
 ?Mathematical formulae have been encoded as MathML and are displayed in this HTML version using MathJax in order to improve their display. Uncheck the box to turn MathJax off. This feature requires Javascript. Click on a formula to zoom.
?Mathematical formulae have been encoded as MathML and are displayed in this HTML version using MathJax in order to improve their display. Uncheck the box to turn MathJax off. This feature requires Javascript. Click on a formula to zoom.Abstract
Introduction: Bispecific antibody (BiAb)-armed activated T cells (BATs) comprise an adoptive T cell therapy platform for treating cancer. Arming activated T cells (ATC) with anti-CD3 x anti-tumour associated antigen (TAA) BiAbs converts ATC into non-major histocompatibility complex (MHC)-restricted anti-tumour cytotoxic T lymphocytes (CTLs). Binding of target antigens via the BiAb bridge enables specific anti-tumour cytotoxicity, Th1 cytokines release, and T cell proliferation. Clinical trials in breast, prostate, and pancreatic cancer using BATs armed with chemically heteroconjugated BiAbs demonstrated safety, feasibility, induction of anti-tumour immune responses and potential increases in overall survival (OS).
Objectives: The primary objective of this study was to develop a recombinant BiAb that confers enhanced anti-tumour activity of BATs against a broad range of solid tumours.
Methods: A recombinant anti-epidermal growth factor receptor (EGFR) x anti-CD3 (OKT3) BiAb (rEGFRBi) was designed and expressed in CHO cells, used to arm ATC (rEGFR-BATs), and tested for specific cytotoxicity against breast, pancreatic and prostate cancers and glioblastoma.
Results: rEGFR-BATs exhibit remarkably enhanced specific cytotoxicity and T1 cytokine secretion against a wide range of solid tumour cell lines vs. their respective chemically-heteroconjugated BATs.
Conclusion: rEGFR-BATs may provide a “universal” T cell therapy for treating a wide range of solid tumours.
A (Gly4Ser)6 linker between the variable light and heavy chains of an scFv fused to the N-terminus of a heavy chain antibody confers unexpected stability to the heavy chain fusion protein and supports the efficient expression of the bispecific antibody.
Arming of activated T cells with the rEGFRBi greatly enhances the relative cytotoxicity and Th1 cytokine secretion of theT cells relative to a chemically heteroconjugated BiAbs.
rEGFR-BATs are promising candidates for the treatment of a broad range of solid tumours.
KEY MESSAGE
Introduction
Chemotherapy (chemoT) resistance and immunosuppression provide major challenges to cancer therapy. Despite recent advances in chemoT and immunotherapy (IT), an estimated 1,762,450 new cases of cancer and 606,880 cancer deaths are projected in the United States in 2019 [Citation1]. Cancer cells in the tumour microenvironment (TME) develop resistance to chemoT and/or IT. While redirected T cells offer a promising new avenue in the anti-cancer treatment landscape, major challenges remain, including variable expression of TAA, clonal escape, structural and physical barriers in the tumour stroma that inhibit migration of effector cells and chemotherapeutics, tumour-promoting factors produced by tumours, myeloid-derived suppressor cells (MDSC) and T regulatory cells (TREGs). Chimeric antigen receptor (CAR)-T cells not only cause cytokine release syndrome (CRS) but also become exhausted in the solid TME [Citation2,Citation3]. There are numerous trials using BiAbs for solid tumour and haematologic malignancies but only BLINCYTO® (blinatumomab) directed at CD19 is FDA approved. These results underscore the need to develop non-toxic and more effective BiAbs for retargeting T cells to solid tumours.
EGFR (ERBB-1 Receptor, HER1) and the human epidermal growth factor receptor 2 (HER2) are receptor tyrosine kinases belonging to the EGF family of growth factors that play a central role in the generation of several carcinomas. EGFR is overexpressed in head and neck cancer, genito-urinary cancers, gastrointestinal malignancies, prostate carcinoma, breast carcinoma, thyroid carcinomas, salivary gland carcinomas, melanoma gastro-esophageal cancer, non-small cell lung cancer (NSCLC), and endometrial cancers [Citation4–7]. EGFR and HER2 overexpression is seen in the advanced disease stage and is associated with poor prognosis [Citation8–10]. Interventions targeting EGFR and HER2 pathways in solid tumours have shown improved patient outcomes. Several therapies including the monoclonal antibodies (mAbs) Erbitux® (cetuximab) and Herceptin® (trastuzumab) and small-molecule tyrosine kinase inhibitors targeting HER2 and EGFR signalling pathways are now FDA approved. Although cetuximab was approved by the FDA for the treatment of metastatic colon cancer and head and neck cancer with concurrent radiation therapy, results in pancreatic cancer were disappointing [Citation11].
Arming anti-CD3-ATC with chemically heteroconjugated anti-CD3 × TAA BiAb converts every ATC into a non-MHC-restricted serial killer CTL [Citation12,Citation13]. In vitro data show that BATs (i) proliferate, (ii) kill tumour cells releasing tumour antigens and (iii) secrete Th1 cytokines/chemokines leading to the recruitment and activation of endogenous immune cells [Citation14] and subsequent in situ vaccination of the patient with antigen and epitope spreading [Citation15].
We selected a recombinant tetravalent BiAb (rTBiAb) format to engineer and produce a homogeneous recombinant anti-CD3 × anti-EGFR BiAb. This study presents the characterization of rEGFRBi and the in vitro activity of rEGFR-BATs versus first-generation chemically heteroconjugated BiAb-armed ATC against a broad range of solid tumour cell lines.
Results
Expression of recombinant BiAb
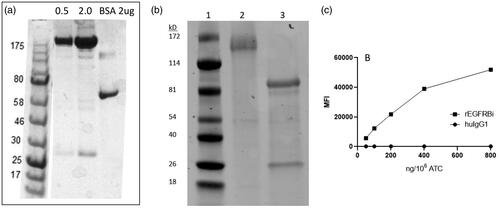
High expressing clones were identified to produce rEGFRBi which was purified by Protein A column chromatography and characterized by SDS PAGE. rEGFRBi is expressed as a single band of the expected size () that comprises full-length light chain and heavy chain fusion chain fragments (). Clone 1E2 was chosen for functional characterization.
Figure 1. (a) SDS PAGE of rEGFRBi clone 1E2. Lanes contain 0.5 or 2 µL of purified BiAb adjacent to 2 µg BSA. Molecular weight markers are indicated in kilodaltons. (b) SDS PAGE of rEGFRBi clone 1E2 under non-reducing (Lane 2) and reducing conditions (Lane 3). (c) Binding of rEGFRBi to normal donor ATC. Concentrations of rEGFRBi ranging from 50–800 ng/106 ATC were incubated with ATC and stained for bound BiAb with PE-conjugated anti-human IgG by flow cytometry. MFI: median fluorescence intensity.
Binding of rEGFRBi to ATC and EGFR-expressing cell lines
As rEGFRBi comprises an anti-CD3 single-chain variable fragment (scFv) linked to the N-terminus of the heavy chain of an Erbitux-human IgG1-Fc construct, the binding of rEGFRBi to ATC, and rEGFRBi and Erbitux to solid tumour targets were compared using flow cytometry. For binding to ATC, a concentration range of 50 to 800 ng/106 cells was used to arm ATC from a normal donor. The median fluorescence intensity (MFI) of phycoerythrin (PE)-anti-human IgG secondary antibody staining was plotted for the rEGFRBi vs. the same concentration of human IgG1. rEGFRBi showed a linear increase in binding to ATC up to 400 ng of rEGFRBi/106 ATC and began to plateau at 800 ng/106 ().
All cell lines express easily detectable levels of EGFR except for MCF7, which expresses very low levels of EGFR. rEGFRBi and cetuximab were incubated with SK-BR-3, MIA PaCa-2 and MCF7 cells at between 0.5 and 4 µg/mL followed by a PE-anti-human IgG secondary antibody. Both antibodies showed similar MFI at each concentration for all three cell lines (). rEGFRBi clearly binds to CD3 on ATC and to EGFR on the target cells.
Table 1. Median fluorescence intensity (1000) of Erbitux and rEGFRBi bound to tumour cells.
Effective arming doses of BiAb on rEGFR-BATs, chemically heteroconjugated HER2Bi-armed ATC (HER2-BATs) or heteroconjugated EGFRBi-armed ATC (EGFR-BATs)
Both preclinical and clinical data show that HER2-BATs and EGFR-BATs armed with 50 ng of BiAb/106 ATC show cytotoxic activity as measured by specific 51Cr release assays and the induction of Th1 cytokines when the BATs engage tumour targets [Citation13,Citation14]. Because the chemically heteroconjugated BiAb reaction is comprised of 20 ∼ 30% dimer, in addition to multimer BiAb and unconjugated monomers, both free OKT3 and BiAbs bind to ATC during arming. On a molecular weight basis, the rEGFRBi represents approximately 3-fold more BiAb vs. the heteroconjugated BiAb mixture. Therefore, a range of rEGFRBi arming concentrations from between 1 to 400 ng of BiAb/106 ATC was used for comparison to the ATC armed with 50 ng of the respective heteroconjugated BiAb/106 ATC. A range of E:T (2:1–10:1) was tested. All comparisons among the different BiAbs were made using the same normal donor cells armed under the same conditions.
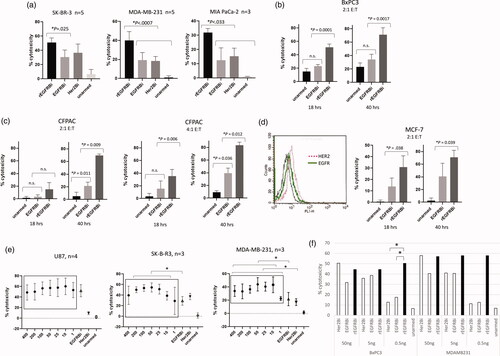
At E:T of 10:1, rEGFR-BATs showed significantly greater cytotoxicity vs. EGFR-BATs against the breast cancer cell lines SK-BR-3 and MDAMB-231, and the pancreatic cancer line MIA PaCa-2 in an 18 h 51Cr-release assay (); they were also significantly greater than HER2-BATs against MDA-MB-231 and MIA-PACA-2 targets. When cytotoxicity was measured in the xCELLigence RTCA MP system, rEGFR-BATs were approximately twice as potent as EGFR-BATs against the pancreatic cell lines BxPC3 and CFPAC-1 at the lower E:T’s of 2:1 and/or 4:1 over the course of 40 h () and the EGFR low expressing cell line MCF7 ().
Figure 2. Cytotoxicity by BATs incubated with solid tumour cell lines. (a) Cytotoxicity of rEGFR-, HER2-, and EGFR-BATs from healthy donors against SK-BR-3, MDA MB231 and MIA PaCa cells at 10:1 E:T in 18 h 51Cr-release assays. *p < .05. (b) Cytotoxicity of rEGFR- and EGFR-BATs against BxPC3 cells at 2:1 E:T using xCELLigence RTCA system at 18 and 40 h (n = 4). (c) Cytotoxicity of rEGFR- and EGFR-BATs against CFPAC-1 cells at 2:1 and 4:1 E:T using xCELLigence RTCA system at 18 and 40 h (n = 3). n.s.: not significant (p > .05). (d) Cytotoxicity of rEGFR- and EGFR-BATs against MCF-7 cells at 2:1 E:T over 40 h (right panel). MCF7 cells express low levels of both HER2 and EGFR when stained with Herceptin and Erbitux, respectively (left panel). (e) Cytotoxicity of rEGFR-BATs armed at 1–400 ng BiAb/106 cells vs. EGFR- and HER2-BATs armed at 50 ng BiAb/106 cells. For U87, there was no significant differences among any of the rEGFR arming concentrations nor those conditions and 50 ng of EGFRBi. For SK-BR-3, concentrations of 10 ng to 200 ng rEGFRBi were significantly greater than EGFRBi (50 ng) but not HER2 (50 ng). For MDA-MB-231, concentrations of rEGFRBi between 10 and 400 ng were significantly greater than EGFRBi (50 ng), whereas all concentrations of rEGFRBi were significantly greater than HER2 (50 ng). *p < .05. (f) Average cytotoxicity from 2 healthy donors’ ATC armed with 50, 5 or 0.5 ng BiAb/106 cells in an 18 h 51Cr-release assay at 10:1 E:T. *p < .05.
rEGFR-BATs maintain cytotoxicity over a broad range of arming concentrations
Normal donor ATC were armed with rEGFRBi with 1, 10, 25, 50, 100, 200 or 400 ng of rEGFRBi/106 cells and tested against MDA-MB-231, SK-BR-3 and U87 cells in an 18 h 51Cr-release assay at 10:1 E:T. For U87 cells there was no significant difference in cytotoxicity among the different rEGFRBi arming conditions nor between any of the rEGFR-BATs conditions and EGFRBi-BATs armed at 50 ng of EGFRBi/106cells (). And, as demonstrated earlier by Zitron et al. [Citation16], HER2-BATs did not kill U87 cells. With SK-BR-3 targets, there was no significant difference among rEGFR-BATs armed at 400 to 10 ng of rEGFRBi, nor any of those rEGFR-BATs conditions and HER2-BATs. However, rEGFR-BATs armed between 10 and 200 ng were significantly higher than EGFR-BATs. For MDA-MB-231, there was no significant difference among rEGFR-BATs between 10 and 100 ng, although all of those were significantly greater than rEGFRBi at 200 and 400 ng. rEGFR-BATs at 25, 50 and 100 ng were significantly higher than at 1 ng, while those at 10, 200 and 400 were not different than 1 ng. However, rEGFRBi-BATs armed from 10 to 400 ng were significantly higher than for EGFR-BATs, while rEGFR-BATS from 1 to 400 were significantly higher than for HER2-BATs (). When 2 normal donors were armed over a narrower range of 50, 5 and 0.5 ng and tested against BXPC-3 and MDA-MB-231 cells, rEGFR-BATs maintained the same level of killing across all three concentrations, while EGFR- and HER2-BATs dropped off at 0.5 ng (). The same pattern was seen with a normal donor against MIA PaCa-2 cells (pancreatic cancer, data not shown).
rEGFR-BATs release greater levels of Th1 cytokines against breast, pancreatic and prostate cancer
rEGFR-BATs were tested for the release of Th1 cytokines granulocyte-macrophage colony-stimulating factor (GM-CSF), interferon gamma (IFN-γ), tumour necrosis factor-alpha (TNF-α), and Granzyme B (Gr B) after overnight culture with tumour cells at 6:1 E:T vs. the respective chemically heteroconjugated BATs that are used clinically for the given tumour cell types (). Thus, rEGFR-BATs were compared to EGFR-BATs against pancreatic cancer lines MIA-PACA-2, ASPC1 and CFPAC-1 and to HER2-BATs against breast cancer line MDA-MB-231 and prostate cancer line PC3. The relative fold increases in Th1 cytokine expression were much higher than the relative increases in cytotoxicity for all the cell lines, ranging between 2.9 and 7.2 for GM-CSF, 3.6 and 8.2 for IFN-γ, 3.3 and 5.7 for TNF-α and 1.7 and 3.6 for Gr B. summarizes the relative fold-increase in secretion levels for the six cell lines. In a separate experiment, two normal donors (VA-07 and VA-53) were armed with rEGFRBi at 50 ng/106 cells and incubated without target cells for 18 h with a cell number equivalent to a 10:1 E:T. They produced low or not detected (ND) levels of IFN-γ, GM-CSF, TNF-α or Granzyme B relative to that produced in the presence of EGFR-positive SKBR3 breast cancer cell targets (). Together, these data demonstrate that the release of Th1 cytokines requires specific engagement with EGFR-expressing target cells.
Figure 3. Cytokine production from rEGFR- and either HER2- or EGFR-BATs against solid tumour lines incubated at 6:1 E:T for 18 h. For (a–e), values are the means of n = 3 donors with ATC armed with 50 ng BiAb/106 cells (a) MIA PaCa-2. (b) ASPC1. (c) CFPAC-1. (d) MDA-MB-231. (e) PC3. p-Values are indicated for each BATs pair. (f) Relative expression of Th1 cytokines by rEGFR-BATs armed at 25 ng and 8 ng/106 ATC and HER2-BATs armed at 50 ng/106 ATC against PC3 cells at 12:1, 6:1 and 3:1 E:T. Values are the means of n = 2. *p < .05.
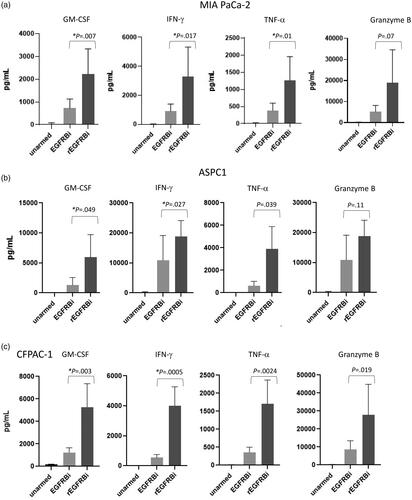
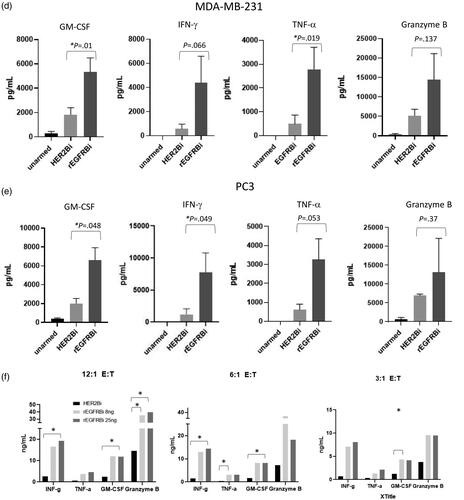
Table 2. Ratio of increased cytokine secretion by rEGFR-BATs vs. EGFR- BATs (pancreatic cancer), HER2-BATs (breast and prostate cancer) and GD2 BATs (neuroblastoma).
Table 3. Th1 cytokine production by rEGFRBi-armed ATC alone (ng/mL).
rEGFR-BATs release greater levels of Th1 cytokines over a range of E:T
rEGFR-BATs were armed at 8 and 25 ng rEGFRBi/106 ATC (n = 2 normal donors) and compared with HER2-BATs armed at 50 ng BiAb/106 cells against PC3 cells. At E:T’s of 12-, 6- and 3:1, rEGFR-BATs produce 5- to 10-fold higher levels of IFN-γ, TNF-α, GM-CSF and multifold levels of Gr B at each E:T, and more impressively, multifold higher levels of IFN-γ, TNF-α, GM-CSF at 3:1 than the HER2-BATs at 12:1 ().
In summary, rEGFR-BATs not only killed better at lower arming doses and E:T, they killed every cell line tested. In particular, U87 GBM cells were highly susceptible to rEGFR-BATs even though they cannot be killed by HER2-BATs.
Discussion
Our data demonstrate the ability to manufacture stable, full-length tetravalent rEGFRBi from stably expressing CHO-S cells. This is a surprising result given the inability to express full-length BiAbs when (Gly4Ser)3 or (Gly4Ser)4 linkers are present in an scFv similarly fused to the N-terminus of a second antibody [Citation17]. rEGFR-BATs were shown to exhibit greater potency than both EGFR- and HER2-BATs in terms of cytotoxicity and/or Th1 cytokine secretion against breast, pancreatic, prostate, and glioblastoma cell lines. They maintain high levels of killing across a wide range of arming concentrations and release higher levels of cytokines and chemokines even at lower E:T than chemically heteroconjugated EGFRBi- and HER2Bi-armed ATC. Bivalency for each variable region specificity is important to maintain attachment of the BiAbs to ATC and optimize activation upon engagement with TAA. Monovalent formats may lead to faster off-rates from the ATC that would reduce or preclude the activity of ex-vivo armed products. The BATs strategy permits optimization of the anti-tumour activity of ATC alone that are only triggered in vivo upon engagement of target antigen. In contrast, the intravenous injection of anti-CD3-containing BiAbs must interact with the different populations of circulating T cells to deliver an effective arming concentration without causing systemic activation. The binding affinity of anti-EGFR in the rEGFRBi is similar to that of Erbitux with respect to binding to tumour lines MCF7, MIA PaCa-2 and SK-BR-3. However, there is a significant difference in the relative binding of EGFRBi and rEGFRBi to ATC. The former is limited by the presence of unconjugated OKT3 in the arming mixture, which competes for binding to the ATC and may influence activation via the TCR separate from the BiAb; furthermore, the relative configuration of the two paratopes is determined by the nature of the chemical heteroconjugation of the two input antibodies. The rEGFRBi is capable of at least 20-fold greater binding capacity to T cells, although the optimal arming concentration is well below this. Functionally, the configuration of rEGFRBi leads to enhanced cytotoxicity and Th1 cytokine secretion by BATs. This improvement may due to shorter spacing between the anti-CD3 and anti-EGFR variable domains in the recombinant vs. the chemically heteroconjugated BiAbs, which would be an effect similar to that observed with CAR T cells transduced with scFvs containing linkers of differing lengths [Citation18,Citation19]. Upon engagement with tumour cells, the ATC may be brought closer to the tumour cell surface which may lead to more efficient binding of adhesion molecules, TCR crosslinking, or both, that may more efficiently activate T cell responses and/or cell division. Further experiments are required to determine the relative contribution of these differences. The larger ratios of cytokines released by rEGFRBi-BATs engagement are much more pronounced than the fold-increases in cytotoxicity assays and may more accurately reflect the relative cytotoxic activity as well as the potential for promoting anti-tumour immune responses within the TME.
Clinical trials of HER2- and EGFR-BATs armed with chemically heteroconjugated BiAbs demonstrated the safety and feasibility of multiple BATs infusions for a variety of solid tumours [Citation20–22]. BATs are produced using BiAbs to arm ex vivo expanded ATCs, which creates an “army” of non-MHC-restricted serial killers that can flexibly be adapted to target one or more TAA. Unlike CAR-T cells, BATs are non-transgenic and are thus self-limiting due to the cell-surface attachment of BiAbs; the concentration of BiAbs decreases as the cells divide and eventually lose activity against their target. BATs are therefore inherently safer than CAR-T, T cell receptor engineered-T cells or CAR-natural killer cells. More importantly, BATs have not been associated with CRS even at multiple doses of up to 40 billion/cells per infusion.
Immune evaluations in BATs clinical trials support the concept that multiple BATs infusions provide a direct anti-tumour effect as well as modifying the TME to promote an endogenous anti-tumour immune response upon release of TAA and Th1 cytokines [Citation15,Citation20]. In the Phase I trial of metastatic breast cancer patients treated with HER2-BATs, there was a marked increase in IL-12 that occurred after the fourth BATs infusion, indicating the activation of macrophages/dendritic cells over time. There was also an increase in anti-tumour cellular cytotoxicity of unarmed ATC that were expanded after the eighth infusion. We also previously showed that EGFR-BATs co-cultured with MIA PaCa-2 pancreatic cells in a 3 D co-culture model of PBMC decreased the differentiation of MDSC in the presence of Th1 cytokines [Citation23]. Therefore, multiple BATs infusions are likely to increase overall cytotoxicity, modulating the TME and promoting in situ immunization more efficiently than a single infusion of cells due to accumulated benefits from “waves” of tumour engagement.
The higher activity of rEGFR-BATs has the potential to increase toxicity due to either direct cytotoxicity against tumour tissue and cells of the TME, and/or the release of Th1 cytokines. The ability to arm ATC over a wide range of concentrations provides an important mechanism to control potency, and because BATs are inherently self-limiting, further ways to regulate their activity include modifying the number of BATs per infusion and the overall number and frequency of infusions. In this manner, BATs can be administered more like a conventional drug vs. a long-lived replicating, gene-modified cell.
The expression of EGFR on a broad range of solid tumours, the role of EGFR signalling in cancer stem cell biology, the upregulation of EGFR on Tregs in cancer patients, and its expression on other tumour promoting cells of the TME, make the rEGFR-BATs product suitable for many solid tumour indications. If broadly effective, they would preclude the need for extensive preclinical screening and safety testing of novel anti-TAA antibodies, some of which have caused serious side effects in Phase 1 studies as CAR T cells. The improved potency demonstrated in this study supports the testing of rEGFR-BATs in clinical trials for indications including pancreatic, breast, and prostate cancers, glioblastoma, and others.
Methods
Recombinant anti-EGFR light chain and OKT3 scFv-anti-EGFR heavy chain gene fusion constructs
The variable light and heavy gene sequences from the DrugBank amino acid entry for Erbitux were back-translated using the most common homologous mouse sequences in the IMGT database. The variable light and heavy chain genes for humanized muromonab-CD3 were designed based on amino acid sequences in US patent 5,885,573. The OKT3 scFv-Erbitux-heavy chain fusion gene was assembled in the order: variable light chain – (G4S)6 linker-variable heavy chain – (G4S)5 (with a change in repeat 3 to introduce a G to T substitution) linker – Erbitux heavy chain variable sequence - human IgG1 Fc. Coding regions for the Erbitux light and OKT3scFv-Erbitux fusion genes were then subcloned into the proprietary expression vector SwiMR (US9910038B2) which contains selectable markers for puromycin or neomycin. The amino acid sequences for the Erbitux light chain and humanized OKT3-Erbitux heavy chain fusion chain expression cassettes are shown in .
Figure 4. Amino acid sequences of the rEGFRBi variable regions. Amino acid sequences for Erbitux variable heavy and light chain genes were obtained from the DRUGBANK Online database, accession number DB00002, and back translated to DNA sequences VS327 & 328 used in the construction of the OKT3-EGFR BiAb. The cloned plasmids were sequenced and translated to confirm the correct amino acid match to the DRUGBANK database.

Selection of stably expressing rEGFRBi clones
The plasmid vectors were linearized and used to transfect CHO suspension cells (CHO-S). The clones were selected in the presence of puromycin and G418 and screened for antibody expression by enzyme-linked immunosorbent assay (ELISA). The highest expressing clones were expanded into shake flask cultures and cryopreserved.
BiAb purification
rEGFRBi was purified from cell-free supernatants via Protein A chromatography. The BiAb samples were stored in 1X phosphate buffered saline (PBS) at −80 °C or 4 °C.
Cell lines
The following cell lines were obtained from ATCC: (1) breast cancer lines SK-BR-3, MCF7, MDA-MB-231; (2) pancreatic cancer lines MIA PaCa-2, CFPAC-1, AsPC-1, BxPC-3; (3) prostate cancer line PC-3; and (4) glioblastoma line U87.
Preparation of heteroconjugated BiAbs
Trastuzumab, cetuximab and OKT3 (Miltenyi or BioXcell) were purchased. Antibodies were heteroconjugated with OKT3 as described [Citation13,Citation24]. Briefly, OKT3 was cross-linked with Traut’s reagent (Thermo Scientific), and anti-tumour antibodies cross-linked with sulfo-SMCC (Thermo Scientific) per the manufacturer’s instructions. After buffer exchange, equal amounts of each antibody pair (OKT3 and anti-tumour antibody) were mixed and rocked overnight at 4 °C to produce anti-CD3 × anti-HER2 BiAb (HER2Bi) and anti-CD3 × anti-EGFR BiAb (EGFRBi). The BiAbs were characterized sodium by dodecyl sulphate-polyacrylamide gel electrophoresis (SDS PAGE), quantitated using Bio-Rad Protein Assay Kit, and stored at 4 °C.
Flow cytometry
Approximately 0.5 × 106 tumour cells or ATC were incubated with rEGFRBi, Erbitux, Herceptin or human IgG1 antibodies in 100 µL flow wash buffer (PBS, 0.2% BSA) for 30 min at 4 °C. The cells were washed and incubated with Phycoerythrin (PE)-conjugated anti-human IgG (BioLegend 409303). Cells were analyzed by flow cytometry using an ACEA Biosciences (San Diego, CA) NovoCyte flow cytometer and analyzed with NovoExpress® software.
Activated T lymphocytes
Peripheral blood mononuclear cells (PBMC) from normal subjects were obtained under University of Virginia (UVA) IRB#18904 under informed and written consent. PBMC were isolated by Ficoll-Hypaque (Lymphocyte Separation Medium from Corning) and stimulated with OKT3 at 20 ng/mL and expanded in RPMI-1640 containing 10% foetal calf serum and IL-2 (100 IU/mL) [Citation14]. ATC was armed with the designated concentration of BiAbs for 15 min under rocking conditions at room temperature, washed with a medium containing 10% foetal bovine serum and resuspended at the required concentration for a given E:T.
Cytotoxicity assays
51Cr-release assays were performed as previously described [Citation24]. Briefly, between 10 and 20,000 target cells were seeded per well of 96-well plates in triplicate in 100 µL media and adhered overnight at 37 °C. The cells were radioactively labelled with 2µCi/well of 51Cr for 4–6 h at 37 °C. BATs were added in triplicate 24 or 48 h after seeding at the designated E:T ratios in 100 µL media. After 16–20 h, 100 µL of cell-free supernatant was added to 100 µL liquid scintillation cocktail and counted in a Perkin Elmer MicroBeta scintillation counter. Wells without added effector cells were used as spontaneous release controls. Wells treated with 100 µL of 2% SDS were used as maximum release controls. Percent cytotoxicity was calculated as [(Experimental cpm − Spontaneous release cpm)/(Maximum release cpm)] × 100.
Cytotoxicity assays using the xCELLigence Real Time Cell Analysis Multiple Plate (RTCA MP) system (ACEA Biosciences, Agilent) were performed with the pancreatic cancer cell lines BxPC3 and CFPAC-1 and breast cancer line MCF7. A media-only impedance check was performed first by adding 50 uL of cell culture media to every well of the 96-well E-Plates (ACEA Biosciences, Agilent) and then measuring the background impedance as a unit of Cell Index. The cell lines were trypsinized, washed, and then seeded at 8 × 103 cells/well. Seeded plates were left at ambient temperature for 30 min to allow for even distribution and settling of tumour cells. Afterwards, the plates were transferred to the xCELLigence RTCA MP system in a humidified 37 °C CO2 incubator and incubated overnight. Cell Index data points were collected at 15 min intervals. The next day, cryopreserved OKT3-activated T cells were thawed, washed, and armed with either a chemically conjugated OKT3 × EGFR BiAb or rEGFRBi at 50 ng/1 × 106 cells for 15 min to generate EGFR- and rEGFR-BATs. Three wells of each target cell type were trypsinized and pooled for counting by haemacytometer or by flow cytometry (ACEA Biosciences Novocyte, Agilent). The BATs were washed with culture media and the cell density was adjusted for the desired E:T ratio based on the target cell counts. The BATs and unarmed ATC were seeded onto the E-Plate at E:T ratios of 2:1 or 4:1 in quadruplicate and returned to the RTCA MP. Data acquisition was resumed, and the E-Plate was incubated for continuous monitoring for over 40 h. Data output was normalized to a common Delta Time, which converted Cell Index to Delta Cell Index (DCI) and established a common starting point for each condition. The % cytotoxicity was calculated for 18 and 40 h time points using the following equation:
where: DCITumour Alone is the average Delta Cell Index between replicates of untreated tumour cells;
DCITreated is the average Delta Cell index between replicates of BATs- or ATC-treated wells.
Cytokine assays
Cell-free supernatants from cytotoxicity assays performed without 51Cr were collected and frozen at −20 °C until analyzed. Samples were diluted and analyzed using R&D Luminex kits.
Author contributions
MTFH, VS, AM, JWL, BY and LGL were involved in the conception and design, or analysis and interpretation of the data; the drafting of the paper, revising it critically for intellectual content; and the final approval of the version to be published; all authors agree to be accountable for all aspects of the work.
Disclosure statement
LGL, JWL and MH are co-founders of TransTarget, Inc. JWL and BY are co-founders of Larix, Inc. The data presented in this manuscript are original and have not been published elsewhere except in the form of abstracts and poster presentations at symposiums and meetings.
Data availability statement
The raw data supporting the conclusions of this article will be made available by the authors, without undue reservation, to any qualified researcher.
Additional information
Funding
References
- Siegel RL, Miller MD, Jemal A. Cancer statistics, 2019. CA Cancer J Clin. 2019;69(1):7–34.
- Heyman B, Yang Y. Chimeric antigen receptor T cell therapy for solid tumors: current status, obstacles and future strategies. Cancers. 2019;11(2):191.
- Schmidts A, Maus MV. Making CAR T cells a solid option for solid tumors. Front Immunol. 2018;9:2593.
- Salomon DS, Brandt R, Ciardiello F, et al. Epidermal growth factor-related peptides and their receptors in human malignancies. Crit Rev Oncol Hematol. 1995;19(3):183–232.
- Clauditz TS, Gontarewicz A, Bokemeyer C, et al. Abundant expression of mTOR kinase in salivary gland tumors – potentials as therapy target? J Oral Pathol Med. 2013;42(10):769–773.
- Yan M, Schwaederle M, Arguello D, et al. HER2 expression status in diverse cancers: review of results from 37,992 patients. Cancer Metastasis Rev. 2015;34(1):157–164.
- Chiosea SI, Williams L, Griffith CC, et al. Molecular characterization of apocrine salivary duct carcinoma. Am J Surg Pathol. 2015;39(6):744–752.
- Yasui W, Sumiyoshi H, Hata J, et al. Expression of epidermal growth factor receptor in human gastric and colonic carcinomas. Cancer Res. 1988;48(1):137–141.
- Chung CK, Antoniades HN. Expression of c-sis/platelet-derived growth factor B, insulin-like growth factor I, and transforming growth factor alpha messenger RNAs and their respective receptor messenger RNAs in primary human gastric carcinomas: in vivo studies with in situ hybridization and immunocytochemistry. Cancer Res. 1992;52(12):3453–3459.
- Tan M, Yu D. Molecular mechanisms of erbB2-mediated breast cancer chemoresistance. Adv Exp Med Biol. 2007;608:119–129.
- Cunningham D, Humblet Y, Siena S, et al. Cetuximab monotherapy and cetuximab plus irinotecan in irinotecan-refractory metastatic colorectal cancer. N Engl J Med. 2004;351(4):337–345.
- Lum LG, Sen M. Activated T-cell and bispecific antibody immunotherapy for high-risk breast cancer. Bench to bedside. Acta Haematol. 2001;105(3):130–136.
- Reusch U, Sundaram M, Davol PA, et al. Anti-CD3 x anti-EGFR bispecific antibody redirects T cell cytolytic activity to EGFR-Positive cancers in vitro and in an animal model. Clin Cancer Res. 2006;12:183–190.
- Grabert RC, Cousens LP, Smith JA, et al. Human T cells armed with Her2/neu bispecific antibodies divide, are cytotoxic, and secrete cytokines with repeated stimulation. Clin Cancer Res. 2006;12(2):569–576.
- Thakur A, Rathore R, Kondadasula SV, et al. Immune T cells can transfer and boost anti-breast cancer immunity. Oncoimmunology. 2018;7(12):e1500672.
- Zitron IM, Thakur A, Norkina O, et al. Targeting and killing of glioblastoma with activated T cells armed with bispecific antibodies. BMC Cancer. 2013;13(1):83.
- Michaelson JS, Demarest SJ, Miller B, et al. Anti-tumor activity of stability-engineered IgG-like bispecific antibodies targeting TRAIL-R2 and LTbetaR. MAbs. 2009;1(2):128–141.
- Srivastava S, Riddell SR. Engineering CAR-T cells: design concepts. Trends Immunol. 2015;36(8):494–502.
- Stoiber S, Cadilha BL, Benmebarek MR, et al. Limitations in the design of chimeric antigen receptors for cancer therapy. Cells. 2019;8(5):472.
- Lum LG, Thakur A, Al-Kadhimi Z, et al. Targeted T-cell therapy in stage IV breast cancer: a phase I clinical trial. Clin Cancer Res. 2015;21(10):2305–2314.
- Vaishampayan U, Thakur A, Rathore R, et al. Phase I study of anti-CD3 x anti-Her2 bispecific antibody in metastatic castrate resistant prostate cancer patients. Prostate Cancer. 2015;2015:285193.
- Lum LG, Thakur A, Choi M, et al. Clinical and immune responses to anti-CD3 x anti-EGFR bispecific antibody armed activated T cells (EGFR BATs) in pancreatic cancer patients. Oncoimmunology. 2020;9(1):1773201.
- Thakur A, Schalk D, Tomaszewski E, et al. Microenvironment generated during EGFR targeted killing of pancreatic tumor cells by ATC inhibits myeloid-derived suppressor cells through COX2 and PGE2 dependent pathway. J Transl Med. 2013;11(1):35.
- Sen M, Wankowski DM, Garlie NK, et al. Use of anti-CD3 x anti-HER2/neu bispecific antibody for redirecting cytotoxicity of activated T cells toward HER2/neu + tumors. J Hematother Stem Cell Res. 2001;10(2):247–260.