
Abstract
Aim
Endometriosis is one of the most common reproductive system diseases, but the mechanisms of disease progression are still unclear. Due to its high recurrence rate, searching for potential therapeutic biomarkers involved in the pathogenesis of endometriosis is an urgent issue.
Methods
Due to the similarities between endometriosis and ovarian cancer, four endometriosis datasets and one ovarian cancer dataset were downloaded from Gene Expression Omnibus (GEO) database. Differentially expressed genes (DEGs) were identified, followed by gene ontology (GO), Kyoto Encyclopedia of Genes and Genomes (KEGG) pathway and protein–protein interaction (PPI) analyses. Then, we validated gene expression and performed survival analysis with ovarian serous cystadenocarcinoma (OV) datasets in TCGA/GTEx database, and searched for potential drugs in the Drug-Gene Interaction Database. Finally, we explored the miRNAs of key genes to find biomarkers associated with the recurrence of endometriosis.
Results
In total, 104 DEGs were identified in the endometriosis datasets, and the main enriched GO functions included cell adhesion, extracellular exosome and actin binding. Fifty DEGs were identified between endometriosis and ovarian cancer datasets including 11 consistently regulated genes, and nine DEGs with significant expression in TCGA/GTEx. Only IGHM had both significant expression and an association with survival, three module DEGs and two significantly expressed DEGs had drug associations, and 10 DEGs had druggability.
Conclusions
ITGA7, ITGBL1 and SORBS1 may help us understand the invasive nature of endometriosis, and IGHM might be related to recurrence; moreover, these genes all may be potential therapeutic targets.
This manuscript used a bioinformatics approach to find target genes for the treatment of endometriosis.
This manuscript used a new approach to find target genes by drawing on common characteristics between ovarian cancer and endometriosis.
We screened relevant therapeutic agents for target genes in the drug database, and performed histological validation of target genes with both expression and survival analysis difference in cancer databases.
KEY MESSAGE
Introduction
Endometriosis is defined by endometrial tissue located outside of the uterine cavity [Citation1,Citation2]. Approximately, 6–10% of women of reproductive age are affected by this disease, and infertility and pelvic pain are the primary symptoms [Citation3]. Dysmenorrhoea, irregular uterine bleeding and dyspareunia also occur frequently in those patients. Endometriosis mainly occurs in the ovary, followed by the ligaments of the pelvic, the fallopian tract, the umbilicus, the abdominal wall, the cervical-vaginal area, the urinary tract, and the eyes, lung and brain. This characteristic of distant metastasis is similar to that of tumours, but the pathogenesis has yet to be fully elucidated. Influencing factors are extensive and include environmental, genetic, stem cell, immunogenicity, lymphatic and vascular dissemination factors [Citation4,Citation5]. Gynaecologic surgery is the main treatment, while other treatments include nonsteroidal anti-inflammatory drugs, progestins, combined oral contraceptives and GnRH-a injection [Citation6]. Regardless of these treatments, endometriosis has a high recurrence rate.
Ovarian cancer is one of the three major malignant tumours in obstetrics and gynaecology, and the diagnosis and treatment of ovarian cancer are relatively mature and prevalent, in particular, nanomedicines offer new prospects for ovarian cancer treatment [Citation7]. Endometriosis and ovarian cancer have certain similarities in terms of invasion, angiogenesis and adhesion, but the difference is that endometriosis does not have the infinite proliferation observed in ovarian cancer. Several studies have shown that endometriosis is one of the risk factors for ovarian cancer [Citation8], and a proportion of ovarian cancers have been shown to originate from 0.5 to 1% of cases of ovarian endometriosis [Citation9,Citation10]. Ovarian endometriosis may present a risk for ovarian malignant lesions according to gene expression and miRNA alterations [Citation11,Citation12], and is always managed with the prevention of carcinogenesis [Citation13]. Immunity and inflammation are thought to be strongly associated with carcinogenicity [Citation14,Citation15]; however, no studies have shown how long ovarian cancer takes to develop from ovarian endometriosis. All evidence shows relationships between endometriosis and ovarian cancer; thus, screening differentially expressed genes (DEGs) between ovarian cancer and endometriosis may provide an alternative route to identify the mechanisms involved in the carcinogenesis and recurrence of endometriosis.
In recent years, microarrays have been widely used to identify therapeutic targets and candidate biomarkers by investigating the alteration of gene expression at a genome-wide level [Citation16,Citation17]. With the integration of bioinformatics technology and clinical treatment [Citation18–21], a number of studies have been published, including studies on endometriosis. DEGs such as NR4A1 [Citation22], ITPR1 [Citation23], CXCL12 [Citation24], HSPA5, ENO2 and TJP1 [Citation25] have been proven important in the progression of endometriosis. miRNAs, such as miR-200b-3p [Citation26], miR-1266-5p, and miR-200a-3p [Citation27], and even circular RNAs (circRNAs), for example, has-circ-0003380, has-circ-0020093 and has-circ-0008016, were all significantly overexpressed in endometriosis [Citation28]. In our study, we drew on the common features of two different diseases to identify key DEGs, which may provide a new direction for treatment.
Materials and methods
Data collection
The Gene Expression Omnibus (GEO, http://www.ncbi.nlm.nih.gov/geo/) is a freely available international public repository for next-generation sequencing-based functional genomic datasets and high-throughput microarrays. It also provides users with several web-based tools to query, analyse and visualize data [Citation29], such as GEO2R. Four endometriosis datasets, GSE5108, GSE7305, GSE11691 and GSE25628, and one ovarian cancer dataset, GSE14407 were obtained from GEO. The GSE5108 dataset contained 11 ectopic endometrium samples and 11 eutopic endometrium samples. GSE7305 contained 10 ectopic endometrium samples and 10 normal endometrium samples. GSE11691 contained nine ectopic endometrium samples and nine normal endometrium samples. GSE25628 contained eight ectopic endometrium samples and eight normal endometrium samples. GSE14407 contained 12 normal samples and 12 tumour samples.
Identification of DEGs
GEO2R (http://www.ncbi.nlm.nih.gov/geo/geo2r/) [Citation29] is an R-based website that helps users perform GEO data analysis, and identify genes that are differentially expressed [Citation30,Citation31]. The four endometriosis datasets described above were analysed using GEO2R, and GSE14407 was analysed by RStudio (version 4.0.4). The limma package was applied to identify the DEGs between cancer and normal groups, with the GPL570 [HG-U133_Plus_2] Affymetrix Human Genome U133 Plus 2.0 Array. The statistically significant settings were | log (fold change) | >1 and p value <.05.
Gene ontology (GO), signalling pathway and protein–protein interaction (PPI) networks
GO (http://geneontology.org) is the most widely used knowledge base and provides structured knowledge regarding the functions of genes and gene products [Citation32], including biological processes (BPs), cellular components (CCs) and molecular functions (MFs) [Citation33]. GO and Kyoto Encyclopedia of Genes and Genomes (KEGG) pathway analyses were performed using the web-based DAVID tool (version 6.8, http://www.david.niaid.nih.gov), which is for the functional annotation of DEGs [Citation34]. In addition, we also used R to perform GO analysis of 104 DEGs, and to ensure the reliability of our results. Next, PPI networks were predicted using by STRING (version 11.0, https://string-db.org/), which was applied to explore the physical and functional associations between the DEGs [Citation35], with a combined score >0.4 (medium confidence). PPIs were visualized using Cytoscape software (version 3.8.1) [Citation36], and the Molecular Complex Detection plugin (MCODE, version 2.0.0) was used to find the most significant modules, with the following settings: degree cut-off = 2, node score cut-off = 0.2, max depth = 100 and k-score = 2.
Validation of DEGs between endometriosis and ovarian cancer on GEPIA in TCGA/GTEx databases
To further select for precise biomarkers, we performed gene expression level and survival analysis with Gene Expression Profiling Interactive Analysis (GEPIA, http://gepia.cancer-pku.cn/), a web-based tool to deliver fast and customizable functionalities based on Cancer Genome Atlas (TCGA) and Genotype-Tissue Expression (GTEx) data, and provided key interactive and customizable functions [Citation37]. Gene expression validation involved 514 samples of ovarian serous cystadenocarcinoma (OV) datasets built in TCGA/GTEx database (tumour: 426 normal: 88), with thresholds |log2FC| ≥1 and p value <.01, setting jitter size =0.4. Overall survival (OS) and disease-free survival (DFS) were assessed in OV datasets, and time data were sorted into low-expression and high-expression groups by the median transcripts per kilobase (TPM).
Possible drugs for target genes
The Drug-Gene Interaction Database (DGIdb, http://www.dgidb.org) is a web resource that helps users interpret the results of genome-wide studies in the context of the druggable genome [Citation38]. DGIdb organizes genes of the druggable genome into known drug interactions and potentially druggable targets [Citation38]. We input module DEGs of endometriosis and significantly evaluated DEGs in DGIdb to find potentially druggable DEGs.
Immunofluorescence
Ectopic endometrium, eutopic endometrium and normal endometrium were fixed, embedded and sliced. After deparaffinizing and rehydrating the paraffin sections [Citation39,Citation40], they were placed in a repair box filled with citric acid antigen retrieval buffer (pH 6.0) for antigen retrieval. Next, sections were placed in 3% hydrogen peroxide and incubated at room temperature for 25 min to block endogenous peroxidase activity, followed by serum blocking with 3% BSA (Servicebio G5001, Wuhan, China) for 30 min at room temperature. Anti-human IgM rabbit monoclonal antibody (1:1000 dilution; HUABIO, Cambridge, MA) was incubated overnight at 4 °C, followed by an incubation with secondary antibody at room temperature for 50 min. After the addition of secondary antibody, the sections were incubated with DAPI (Servicebio G1012, Wuhan, China) solution for 10 min at room temperature, and then spontaneous fluorescence quenching reagent was added and incubated for 5 min. Then, cover slips were mounted with anti-fade mounting medium, and images were captured by fluorescence microscopy.
Results
DEG identification
After standardization, DEGs associated with endometriosis (1846 in GSE5108, 2633 in GSE7305, 1513 in GSE11691 and 509 in GSE25628) were identified, as were DEGs associated with ovarian cancer (6887 in GSE14407). There were 104 genes among the four endometriosis datasets as shown in the Venn diagram (), including 84 consistently upregulated DEGs and 19 consistently downregulated DEGs. The DEGs behaved differently due to the heterogeneity of humans (). The overlap contained 50 DEGs, and only 11 DEGs had consistent regulation, including 10 upregulated DEGs and one downregulated DEG ().
Figure 1. Venn diagram. (A) DEGs of endometriosis were selected with a fold change >1 and p value <.05 among the expression profiling sets GSE5108, GSE7305, GSE11691 and GSE25628. The four datasets showed an overlap of 104 genes. (B) DEGs of endometriosis and ovarian cancer datasets showed an overlap of 50 genes including 10 up-regulated and one down-regulated DEGs.
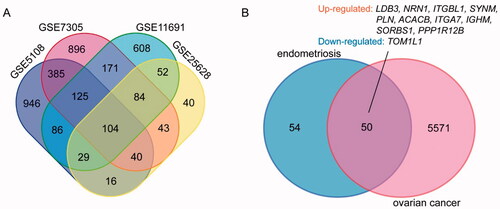
Table 1. Identified DEGs in four endometriosis datasets, but PLA2G5 had the different regulatory.
GO and KEGG enrichment analyses of DEGs in endometriosis
We identified the top five significant GO and signalling pathways with the criterion of a p value <.05 (). Then, we analysed the most enriched GO functions (). Among the upregulated, BP was mostly enriched in cell adhesion, muscle contraction and positive regulation of inflammatory response; CC was mainly enriched in extracellular exosome, plasma membrane and extracellular space; and MF was significantly enriched in actin binding, calmodulin binding and structural constituent of muscle. KEGG pathway analysis revealed that DEGs were mainly enriched in vascular smooth muscle contraction and the cGMP-PKG signalling pathway. The downregulated DEGs were mainly involved in response to osmotic stress and metabolic pathways. GO function analysis was also performed by R, and more results for BP, MF and CC were obtained, but we only showed the top functions in the diagram (). The main functions were roughly the same for the two methods, but we could not obtain KEGG results in the R analysis, as the gene number was too small. Thus, it seemed that DAVID was more advantageous, but the key DEGs involved in cell adhesion in the two methods were consistent.
Figure 2. Most significant GO analysis of 104 DEGs by using DAVID (A) and R (B).
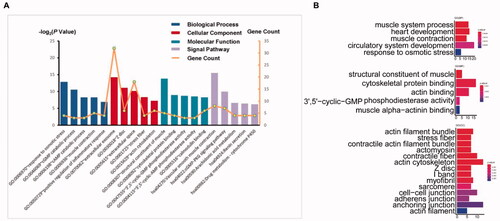
Table 2. Top three most enriched BP, CC, MF and signal pathway analysis of 104 DEGs.
PPI networks and the most significant modules of endometriosis
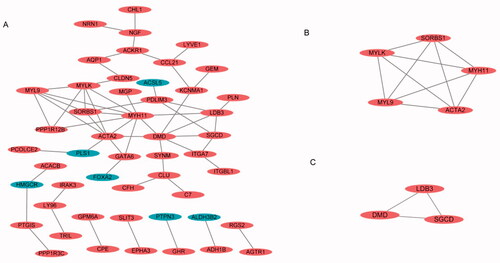
A total of 104 DEGs were uploaded into the STRING website (https://string-db.org/cgi/) and analysed by Cytoscape, with a setting score >0.4 (medium confidence), with options such as hiding disconnected nodes and showing input protein names selected for the construction (). The two most significant modules were clustered via MCODE. Module 1 was made up of five upregulated DEGs, and module 2 consisted of three upregulated DEGs ().
Figure 3. PPI network and the most significant module of DEGs of four endometriosis datasets. (A) The PPI network of DEGs was constructed using Cytoscape. (B, C) The most significant module was obtained by MCODE.
Validation of the 11 DEGs in TCGA/GTEx
The 11 DEGs were significantly enriched in protein binding and cell adhesion, and the signalling pathways were mainly enriched in dilated cardiomyopathy and the insulin signalling pathway (). For validation of the OV build in TCGA/GTEx, we found that only nine DEGs had significant expression in OV (). In addition, only IGHM had a significant difference between high expression and low expression in OS and DFS (). This candidate gene was significantly enriched in the regulation of extracellular exosomes and extracellular space ().
Figure 4. (A) Significant expression of nine DEGs with OV built in TCGA/GTEx datasets. (B) Significant OS and DFS analysis of IGHM between high expression and low expression on GEPIA. (C) Drug–gene interactions of three module DEGs and two significant expressed DEGs in DGIdb database; druggability of 10 DEGs and their nine kinds of drug categories.
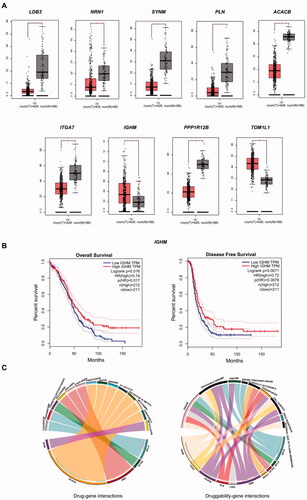
Table 3. Most significantly GO and signalling pathway of 11 DEGs.
Possible drugs for target genes
We input eight module DEGs and nine significantly expressed DEGs involved in the DGIdb database to identify drug–gene interactions and potential druggable gene targets. MYLK, ACTA2 and DMD were associated with six kinds of drugs for endometriosis, and four of which had been validated by researchers, ACACB and IGHM were associated with eight kinds of drugs, three of which had been approved by researchers (). Ten of drugs were present in nine drug categories ().
Table 4. Drugs corresponding to DEGs in DGIdb database.
Immunofluorescence
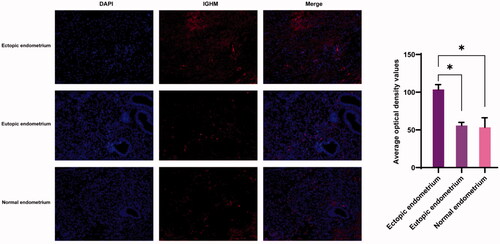
Three sets of human tissue were collected for verification. In , the expression of IGHM in endometrium, eutopic endometrium and normal endometrium was labelled by red fluorescence. The expression of IGHM was significantly higher in ectopic endometrium than in eutopic and normal endometrium, while there was no significant difference in its expression between eutopic and normal endometrium.
Figure 5. Immunofluorescence of ectopic endometrium, eutopic endometrium and normal endometrium.
Discussion
Endometriosis is an oestrogen-dependent disease associated with pelvic pain and reduced fertility [Citation41,Citation42], and has a complex aetiology, influenced by both genetic and environmental factors [Citation43]. Relationships between endometriosis and ovarian cancer have been established, such as inflammatory response, vascular proliferation, distant invasion and associated increased levels of serum CA125. Endometriosis is a risk factor for ovarian cancer [Citation44] and can transform to an atypical form and even to malignancy in 0.7–2.5% of cases [Citation45]. In this study, the similarity in distant progression between endometriosis and ovarian cancer was used to find targets for the treatment of endometriosis.
The datasets used in our study have been widely used in other studies, suggesting that the results analysed with these datasets are supported by credibility. Research involving GSE5108 [Citation46] is the most original sequencing analysis in this dataset, which only lists the genes with large variations in fold, and it indicated that cell adhesion associated genes may contribute to the adhesive and invasive properties of ectopic endometrium, consistent with our study. The GSE11691 [Citation24,Citation25], GSE7305 [Citation24], GSE25628 [Citation25] and GSE14407 had all been submitted to GO, KEGG and PPI analyses. These studies all selected the functions of DEGs ranked at the top by |log2FC|. Of course, this selection helped to obtain certain key functions, whereas in our study, we did not focus only on the magnitude with |log2FC|. As expression was not limited to |log2FC|, we focussed on the common DEGs of the two diseases with similar properties, and then selected the DEGs associated with adhesion function, to more precisely screen the target DEGs for our corresponding studies.
By taking the intersection of the four endometriosis datasets, we obtained 104 DEGs, and we obtained two clusters on endometriosis through the construction of PPI networks. SORBS1, MYLK, MYH11, MYL9 and ACAT2 were involved in cluster 1, and LDB3, DMD and SGCD were involved in cluster 2 (). Eight DEGs may play important roles in the development of endometriosis, and three of the eight DEGs were associated with drugs in the drug database DGIdb (, ). To identify target DEGs that may be involved in the recurrence of endometriosis, 11 DEGs with consistent up- and downregulation were identified among the 50 DEGs shared by endometriosis and ovarian cancer, and validations were performed in TCGA/GTEx. Nine DEGs had significant expressions; LDB3, NRN1, SYNM, PLN, ACACB, ITGA7, IGHM, PPP1R12B and TOM1L1, all of which were involved in the function of protein binding, and only ACACB and IGHM were identified as druggable targets in DGIdb (, ). These druggable targets are pending future cellular and animal studies.
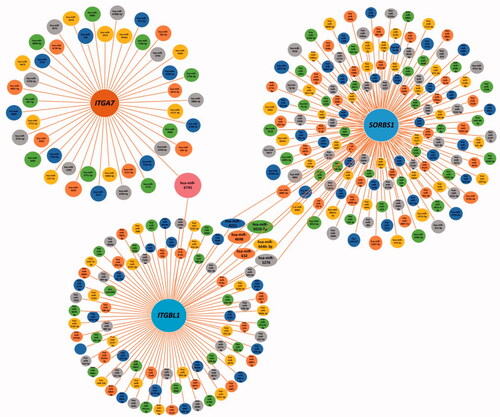
By analysing the GO functions of these 11 DEGs, we found that ITGA7, ITGBL1 and SORBS1 were mainly involved in cell adhesion (). We observed that ITGA7 had a direct interaction with ITGBL1 in PPI network (), which suggested that they might be coexpressed, and all three genes were upregulated DEGs that regulated cell proliferation, invasion and migration in cancers [Citation47–51]. It is of great significance to analyse their survival in obstetrics- and gynaecology-related tumours (). ITGA7 regulates cell proliferation via the PTK2-PI3K-Akt signalling pathway and is negatively associated with clinical outcomes in hepatocellular carcinoma [Citation52], and via the laminin-integrin α7β1 signalling pathway in mechanical ventilation-induced pulmonary fibrosis [Citation53]. Upregulation of ITGBL1 predicted poor prognosis and promoted chemoresistance in ovarian cancer [Citation54] and activated fibroblasts using extracellular vesicles (EVs) via NF-κB signalling. Moreover, it promoted epithelial-to-mesenchymal transition (EMT) of colorectal cancer (CRC) cells [Citation55] and had the same characteristics in ovarian cancer [Citation51] and prostate cancer [Citation50]. SORBS1 overexpression promoted CRC growth and migration via inhibition of AHNAK expression [Citation56], while SORBS1 was downregulated in breast cancer and led to poor prognosis [Citation47]. Silencing of SORBS1 promoted the EMT process and attenuated chemical drug sensitivity, and it is a potential inhibitor of metastasis in cancer [Citation57]. We inputted the tree DEGs in the miRDB, TargetScan and miWalk databases to identify the key miRNAs for the prognosis of endometriosis. As shows, ITGA7 had 45 miRNAs, ITGBL1 had 92 miRNAs, SORBS1 had 159 miRNAs, hsa-miR-6745 was the only expressed miRNA between ITGA7 and ITGBL1, and there were six expressed miRNAs between ITGBL1 and SORBS1. We conjectured that overexpressed hsa-miR-6745 may be associated with poor outcomes and high recurrence of endometriosis. Although all three genes were upregulated, through literature data, we found that silencing of SORBS1 may promote the progression of disease in most cancers; thus there may be some other regulatory relationship between ITGBL1 and SORBS1. We cannot say that the six miRNAs may have a certain relationship with the prognosis of endometriosis.
Figure 6. Significant OS and DFS analysis of ITGA7, ITGBL1 and SORBS1 between high expression and low expression among breast invasive carcinoma (BRCA), cervical squamous cell carcinoma and endocervical adenocarcinoma (CESC), ovarian serous cystadenocarcinoma (OV), uterine corpus endometrial carcinoma (UCEC) and uterine carcinosarcoma (UCS) on GEPIA.
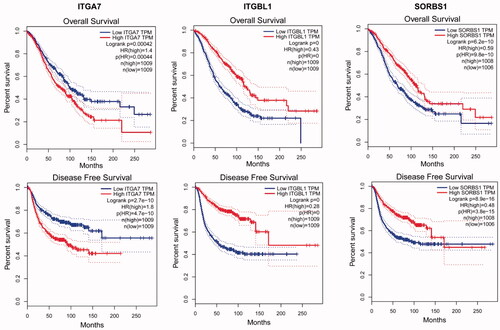
Figure 7. miRNAs and overlap of ITGA7, ITGBL1 and SORBS1 in miRDB, TargetScan and miWalk databases.
In survival analysis, only IGHM had significance in OS and DFS. IGHM is a protein-coding gene. IgM antibodies are involved in the recognition and elimination of precancerous and cancerous lesions, have been found to be upregulated in breast cancer [Citation58] and were considered a biomarker for recurrence [Citation59]. IGHM also retained a significant prognostic impact on the density of intratumoural CD20+ B cells [Citation60] and was associated with type 1 diabetes [Citation61]. IGHM is involved in oxidative stress and in skin regeneration [Citation62], suggesting that it may be involved in cell proliferation. We tried to find relevant IGHM-regulated cascade response signals, similar to other studies [Citation63–67]. Transcription factor binding to IGHM enhancer 3 (TFE3) is related to renal cell carcinoma [Citation68,Citation69]. PRCC-TFE3 tRCC is a TFE3 Xp11.2 translocation renal cell carcinoma (TFE3-tRCC) that promotes cell survival and proliferation by implicating in PINK1-PRKN/parkin-dependent mitophagy and activating the expression of the E3 ubiquitin ligase PRKN, leading to rapid PINK1-PRKN-dependent mitophagy that promotes cell survival under mitochondrial oxidative damage as well as cell proliferation by decreasing mitochondrial ROS formation [Citation68], suggesting that there are similar regulatory mechanisms in endometriosis. In our study, IGHM was significantly involved in the CC of extracellular exosomes. Exosomes are released following the fusion of multivesicular bodies with the plasma membrane and the extracellular release of intraluminal vesicles [Citation70]. Exosomes are EVs 50–100 nm in size that deliver proteins, lipids and nucleic acids [Citation71,Citation72] to target cells, and their main functions include antigen presentation, pathogen spread, proliferation, differentiation, apoptosis, migration and invasion [Citation73–75]. In our immunofluorescence validation, the expression of IGHM was highest in ectopic endometrium, and differed from eutopic endometrium and normal endometrium (), which is consistent with our analysis. Therefore, regulating IGHM may be another method for endometriosis. We could not find any miRNAs that had been confirmed to interact with IGHM in the three miRNA databases, possibly indicating that IGHM may be a new biomarker for us to explore in the future.
Conclusions
Above all, ITGA7, ITGBL1 and SORBS1 may be associated with cell proliferation, invasion and migration of endometriosis, hsa-miR-6745 may be a potential miRNA biomarker, and its high expression may be associated with poor prognosis. IGHM might be a potential target gene for the recurrence of endometriosis; however, to date, there have been no studies on IGHM in the reproductive system. Further research is needed to elucidate the role of this new target gene in endometriosis, and ITGA7, ITGBL1, SORBS1 and IGHM may be therapeutic target genes. All drugs need to be validated by molecular biology or animal studies in future research.
Ethics approval
All the human tissue collection was approved by the Ethics Committee of Tongji Medical College, Huazhong University of Science and Technology (ethical approval number for human research: s1056) and were based on the World Medical Association Declaration of Helsinki.
Author contributions
We certify that Prof. Ying Gao has participated sufficiently in the intellectual content, and Zhenzhen Lu involved in work of conception and design of this research or the analysis and interpretation of the data, as well as the writing of the manuscript. All author contributed to editorial changes in the manuscript. All authors read and approved the final manuscript.
Acknowledgements
Thanks to Dr. Yanfang Lu and Dr. Bin Zhao for their help in the data analysis process.
Disclosure statement
No potential conflict of interest was reported by the author(s).
Data availability statement
The datasets that support the findings of this study are openly available in GEO database at http://www.ncbi.nlm.nih.gov/geo/.
References
- Wang Y, Nicholes K, Shih IM. The origin and pathogenesis of endometriosis. Annu Rev Pathol. 2020;15:71–95.
- Ng SW, Norwitz SG, Taylor HS, et al. Endometriosis: the role of iron overload and ferroptosis. Reprod Sci. 2020;27(7):1383–1390.
- Vallee A, Lecarpentier Y. Curcumin and endometriosis. Int J Mol Sci. 2020;21;2440.
- Reis FM, Coutinho LM, Vannuccini S, et al. Is stress a cause or a consequence of endometriosis? Reprod Sci. 2020;27(1):39–45.
- Prins JR, Marissen LM, Scherjon SA, et al. Is there an immune modulating role for follicular fluid in endometriosis? A narrative review. Reproduction. 2020;159(1):R45–R54.
- Schwartz K, Llarena NC, Rehmer JM, et al. The role of pharmacotherapy in the treatment of endometriosis across the lifespan. Expert Opin Pharmacother. 2020;21(8):893–903.
- Barani M, Bilal M, Sabir F, et al. Nanotechnology in ovarian cancer: diagnosis and treatment. Life Sci. 2021;266:118914.
- Anglesio MS, Yong PJ. Endometriosis-associated ovarian cancers. Clinical Obstetrics and Gynecology. 2017;60:711–727.
- Kobayashi H, Sumimoto K, Moniwa N, et al. Risk of developing ovarian cancer among women with ovarian endometrioma: a cohort study in Shizuoka, Japan. Int J Gynecol Cancer. 2007;17(1):37–43.
- Brinton LA, Sakoda LC, Sherman ME, et al. Relationship of benign gynecologic diseases to subsequent risk of ovarian and uterine tumors. Cancer Epidemiol Biomarkers Prev. 2005;14(12):2929–2935.
- Yachida N, Yoshihara K, Yamaguchi M, et al. How does endometriosis lead to ovarian cancer? The molecular mechanism of endometriosis-associated ovarian cancer development. Cancers. 2021;13:1439.
- Gaia-Oltean AI, Braicu C, Gulei D, et al. Ovarian endometriosis, a precursor of ovarian cancer: histological aspects, gene expression and microRNA alterations (review). Exp Ther Med. 2021;21(3):243.
- Murakami K, Kotani Y, Shiro R, et al. Endometriosis-associated ovarian cancer occurs early during follow-up of endometrial cysts. Int J Clin Oncol. 2020;25(1):51–58.
- Oda K, Hamanishi J, Matsuo K, et al. Genomics to immunotherapy of ovarian clear cell carcinoma: unique opportunities for management. Gynecol Oncol. 2018;151(2):381–389.
- Vercellini P, Vigano P, Somigliana E, et al. Endometriosis: pathogenesis and treatment. Nat Rev Endocrinol. 2014;10(5):261–275.
- Gerhold DL, Jensen RV, Gullans SR. Better therapeutics through microarrays. Nat Genet. 2002;32(Suppl.):547–551.
- Cheung VG, Spielman RS. The genetics of variation in gene expression. Nat Genet. 2002;32(Suppl.):522–525.
- Udhaya Kumar S, Thirumal Kumar D, Bithia R, et al. Analysis of differentially expressed genes and molecular pathways in familial hypercholesterolemia involved in atherosclerosis: a systematic and bioinformatics approach. Front Genet. 2020;11:734.
- Udhaya Kumar S, Bithia R, Thirumal Kumar D, et al. Involvement of essential signaling cascades and analysis of gene networks in diabesity. Genes. 2020;11:25.
- Fu D, Zhang B, Yang L, et al. Development of an immune-related risk signature for predicting prognosis in lung squamous cell carcinoma. Front Genet. 2020;11:978.
- Wan J, Jiang S, Jiang Y, et al. Data mining and expression analysis of differential lncRNA ADAMTS9-AS1 in prostate cancer. Front Genet. 2019;10:1377.
- Zeng X, Yue Z, Gao Y, et al. NR4A1 is involved in fibrogenesis in ovarian endometriosis. Cell Physiol Biochem. 2018;46(3):1078–1090.
- Yin M, Zhang J, Zeng X, et al. Target identification and drug discovery by data-driven hypothesis and experimental validation in ovarian endometriosis. Fertil Steril. 2021;116(2):478–492.
- Chen M, Zhou Y, Xu H, et al. Bioinformatic analysis reveals the importance of epithelial-mesenchymal transition in the development of endometriosis. Sci Rep. 2020;10(1):8442.
- Dai FF, Bao AY, Luo B, et al. Identification of differentially expressed genes and signaling pathways involved in endometriosis by integrated bioinformatics analysis. Exp Ther Med. 2019;19:264–272.
- Hu W, Xie Q, Xu Y, et al. Integrated bioinformatics analysis reveals function and regulatory network of miR-200b-3p in endometriosis. Biomed Res Int. 2020;2020:3962953.
- Khalaj K, Miller JE, Lingegowda H, et al. Extracellular vesicles from endometriosis patients are characterized by a unique miRNA-lncRNA signature. JCI Insight. 2019;4:e128846.
- Wang D, Luo Y, Wang G, et al. Circular RNA expression profiles and bioinformatics analysis in ovarian endometriosis. Mol Genet Genomic Med. 2019;7(7):e00756.
- Barrett T, Wilhite SE, Ledoux P, et al. NCBI GEO: archive for functional genomics data sets-update. Nucleic Acids Res. 2013;41(Database issue):D991–D995.
- Lou W, Ding B, Xu L, et al. Construction of potential glioblastoma multiforme-related miRNA-mRNA regulatory network. Front Mol Neurosci. 2019;12:66.
- Li MX, Jin LT, Wang TJ, et al. Identification of potential core genes in triple negative breast cancer using bioinformatics analysis. Onco Targets Ther. 2018;11:4105–4112.
- The Gene Ontology Consortium. The gene ontology resource: 20 years and still GOing strong. Nucleic Acids Res. 2019;47:D330–D338.
- Thomas PD, Hill DP, Mi H, et al. Gene ontology causal activity modeling (GO-CAM) moves beyond GO annotations to structured descriptions of biological functions and systems. Nat Genet. 2019;51(10):1429–1433.
- Dennis G Jr, Sherman BT, Hosack DA, et al. DAVID: database for annotation. Genome Biol. 2003;4(9):P3.
- Zhao L, Fu X, Han X, et al. Tumor mutation burden in connection with immune-related survival in uterine corpus endometrial carcinoma. Cancer Cell Int. 2021;21(1):80.
- Rabbani G, Baig MH, Ahmad K, et al. Protein–protein interactions and their role in various diseases and their prediction techniques. Curr Protein Pept Sci. 2018;19(10):948–957.
- Tang Z, Li C, Kang B, et al. GEPIA: a web server for cancer and normal gene expression profiling and interactive analyses. Nucleic Acids Res. 2017;45(W1):W98–W102.
- Griffith M, Griffith OL, Coffman AC, et al. DGIdb: mining the druggable genome. Nat Methods. 2013;10(12):1209–1210.
- Rudnicka L, Varga J, Christiano AM, et al. Elevated expression of type VII collagen in the skin of patients with systemic sclerosis. Regulation by transforming growth factor-beta. J Clin Invest. 1994;93(4):1709–1715.
- Ryynanen M, Ryynanen J, Sollberg S, et al. Genetic linkage of type VII collagen (COL7A1) to dominant dystrophic epidermolysis bullosa in families with abnormal anchoring fibrils. J Clin Invest. 1992;89(3):974–980.
- Zondervan KT, Becker CM, Koga K, et al. Endometriosis. Nat Rev Dis Primers. 2018;4(1):9.
- Mizutani S, Matsumoto K, Kato Y, et al. New insights into human endometrial aminopeptidases in both implantation and menstruation. Biochim Biophys Acta Proteins Proteom. 2020;1868(2):140332.
- Montgomery GW, Mortlock S, Giudice LC. Should genetics now be considered the pre-eminent etiologic factor in endometriosis? J Minim Invasive Gynecol. 2020;27(2):280–286.
- Pearce CL, Stram DO, Ness RB, et al. Population distribution of lifetime risk of ovarian cancer in the United States. Cancer Epidemiol Biomarkers Prev. 2015;24(4):671–676.
- Munksgaard PS, Blaakaer J. The association between endometriosis and ovarian cancer: a review of histological, genetic and molecular alterations. Gynecol Oncol. 2012;124(1):164–169.
- Eyster KM, Klinkova O, Kennedy V, et al. Whole genome deoxyribonucleic acid microarray analysis of gene expression in ectopic versus eutopic endometrium. Fertil Steril. 2007;88(6):1505–1533.
- Yu W, Li D, Zhang Y, et al. MiR-142-5p acts as a significant regulator through promoting proliferation, invasion, and migration in breast cancer modulated by targeting SORBS1. Technol Cancer Res Treat. 2019;18:1533033819892264.
- Bhandari A, Xia E, Zhou Y, et al. ITGA7 functions as a tumor suppressor and regulates migration and invasion in breast cancer. Cancer Manag Res. 2018;10:969–976.
- Huang W, Yu D, Wang M, et al. ITGBL1 promotes cell migration and invasion through stimulating the TGF-β signalling pathway in hepatocellular carcinoma. Cell Prolif. 2020;53(7):e12836.
- Li W, Li S, Yang J, et al. ITGBL1 promotes EMT, invasion and migration by activating NF-κB signaling pathway in prostate cancer. Onco Targets Ther. 2019;12:3753–3763.
- Sun L, Wang D, Li X, et al. Extracellular matrix protein ITGBL1 promotes ovarian cancer cell migration and adhesion through wnt/PCP signaling and FAK/SRC pathway. Biomed Pharmacother. 2016;81:145–151.
- Ge JC, Wang YX, Chen ZB, et al. Integrin alpha 7 correlates with poor clinical outcomes, and it regulates cell proliferation, apoptosis and stemness via PTK2-PI3K-Akt signaling pathway in hepatocellular carcinoma. Cell Signal. 2020;66:109465.
- Liao HD, Mao Y, Ying YG. The involvement of the laminin-integrin alpha7beta1 signaling pathway in mechanical ventilation-induced pulmonary fibrosis. J Thorac Dis. 2017;9(10):3961–3972.
- Song J, Yang P, Lu J. Upregulation of ITGBL1 predicts poor prognosis and promotes chemoresistance in ovarian cancer. Cancer Biomark. 2020;27(1):51–61.
- Ji Q, Zhou L, Sui H, et al. Primary tumors release ITGBL1-rich extracellular vesicles to promote distal metastatic tumor growth through fibroblast-niche formation. Nat Commun. 2020;11(1):1211.
- Cho WC, Jang JE, Kim KH, et al. SORBS1 serves a metastatic role via suppression of AHNAK in colorectal cancer cell lines. Int J Oncol. 2020;56(5):1140–1151.
- Song L, Chang R, Dai C, et al. SORBS1 suppresses tumor metastasis and improves the sensitivity of cancer to chemotherapy drug. Oncotarget. 2017;8(6):9108–9122.
- Qi F, Qin WX, Zang YS. Molecular mechanism of triple-negative breast cancer-associated BRCA1 and the identification of signaling pathways. Oncol Lett. 2019;17(3):2905–2914.
- Hsu HM, Chu CM, Chang YJ, et al. Six novel immunoglobulin genes as biomarkers for better prognosis in triple-negative breast cancer by gene co-expression network analysis. Sci Rep. 2019;9(1):4484.
- Yeong J, Lim JCT, Lee B, et al. High densities of tumor-associated plasma cells predict improved prognosis in triple negative breast cancer. Front Immunol. 2018;9:1209.
- Rolim I, Duarte N, Barata G, et al. Immunoglobulin M gene association with autoantibody reactivity and type 1 diabetes. Immunogenetics. 2017;69(7):429–437.
- Ceballos-Francisco D, Cordero H, Guardiola FA, et al. Healing and mucosal immunity in the skin of experimentally wounded gilthead seabream (Sparus aurata L). Fish Shellfish Immunol. 2017;71:210–219.
- Udhaya Kumar S, Saleem A, Thirumal Kumar D, et al. A systemic approach to explore the mechanisms of drug resistance and altered signaling cascades in extensively drug-resistant tuberculosis. Adv Protein Chem Struct Biol. 2021;127:343–364.
- Mishra S, Shah MI, Udhaya Kumar S, et al. Network analysis of transcriptomics data for the prediction and prioritization of membrane-associated biomarkers for idiopathic pulmonary fibrosis (IPF) by bioinformatics approach. Adv Protein Chem Struct Biol. 2021;123:241–273.
- Udhaya Kumar S, Thirumal Kumar D, Siva R, et al. Dysregulation of signaling pathways due to differentially expressed genes from the B-cell transcriptomes of systemic lupus erythematosus patients – a bioinformatics approach. Front Bioeng Biotechnol. 2020;8:276.
- Yan H, Zheng G, Qu J, et al. Identification of key candidate genes and pathways in multiple myeloma by integrated bioinformatics analysis. J Cell Physiol. 2019;234(12):23785–23797.
- Kumar SU, Kumar DT, Siva R, et al. Integrative bioinformatics approaches to map potential novel genes and pathways involved in ovarian cancer. Front Bioeng Biotechnol. 2019;7:391.
- Wang B, Yin X, Gan W, et al. PRCC-TFE3 fusion-mediated PRKN/parkin-dependent mitophagy promotes cell survival and proliferation in PRCC-TFE3 translocation renal cell carcinoma. Autophagy. 2020;1–19.
- Gutierrez Olivares VM, Gonzalez Torres LM, Hunter Cuartas G, et al. Immunohistochemical profile of renal cell tumours. Rev Esp Patol. 2019;52(4):214–221.
- Schorey JS, Bhatnagar S. Exosome function: from tumor immunology to pathogen biology. Traffic. 2008;9(6):871–881.
- Zhou W, Lian Y, Jiang J, et al. Differential expression of microRNA in exosomes derived from endometrial stromal cells of women with endometriosis-associated infertility. Reprod Biomed Online. 2020;41(2):170–181.
- Wu J, Huang H, Huang W, et al. Analysis of exosomal lncRNA, miRNA and mRNA expression profiles and ceRNA network construction in endometriosis. Epigenomics. 2020;12(14):1193–1213.
- Shi Y, Zha J, Zuo M, et al. Long noncoding RNA CHL1-AS1 promotes cell proliferation and migration by sponging miR-6076 to regulate CHL1 expression in endometrial cancer. J Cell Biochem. 2020;121(3):2655–2663.
- Xu Z, Zhang L, Yu Q, et al. The estrogen-regulated lncRNA H19/miR-216a-5p axis alters stromal cell invasion and migration via ACTA2 in endometriosis. Mol Hum Reprod. 2019;25(9):550–561.
- Liu Z, Liu L, Zhong Y, et al. LncRNA H19 over-expression inhibited Th17 cell differentiation to relieve endometriosis through miR-342-3p/IER3 pathway. Cell Biosci. 2019;9:84.