;24(4):283–287.
- Brantberg A, Blaas HG, Haugen SE, et al. Characteristics and outcome of 90 cases of fetal omphalocele. Ultrasound Obstet Gynecol. 2005;26(5):527–537.
- Zork NM, Pierce S, Zollinger T, et al. Predicting fetal karyotype in fetuses with omphalocele: the current role of ultrasound. J Neonatal Perinatal Med. 2014;7(1):65–69.
- Chen CP. Chromosomal abnormalities associated with omphalocele. Taiwan J Obstet Gynecol. 2007;46(1):1–8.
- Khalil A, Arnaoutoglou C, Pacilli M, et al. Outcome of fetal exomphalos diagnosed at 11–14 weeks of gestation. Ultrasound Obstet Gynecol. 2012;39(4):401–406.
- Wapner RJ, Martin CL, Levy B, et al. Chromosomal microarray versus karyotyping for prenatal diagnosis. N Engl J Med. 2012;367(23):2175–2184.
- Beygo J, Joksic I, Strom TM, et al. A maternal deletion upstream of the imprint control region 2 in 11p15 causes loss of methylation and familial beckwith-wiedemann syndrome. Eur J Hum Genet. 2016;24(9):1280–1286.
- Shieh HF, Estroff JA, Barnewolt CE, et al. Prenatal imaging throughout gestation in Beckwith-Wiedemann syndrome. Prenat Diagn. 2019;39(9):792–795.
- Brioude F, Kalish JM, Mussa A, et al. Expert consensus document: clinical and molecular diagnosis, screening and management of Beckwith-Wiedemann syndrome: an international consensus statement. Nat Rev Endocrinol. 2018;14(4):229–249.
- Wilkins-Haug L, Porter A, Hawley P, et al. Isolated fetal omphalocele, Beckwith-Wiedemann syndrome, and assisted reproductive technologies. Birth Defect Res A. 2009;85(1):58–62.
- Grover R, Collins AL, Wellesley D. Exomphalos without other prenatally detected anomalies: perinatal outcomes from 22 years of population-based data. Prenat Diagn. 2020;40(10):1310–1314.
- Abbasi N, Moore A, Chiu P, et al. Prenatally diagnosed omphaloceles: report of 92 cases and association with Beckwith-Wiedemann syndrome. Prenat Diagn. 2021;41(7):798–816.
- Ozdemir H, Plamondon J, Gaskin P, et al. A prenatally diagnosed case of Donnai-Barrow syndrome: highlighting the importance of whole exome sequencing in cases of consanguinity. Am J Med Genet A. 2020;182(2):289–292.