
Abstract
Aim
Fibrosis is a common pathological feature of most types of chronic liver injuries. There is no specific treatment for liver fibrosis at present. The liver microenvironment, which fosters the survival and activity of liver cells, plays an important role in maintaining the normal structure and physiological function of the liver. The aim of this review is to deeply understand the role of the liver microenvironment in the dynamic and complicated development of liver fibrosis.
Methods
After searching in Elsevier ScienceDirect, PubMed and Web of Science databases using ‘liver fibrosis’ and ‘microenvironment’ as keywords, studies related to microenvironment in liver fibrosis was compiled and examined.
Results
The homeostasis of the liver microenvironment is disrupted during the development of liver fibrosis, affecting liver cell function, causing various types of cell reactions, and changing the cell-cell and cell-matrix interactions, eventually affecting fibrosis formation.
Conclusion
Liver microenvironment may be important for identifying potential therapeutic targets, and restoring microenvironment homeostasis may be an important strategy for promoting the reversal of liver fibrosis.
The homeostasis of the liver microenvironment is disrupted in liver fibrosis;
A pro-fibrotic microenvironment is formed during the development of liver fibrosis;
Restoring microenvironment homeostasis may be an important strategy for promoting the reversal of liver fibrosis.
KEY MESSAGES
Keywords:
1. Background
Chronic liver disease is a global public health problem. It is estimated that currently, 844 million people suffer from chronic liver disease worldwide, with an annual death rate of about two million [Citation1]. This is mainly because most chronic liver injuries, such as toxic liver disease, alcoholic liver disease, non-alcoholic fatty liver disease, chronic viral hepatitis, and cholestatic liver disease, can develop into liver fibrosis [Citation2,Citation3]. This is a pathological change resulting in increased extracellular matrix (ECM) and decreased parenchymal cells in the liver. Although mild fibrosis is mostly asymptomatic, it eventually progresses to cirrhosis and is often accompanied by serious structural disorders and vascular distortion, which is the leading cause of liver-related morbidity and mortality [Citation4]. Since most patients had already developed obvious liver fibrosis or cirrhosis when they were first clinically identified, anti-fibrosis drugs that can prevent the progression of liver fibrosis or induce cirrhosis regression are urgently needed [Citation5].
2. Liver microenvironment and liver fibrosis
A significant number of literature reviews on the mechanism of liver fibrosis have demonstrated that the development of liver fibrosis is a complex and dynamic process involving a variety of cells and molecules, and the interaction between them is crucial to the development direction of the disease. This complex regulatory process makes the liver microenvironment the focus of research. Multiple components, including not only hepatocytes that account for the largest proportion, but a variety of interstitial liver cells, such as liver sinusoidal endothelial cells (LSEC), liver macrophages, hepatic stellate cells (HSC), and intrahepatic lymphocytes are included in the liver microenvironment. These cells and their secreted cytokines and ECM form a complex interaction network essential for maintaining the liver’s normal physiological functions.
When the liver suffers a persistent injury, microenvironmental homeostasis is disrupted. The imbalances in the microenvironment significantly affect cell function, leading to various cellular responses and changing cell-cell and cell-matrix interactions. Simultaneously, the different components of the liver microenvironment change in this process, resulting in significant changes in the physical and chemical properties as well as the structure of the liver microenvironment. The unique microenvironment created by the above changes will significantly influence the establishment and development of fibrosis. Understanding the above-mentioned alterations in the liver microenvironment may lead to a better understanding of the regulatory effects of the liver microenvironment, which is critical for finding potential therapeutic targets.
2.1. Changes in the liver microenvironment in liver fibrosis
2.1.1. Metabolic changes in the liver microenvironment
The liver is the main metabolic organ of the body and can regulate the metabolism of various nutrients [Citation6]. Liver cells undergo metabolic reprogramming during liver fibrosis development to adapt to the environmental changes [Citation7], which can affect a variety of metabolic processes, including glucose, lipid, and amino acid metabolism [Citation8], and can also lead to mitochondrial dysfunction, the centre of cellular energy metabolism [Citation9].
2.1.1.1. Alteration of glucose metabolism
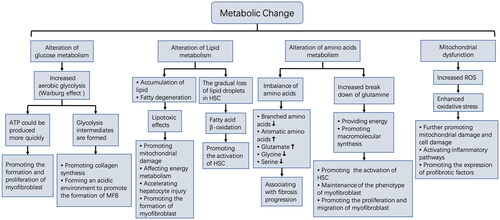
Glucose metabolism alters after chronic liver injury [Citation10]. The energy generation process from glucose will mostly shift to glycolysis [Citation11]. This enhanced glycolysis in an aerobic state is called the Warburg effect [Citation12,Citation13], and this kind of metabolic shift is strongly associated with liver fibrosis development. In liver fibrosis, HSC is the main source of myofibroblasts, and its activation is accompanied by increased glycolysis [Citation14]. It is possible that glycolysis can better meet the activation energy requirements of HSC since it produces ATPs more quickly. Glycolysis inhibition could have toxic effects on activated HSC, demonstrated as reduced cell proliferation [Citation15], and inhibited cell contraction [Citation16]. The above studies suggest glycolysis is necessary for transdifferentiation from HSC to myofibroblasts. In addition to HSC, studies have demonstrated that the mode of energy production of hepatocytes changes into glycolysis in the early stage of hepatic fibrosis. However, this method of glucose metabolism cannot generate enough energy, eventually leading to hepatocyte dysfunction, aggravating hepatocyte damage, and fibrosis development [Citation17]. In addition to the direct influence of glycolysis energy supply on liver fibrosis, the metabolism products in this process will also participate in the fibrosis process. Glycolysis intermediates have been reported to support cell anabolism and promote cell proliferation [Citation18,Citation19]. Recent studies also demonstrated that glycolysis metabolism products could promote collagen synthesis in myofibroblast, and inhibiting the formation of glycolytic intermediates in liver fibrosis mice model can reverse fibrosis [Citation20]. Furthermore, pyruvic acid, a product of glycolysis, can be converted into lactic acid, leading to high lactic acid levels [Citation21,Citation22], thus promoting the formation of an acidic microenvironment, which can promote TGFβ activation and myofibroblast formation [Citation23,Citation24].
Several regulatory factors related to this glucose metabolism pattern have been identified in liver fibrosis. Lactate dehydrogenase-A (LDH-A) is thought to play an important role in this regulation of glycolysis. Its high expression promotes glycolysis, which meets the energy requirements for rapid cell proliferation. A natural compound Oroxylin A has been found to block aerobic glycolysis by inhibiting LDH-A, which inhibited HSC contraction and alleviated liver fibrosis [Citation16]. M2 type pyruvate kinase (PKM2) is also an important regulator of aerobic glycolysis. Studies have found that PKM2 expression is significantly upregulated in fibrotic livers of mice and humans, of which its dimer structure induces HSC activation, while changing it into a tetrameric structure will reduce aerobic glycolysis and inhibit HSC activation [Citation25]. Further studies have demonstrated that induction of this glucose metabolism process is significantly correlated with the Hedgehog signalling pathway and the activity of hypoxia inducer 1α (HIF1α) [Citation15]. Some enzymes in other metabolic pathways also may influence sugar metabolic reprogramming. Geranylgeranyl diphosphate synthase (GGPPS) is a key enzyme in the mevalonate pathway, and it is considered to be the risk factor for the progression of non-alcoholic fatty liver disease (NAFLD) to fibrosis. The study found that the liver specificity lacking GGPPS can enhance aerobic glycolysis process by undermining the mitochondrial function, which can induce liver inflammation and aggravate the progression of fibrosis [Citation26].
2.1.1.2. Alteration of lipid metabolism
Lipid metabolism disorder may occur during liver fibrosis development [Citation27,Citation28]. Hepatocyte damage caused by various harmful factors can lead to fatty acid oxidation disorder, causing an accumulation of free fatty acids, free cholesterol, and other lipids in the liver. Excessive lipid accumulation can promote liver fibrosis through different mechanisms, such as disrupting energy metabolism by promoting mitochondrial and hepatocyte damage, causing hepatocytes to produce several pro-fibrotic mediators, and promoting HSC activation [Citation29]. A study has demonstrated that HSC activation is accompanied by the gradual loss of retinol-containing lipid droplets stored in HSC [Citation30]. It has also been demonstrated that the loss of lipid droplets is related to autophagy activation, which promotes the decomposition of lipids into fatty acids for β-oxidation, and the resultant energy is essential for HSC activation and is closely related to the ECM secretion and contraction function of activated HSC [Citation31].
Studies on the mechanisms of lipid metabolism disorders in liver fibrosis suggest some possible targets for intervention. Dual specificity phosphatase 9 (Dusp9) is a member of the DUSP protein family. Studies have found that Dusp9 expression in liver tissue is significantly decreased during the development of liver fibrosis induced by a high-fat diet; however, Dusp9 overexpression can prevent lipid accumulation and inhibit the development of liver fibrosis by blocking apoptosis signal-regulated kinase 1 (ASK1) related signalling pathway [Citation32]. Fang et al. showed that cathepsin B (CatB) played a role in hepatic lipid accumulation, and inhibition of CatB expression could inhibit lipid production and improve fibrosis by blocking CD36 and PPARγ expression [Citation33]. Adenosine monophosphate-activated protein kinase (AMPK) is also involved in metabolic regulation in the development of chronic liver disease. Its activation can reduce de novo adipogenesis, increase fatty acid oxidation, and reduce steatosis; meanwhile, AMPK activator has been shown to reduce liver fat content and inhibit fibrogenesis in rodents [Citation34]. Milk fat globule-epidermal growth factor 8 (MFG-E8) is a multifunctional glycoprotein, which has an anti-fibrosis effect, and Hu et al. further found that it plays a vital role in the process of non-alcoholic steatohepatitis (NASH). When MFG-E8 is knocked down, liver steatosis and lipid accumulation are aggravated, and fibrosis is promoted, confirming that this is related to the activation of the TLR4/NF-κB signalling pathway after the knockdown of MFG-E8 [Citation35]. Sodium-glucose cotransporter 2 (SGLT2) inhibitors can improve hepatic steatosis and fibrosis in patients with chronic liver disease. Recent studies have shown that the mechanism of the hepatic protective effect of SGLT2 inhibitors is independent of glycemic control but promotes liver phospholipid balance by regulating lipid composition and thus forms the protective lipid structure to play a beneficial role in the liver [Citation36]. Genetic factors may also be involved in the progression of such diseases through lipid metabolism regulation. Two genes, PNPLA3 and TM6SF2, were found to be associated with NAFLD in genome-wide Association Studies (GWAS), and they are considered to be markers of steatosis and more severe fibrosis. Detection of PNPLA3 and TM6SF2 gene variants may enhance the risk assessment of such diseases [Citation37].
Different drugs targeting lipid metabolic disorders in liver fibrosis are being screened. A study has found that Jiaqi Ganxian granules may inhibit collagen deposition in the liver by regulating lipid metabolism [Citation38]. Bile acid receptor (FXR) can inhibit hepatic bile acid synthesis and promote hepatic bile acid transport in vivo. Studies have demonstrated that the FXR agonist GW4064 can significantly reduce liver injury in model animals with cholestatic liver disease [Citation39]. GW4064 may also inhibit the development of liver fibrosis by inhibiting HSC activation [Citation40]. Peroxisome proliferator-activated receptor (PPAR) regulates fatty acid metabolism in the liver, and recent studies have demonstrated that PPARα antagonist GW6471 can improve NASH [Citation41]. Other studies have demonstrated that lipid-regulating drugs fenofibrate and ciprofibrate can reduce pulmonary fibrosis, collagen production, and myofibroblast differentiation in mice [Citation42,Citation43]. Edible insects as a possible dietary supplement may play a role in antifibrosis through liver lipid metabolism regulation. A study found that oral allomyrina dichotoma Larva extract (ADLE) can improve the high-fat diet-induced liver fibrosis in mice, which is closely related to the regulation of lipid metabolism. They further found that ADLE not only inhibits the expression of genes related to adipogenesis but also attenuates hepatic steatosis by increasing AMPK phosphorylation [Citation44].
2.1.1.3. Alteration of amino acids metabolism
Proteins are the building blocks of life. As the basic unit of proteins, amino acids are essential for various life activities of cells. The liver is the main organ responsible for amino acid metabolism. In liver fibrosis, there is an imbalance in the proportion of amino acids, manifested by a decrease in branched amino acids and an increase in aromatic amino acids [Citation45]. Previous studies have demonstrated that a decrease in the ratio of branched amino acids to tyrosine (BTR) is closely related to liver fibrosis progression [Citation46]. Gaggini et al. believed that glutamate was an amino acid more closely related to the severity of liver fibrosis. It was found that the concentrations of glycine and serine in plasma of patients with non-alcoholic fatty liver disease decreased while the concentration of glutamate increased, and combined with liver histological analysis, they proposed that GSG index (glutamate/[serine + glycine]) may be a useful index for evaluating the severity of liver fibrosis [Citation47]. Furthermore, glutamine metabolism was found to be associated with liver fibrosis. Glutamine metabolism can provide cells with energy and raw materials for macromolecular synthesis. Glutamine is metabolised into a – ketone glutaric acid that can be used as amino acid and lipid precursor and can also provide energy support to the Krebs cycle. Synthetic metabolism is also thought to be associated with liver fibrosis. Du et al. found that glutaminase expression in the livers of mice and humans rises significantly in fibrosis, prompting an increase in glutamine metabolism. They further depicted that glutaminase promotes glutamine decomposition in myofibroblasts, which plays an essential role in maintaining cell phenotype and can adjust the proliferation and migration of myofibroblasts [Citation48]. Li et al. also noticed that the expression of genes involved in glutamine metabolism, including glutaminase (GLS), aspartate aminotransferase (GOT1), and glutamate dehydrogenase (GLUD1), were significantly upregulated in the human fibrotic liver, suggesting an increase in glutamine catabolism. Furthermore, in vitro studies have displayed that glutamine breakdown plays an important role in HSC activation and proliferation [Citation49].
Liver metabolic function damage is the main cause of amino acid imbalance in the development of liver fibrosis after liver injury. However, Huang et al. recently reported that the ECM microstructure could regulate amino acid metabolism through the mechanism associated with integrin β1. It has a regulatory effect on tryptophan, branched-chain amino acid and methionine in the hepatocyte, which will provide new ideas for further study of amino acid metabolism in the fibrotic liver [Citation50].
Some drugs targeting changes in amino acid metabolism in liver fibrosis may play a beneficial role. AXA1665 is a novel amino acid component, and research suggests that it can improve amino acid imbalance and has good safety and tolerability when given orally to cirrhotic subjects [Citation51]. As the concentration of branched-chain amino acids (BCAA) decreases in the development of liver fibrosis, it is recommended to use BCAA to improve protein balance. However, clinical trials do not show the significant benefits of BCAA supplements. Researchers have speculated that supplementing the branched chain ketones acid may correct the amino acid imbalance, which still needs further study to evaluate its role in hepatic fibrosis [Citation52].
2.1.1.4. Mitochondrial dysfunction
Mitochondria produce a small number of reactive oxygen species under physiological conditions. When mitochondria are dysfunctional, significant electron leakage occurs, resulting in numerous reactive oxygen species, and the subsequent oxidative stress reaction can cause cell damage [Citation53]. The production of a large number of reactive oxygen species due to mitochondrial dysfunction is considered a key driver of liver fibrosis, and it has been found that liver mitochondrial function is significantly impaired in liver fibrosis development [Citation54–56]. Protein spectrum analysis suggested that oxidative stress and lipid peroxidation-related proteins were upregulated in the rat liver fibrosis model [Citation57]. Proteomic analysis of human fibrotic liver tissue also depicted that oxidative stress, and liver mitochondrial dysfunction was significantly correlated with fibrosis development [Citation58]. In addition to causing mitochondrial dysfunction, excessive reactive oxygen species generated by mitochondria can activate inflammatory pathways and directly promote the expression of pro-fibrosis factor transforming growth factor-beta (TGFβ) [Citation59].
There have been some studies on the mechanism of mitochondrial dysfunction in liver fibrosis. Mitochondrial membrane lipid composition is essential to maintain the structure and function of mitochondria. Studies have found that cardiolipin and phosphatidylethanolamine in mitochondrial lipids gradually decrease in the progression of chronic liver disease, which is associated with increased oxidative stress in mitochondria. The researchers also believe these changes in mitochondrial lipids are the early events of mitochondrial dysfunction and the progression of chronic liver disease [Citation60]. Fernandez-tussy et al. found that glycine-N-methyltransferase (GNMT) is an important regulator of Complex II activity in the mitochondrial electron transport chain. GNMT expression is controlled by Mir-873-5p in hepatocytes of NASH patients, and up-regulation of Mir-873-5p leads to the down-regulation of GNMT, thereby inhibiting mitochondrial function. While inhibition of Mir-873-5p in the corresponding mouse model can improve mitochondrial function and inhibit fibrosis progression [Citation61]. Krishnan et al. found that mitochondrial dysfunction in liver fibrosis is associated with the liver pyruvate kinase (L-PK) expression, which can alter mitochondrial pyruvate flux and its binding to citrate. Silencing L-PK in a mouse fibrosis model improves mitochondrial function and reduces fibrosis [Citation62]. Moreover, Pusec et al. found that hexokinase domain protein 1 (HKDC1), a member of the hexokinase family, was highly expressed in the fibrotic liver of mice, localized to mitochondria of hepatocytes and caused mitochondrial dysfunction, manifested by decreased cellular respiration and decreased mitochondrial membrane potential. Furthermore, this was mainly related to increased mitochondrial dynamic-related protein 1 (DRP1) [Citation63].
A significant amount of data indicates that metabolic changes in the liver microenvironment are strongly associated with the development of liver fibrosis. It is not only characteristic of liver fibrosis but also plays an important role in disease development. The metabolites produced due to the unbalanced metabolic environment can directly damage parenchymal cells and further induce inflammation to aggravate liver injury. More importantly, the metabolic reprogramming process creates an environment conducive to the information of myofibroblast. The process provides the necessary energy for myofibroblast formation, and the metabolites provide the biomolecular raw materials for maintaining phenotype and promoting myofibroblast proliferation. All these changes promote the development of liver fibrosis (). The in-depth study of the above metabolic abnormalities will not only further clarify the complex mechanism of liver fibrosis but may also provide effective new targets for treating liver fibrosis.
Figure 1. Metabolic changes in the liver microenvironment.
2.1.2. The immune microenvironment in liver fibrosis
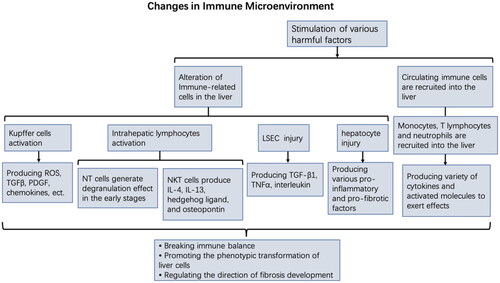
Persistent chronic inflammation is the basis of hepatic fibrosis and continues throughout all stages of fibrosis development. Under various liver injury conditions, various liver innate immune cells, including liver macrophages and lymphocytes, are activated to initiate inflammatory responses [Citation64,Citation65]. Kupffer cells can produce active oxygen species, TGFβ, platelet-derived growth factor (PDGF), and other pro-inflammatory and pro-fibrosis factors, as well as secrete many chemokines to recruit circulating immune cells (monocytes, lymphocytes) to the site of injury [Citation66–70]. Natural killer (NK) cells and natural killer T (NKT) cells are abundant in the liver. NK cells express effector molecules to kill or promote activated HSC apoptosis through the degranulation effect after activation [Citation71]. However, its anti-fibrosis activity is limited to the early stages of liver fibrosis and inhibited in the later stage [Citation72,Citation73]. Activated NKT cells can promote fibrosis by producing pro-fibrotic cytokines such as IL-4, IL-13, hedgehog ligand, and osteopontin [Citation74]. Furthermore, damaged LSECs can secrete pro-inflammatory cytokines, such as TGF-β1, interleukin, and tumour necrosis factor α (TNFα), to promote the recruitment of circulating inflammatory cells [Citation75]. The damaged hepatocytes secrete various pro-inflammatory and pro-fibrotic factors to maintain their survival, thereby directly or indirectly activating HSC [Citation76]. Immune cells recruited to the damaged liver can also continue to synthesize and secrete a variety of pro-inflammatory and pro-fibrotic factors: mononuclear cells will differentiate into macrophages and produce a variety of cytokines, including TGFβ, platelet-derived growth factor (PDGF), TNFα, tumour necrosis factor β (TNFβ) and interleukin-1β (IL-1β), further promoting HSC activation [Citation77]. T helper cell 2 (Th2) stimulates TGFβ synthesis by secreting IL-13 and macrophage differentiation by secreting IL-4 and IL-13 [Citation78]. T helper cell 17 (Th17) can secrete IL-7, which can either directly induce HSC activation or indirectly promote fibrosis by upregulating TGFβ expression to promote neutrophil recruitment to the liver [Citation78,Citation79].
Although the inflammation induced by liver injury at the initial stage promotes tissue repair, the influence of persistent injury factors on liver homeostasis disrupts the liver’s inflammatory balance, leading to excessive inflammation, effectively changing the interaction between liver cells, and driving the liver microenvironment towards promoting fibrosis. The final result is that HSC, the main source of myofibroblasts in liver fibrosis, transforms into an activated state and enters the permanent stage under the stimulation of various cell products [Citation80]. Furthermore, other intrahepatic cells, including fibroblasts, hepatocytes, and extrahepatic bone marrow-derived cells recruited into liver tissue, may also be transformed into myofibroblast phenotype under the above factors [Citation81–85]. Myofibroblasts are the core components of fibrotic lesions; their increased concentration leads to excessive ECM synthesis, thereby promoting fibrosis progression.
The changes of immune microenvironment in the development of fibrosis are caused by innate immune cell activation in the liver, the recruitment of systemic immune cells to the liver tissue, and their interaction with intrahepatic cells, all of which promote the phenotypic transformation of liver cells. The transformed cells can secrete pro-inflammatory factors and continue to receive inflammatory stimulation, which maintains the activated state of myofibroblasts. Chemokines are also secreted to recruit more immune cells to the damaged liver, thereby aggravating the inflammatory damage [Citation86,Citation87]. All these factors lead to an uncontrollable vicious cycle and aggravate the damage to the intrahepatic microenvironment homeostasis ().
Figure 2. The changes of immune microenvironment in liver fibrosis.
However, studies have also found that liver cells may play different roles in the inflammatory response. Regulating cellular interactions by promoting the stability of the liver microenvironment may help restore inflammatory balance and even promote fibrosis regression. Macrophages will form different phenotypes under the influence of the tissue’s microenvironment, and their specific signals can promote or inhibit the fibrosis process. For example, hepatic macrophages can promote fibrosis by activating HSC in liver injury. However, studies have also found that in the CCL4-induced liver injury model, when macrophages develop a recovery phenotype, they can inhibit the fibrosis process by inhibiting inflammation and reducing the formation of myofibroblasts [Citation88] and may promote the regression of fibrosis by secreting a variety of matrix metalloproteinases [Citation89,Citation90]. NKT cells are also plastic, and their differentiation into subtypes depends on the microenvironment in which they are located. Its different subtypes regulate immune processes through different cytokine expression profiles, and studies suggest that such different regulatory effects on inflammation may inhibit the process of fibrosis [Citation91]. Studies have found that NKT cells can relieve liver fibrosis by releasing IFN-γ to kill activated HSC [Citation92]. Other studies have demonstrated that IL-30 can improve liver fibrosis by killing activated HSC through NKT cells [Citation93]. T lymphocytes can also differentiate into different cell phenotypes in response to different microenvironments such when they are differentiated into TH1 cells, interferon-γ released by them can antagonize the activity of TGFβ and thus alleviate liver fibrosis [Citation79].
In summary, the outcome of the inflammatory response is influenced by the local microenvironment, and it is essential to understand how it varies under different disease conditions. Inflammation may act as a double-edged sword in the development of liver fibrosis; uncontrolled inflammation resulting from liver homeostasis imbalance can aggravate the hepatic injury and promote the production of myofibroblasts. However, it may also restore inflammatory equilibrium by changing the liver microenvironment, which may produce an integrated regulatory effect on liver cells, and promote the inflammatory reaction in the direction of tissue repair and fibrogenolysis.
2.1.3. Vascular changes in the liver microenvironment
The development of liver fibrosis is accompanied by changes in the vascular system, primarily manifested as vascular remodelling and angiogenesis (). Vascular remodelling is characterized by hepatic sinusoid capillarization, mainly caused by structural dysfunction of LSEC, which constitutes the wall of the hepatic sinusoid [Citation94–97]. Normal LSEC lacks a basement membrane and has the characteristic structure of fenestration, which plays an important role in regulating blood flow and material exchange in the hepatic sinusoid [Citation98,Citation99]. Studies have depicted that under various liver injury factors, LSEC will lose the characteristic fenestration, resulting in the formation of a continuous basement membrane of hepatic sinusoid [Citation100,Citation101]. This change is also known as the capillarization of LSEC. Capillarized LSECs are also closely related to fibrosis development, and research indicates that capillarized LSECs will lose their ability to inhibit HSC activation [Citation102]. Meanwhile, structural dysfunction of LSEC is considered the leading cause of increased portal vein resistance in patients with liver disease [Citation103,Citation104].
Figure 3. Vascular changes in the liver microenvironment.
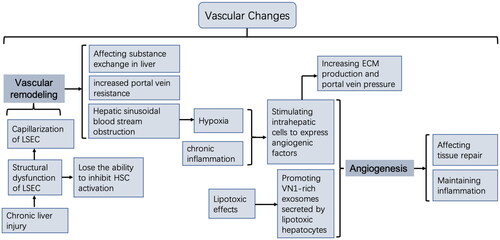
The progression of liver fibrosis is also accompanied by angiogenesis. Due to the massive ECM deposition, the liver structure is disorganized, resulting in the obstruction of hepatic sinusoidal blood flow and eventual hypoxia. The dysfunction of LSEC structure and function will affect oxygen exchange, further aggravating insufficient oxygen supply to the tissues. Hypoxic conditions can lead to the upregulation of angiogenic factors, thus inducing new angiogenesis [Citation66,Citation105]. Furthermore, studies have demonstrated that chronic liver inflammation stimulates epithelial cells, HSC, and LSEC to express a variety of angiogenic factors, such as fibroblast growth factor (FGF), vascular endothelial growth factor (VEGF), platelet-derived growth factor (PDGF), and angiopoietin (Ang) [Citation96,Citation106]. Other studies have demonstrated that VN 1-rich exosomes secreted by lipotoxic hepatocytes can also promote angiogenesis [Citation107]. Different studies have demonstrated that angiogenesis under such pathological conditions is also associated with liver fibrosis progression, which may disrupt normal tissue repair and maintain inflammatory processes to promote fibrosis [Citation108]. Other studies have revealed that, in addition to promoting angiogenesis, angiogenic factors can promote liver fibrosis by increasing ECM production and portal vein pressure [Citation109].
The role of vascular remodelling and angiogenesis in promoting liver fibrosis is closely related to their interaction with the liver microenvironment. LSEC, an important component of liver microcirculation, can regulate the balance between liver regeneration and fibrosis by producing angiocrine factors. However, the angiocrine factors produced by LSEC become pro-fibrotic in the microenvironment of liver fibrosis [Citation110]. Although angiogenesis can provide nutrition for tissues, they become the source of inflammatory factors in the process of fibrosis and promote the vicious cycle by maintaining local chronic inflammation, thus promoting disease progression [Citation111,Citation112].
Due to the association of vascular remodelling and angiogenesis with fibrosis, vascular alterations in liver fibrosis may also be a therapeutic target. Several studies have shown that inhibiting LSEC capillarization can stabilize the HSC phenotype, and inhibiting angiogenesis in liver fibrosis can reduce portal pressure and liver inflammation, both of which can inhibit fibrosis development [Citation106]. Taura et al. reported that blocking angiogenic signalling of HSC in BALB/C mice with liver fibrosis induced by carbon tetrachloride (CCl4) or bile duct ligation (BDL) could inhibit fibrosis [Citation113]. Therefore, targeting hepatic vascular regulation may also be an option for maintaining organ homeostasis in liver fibrosis. However, the timing of vascular intervention must be thoroughly evaluated, as physiological angiogenesis is also necessary for injury repair.
2.1.4. Changes in ECM in the liver microenvironment
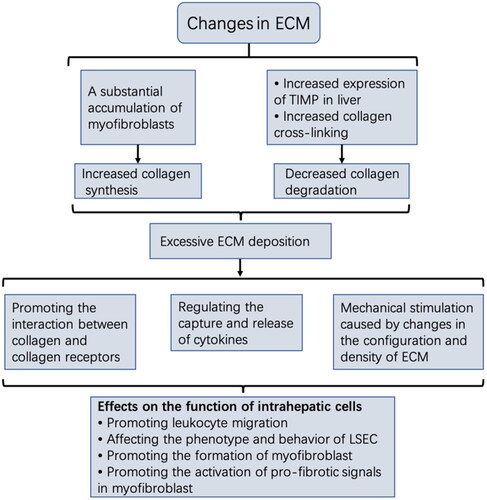
ECM consists of collagen, fibronectin, hyaluronic acid, proteoglycan, and other molecules that undergo highly dynamic changes during synthesis and breakdown. ECM is essential for maintaining the structural integrity of tissues under physiological conditions. During the development of liver fibrosis, persistent inflammation and changes in metabolic and vascular components in the microenvironment lead to the production of a large number of myofibroblasts, the immediate cause of excessive ECM. The expression of tissue inhibitors of matrix metalloproteinases (TIMP), especially TIMP-1, increases in the damaged liver tissue, and it inactivates MMPs by binding to them, resulting in reduced ECM degradation [Citation114]. Furthermore, increased collagen cross-linking hinders collagen degradation, primarily mediated by lysyl oxidase (LOX) and transglutaminase (TGase). Different studies have shown that the activity and expression of LOX and TGase are significantly increased in fibrotic liver, which can increase liver hardness and inhibit the breakdown effect of MMP on ECM by mediating collagen cross-linking to increase its stability [Citation115–120]. Furthermore, as liver fibrosis develops, not only does the amount of ECM increase, but also the composition and properties of ECM change significantly. The main components change from type IV and VI collagen to type I and III collagen, which becomes resistant to degradation [Citation121,Citation122]. Under the influence of the above factors, ECM remodelling is excessive and uncontrollable, leading to a large amount of ECM deposition.
ECM deposition is not only the result of the development of liver fibrosis but also an essential component of the hepatic fibrosis microenvironment. Excessive ECM deposition can significantly alter liver microenvironmental homeostasis and affect various liver cells. Different studies have demonstrated that ECM composition and configuration changes can be used as extracellular signals to activate intracellular signal transduction, influencing cell phenotype and function [Citation123]. The direct interaction between ECM and cells is mainly mediated by cell surface receptor integrin and discoidin domain receptor (DDR). A study showed that the integrin αVβ6 expression was significantly upregulated in both rodent models of liver fibrosis and patients with chronic liver disease [Citation124]. Integrin has been found to play a critical role in latent TGFβ activation, a key regulator of fibrosis [Citation125]. Increased DDR1 expression has also been noticed in liver fibrosis, and research indicates that the interaction between DDR1 and collagen promotes white blood cell migration in liver tissues [Citation126]. Since ECM also serves as a reservoir of cytokines in tissues, its components can regulate cell function by capturing or releasing cytokines [Citation127]. There is evidence that the biological activity of cytokines can be regulated by binding to ECM molecules [Citation128]. Activation of ECM-stored cytokines changes the behaviour of LSEC [Citation129]. Mechanical stimulation caused by changes in ECM configuration and density also impacts the fibrosis process, significantly promoting the differentiation of HSC and portal fibroblasts into myofibroblasts [Citation130,Citation131]. Liu et al. reported that mechanical signals generated by collagen fibres promoted intracellular signal transduction through DDR2 receptors on HSC and were also closely related to HSC activation [Citation132]. Moreover, sclerotic ECM has been revealed to promote the progression of liver fibrosis by activating Rho-associated protein kinase (ROCK) and the YAP/TAZ signalling pathway in myofibroblasts [Citation120].
Changes in the liver microenvironment promote ECM synthesis over ECM degradation, producing pathological ECM in large quantities. However, ECM overproduction is not the end point of fibrosis development but rather a component of the microenvironment to influence the behaviour of liver cells through abnormal physical and chemical signals. This can establish a vicious cycle and lead to persistent fibrosis progression even after the damage factors are removed ().
Figure 4. changes in ECM in the liver microenvironment.
The shortest method for regression of excessive ECM in liver fibrosis is to promote ECM degradation. Regulating the release and activity of collagenase may be advantageous. Research has shown that collagenase encapsulated with nanoparticles can reverse fibrosis after targeted delivery to the fibrotic liver and release it in an active form [Citation133]. Furthermore, research on cellular therapy for liver fibrosis has shown that bone-marrow-derived macrophages differentiated under the condition of CSF-1 stimulation can inhibit fibrosis when injected into mice with liver fibrosis. This is believed to be related to increased MMP expression, promoting collagen degradation [Citation134]. Other studies have suggested that LOXL2 inhibition plays an important role in preventing the development of pathological microenvironment in fibrotic diseases. The inhibitory monoclonal antibody AB0023 against LOXL2 can significantly reduce fibroblast activation and the production of related growth factors and cytokines in fibrotic diseases [Citation135]. Ikenaga et al. also reported that inhibiting LOXL2 in liver fibrosis can effectively inhibit liver fibrosis progression [Citation119]. The regulation of mechanical signals in ECM may also inhibit fibrosis development. Yes-associated protein (YAP) is an essential transcriptional coactivator for mechanical signal transduction. Inhibiting YAP expression or pharmacological inhibition of YAP can prevent HSC activation in vitro, whereas pharmacological inhibition of YAP can inhibit CCL4-induced liver fibrosis in mice [Citation136]. ROCK activity is essential for cellular mechanical perception, and selective ROCK inhibitor Y27632 has been shown to prevent CCL4-induced liver fibrosis, reduce collagen content, and HSC activation in rats [Citation137].
2.2. Therapeutic strategies based on liver fibrosis microenvironment
Liver fibrosis is a complicated heterogeneous disease characterized by forming a pro-fibrotic microenvironment in the liver due to interactions between different types of cells, thereby promoting disease progression. Liver fibrosis was initially thought to be irreversible. However, cholestatic and viral liver disease studies demonstrate that liver fibrosis regression is possible even in advanced stages [Citation138,Citation139]. The strong regenerative ability of the liver further stimulates the research into the reversal of liver fibrosis.
There are no specific drugs against liver fibrosis, and the best treatment for liver fibrosis is still the removal of damage factors [Citation140,Citation141]. However, this is insufficient to improve the prognosis of all patients with liver fibrosis. Based on microenvironmental changes, the pathogenesis of liver fibrosis indicates that its development is a complex dynamic process, and targeting a single pathway in the anti-fibrosis strategy is unlikely to prevent or reverse fibrosis. Similar clinical studies have confirmed that single-target selective drugs cannot successfully treat fibrosis in the human body [Citation142,Citation143]. Since various types of liver cells and molecules are involved in liver fibrosis development, therapeutic strategies to promote microenvironmental homeostasis may have a global regulatory action on the functional conditions of various liver cells, inhibiting cell transition to a pro-fibrotic state, thereby forming a homeostasis feedback loop, a key to reverse fibrosis. It has been demonstrated that hepatocytes from cirrhotic tissues regained their metabolic activity and ability to secrete liver-specific proteins after exposure to a healthy liver microenvironment [Citation144]. Other studies have demonstrated that myofibroblasts could revert to an inactive phenotype as fibrosis regresses and pro-fibrosis signals diminish [Citation145,Citation146]. These studies suggest that regulating the microenvironment in response to the characteristic changes of the fibrotic microenvironment may play an important role in preventing or reversing fibrosis.
Currently, most of the studies on liver fibrosis focus on a specific factor or cell necessary for the occurrence and development of liver fibrosis but is insufficient to explain the multi-step process of continuous progression of liver fibrosis. Changes in the microenvironment during liver fibrosis development suggest that it is a complex multicellular disease. Although the massive production of myofibroblasts is the key factor in liver fibrosis development, it is not the only intervention target. Changes in the microenvironment during this process need to be identified. It is necessary to restore microenvironmental homeostasis to fundamentally inhibit the formation and sustained activation of myofibroblasts in liver fibrosis, thus inhibiting the dynamic progression of the disease. Based on the characteristics of the liver fibrotic microenvironment, a holistic therapeutic strategy for reshaping microenvironmental homeostasis may improve or even reverse fibrosis since it can restore and maintain normal liver cell function. This requires multi-target combination therapy. However, further evaluation of possible interactions between multiple drug combinations is required to improve efficacy.
3. Conclusion
Liver fibrosis is the result of multicellular interaction in a pro-fibrotic microenvironment. The involvement of liver microenvironment changes in its progression suggests that the therapeutic target for liver fibrosis should be identified in the liver microenvironment. Restoration of microenvironmental homeostasis from a holistic perspective can enable all kinds of liver cells to maintain a more stable and long-lasting condition and inhibit the transformation of liver cells into a pro-fibrotic state, which may be an important strategy to promote the reversal of liver fibrosis.
Author contributions
YM and TZ conceived and designed the paper, and reviewed and summarized the related literature; YM wrote the first draft of the manuscript; YM, TZ, ZZ and DZ contributed to interpreting data, revising the manuscript, and approved the published version. All authors agree to be accountable for all aspects of the work.
Disclosure statement
No potential conflict of interest was reported by the author(s).
Additional information
Funding
References
- Marcellin P, Kutala BK. Liver diseases: a major, neglected global public health problem requiring urgent actions and large-scale screening. Liver Int. 2018;38(Suppl 1):2–6.
- Zhou WC, Zhang QB, Qiao L. Pathogenesis of liver cirrhosis. World J Gastroenterol. 2014;20(23):7312–7324.
- Rockey DC, Bell PD, Hill JA. Fibrosis–a common pathway to organ injury and failure. N Engl J Med. 2015;372(12):1138–1149.
- Ginès P, Krag A, Abraldes JG, et al. Liver cirrhosis. Lancet. 2021;398(10308):1359–1376.
- Friedman SL, Sheppard D, Duffield JS, et al. Therapy for fibrotic diseases: nearing the starting line. Sci Transl Med. 2013;5(167):167sr1–167sr1. (
- Rui L. Energy metabolism in the liver. Compr Physiol. 2014;4(1):177–197.
- Henderson J, O’Reilly S. The emerging role of metabolism in fibrosis. Trends Endocrinol Metab. 2021;32(8):639–653.
- Chang ML, Yang SS. Metabolic signature of hepatic fibrosis: from individual pathways to systems biology. Cells. 2019;8(11):1423.
- Li X, Zhang W, Cao Q, et al. Mitochondrial dysfunction in fibrotic diseases. Cell Death Discov. 2020;6:80.
- Bahr MJ, Ockenga J, Böker KH, et al. Elevated resistin levels in cirrhosis are associated with the proinflammatory state and altered hepatic glucose metabolism but not with insulin resistance. Am J Physiol Endocrinol Metab. 2006;291(2):E199–E206.
- Lee NCW, Carella MA, Papa S, et al. High expression of glycolytic genes in cirrhosis correlates with the risk of developing liver cancer. Front Cell Dev Biol. 2018;6:138.
- Vaupel P, Schmidberger H, Mayer A. The warburg effect: essential part of metabolic reprogramming and Central contributor to cancer progression. Int J Radiat Biol. 2019;95(7):912–919.
- Li J, Wang T, Xia J, et al. Enzymatic and nonenzymatic protein acetylations control glycolysis process in liver diseases. Faseb J. 2019;33(11):11640–11654.
- Lian N, Jin H, Zhang F, et al. Curcumin inhibits aerobic glycolysis in hepatic stellate cells associated with activation of adenosine monophosphate-activated protein kinase. IUBMB Life. 2016;68(7):589–596.
- Chen Y, Choi SS, Michelotti GA, et al. Hedgehog controls hepatic stellate cell fate by regulating metabolism. Gastroenterology. 2012;143(5):1319–1329.e11.
- Wang F, Jia Y, Li M, et al. Blockade of glycolysis-dependent contraction by oroxylin a via inhibition of lactate dehydrogenase-a in hepatic stellate cells. Cell Commun Signal. 2019;17(1):11.
- Nishikawa T, Bellance N, Damm A, et al. A switch in the source of ATP production and a loss in capacity to perform glycolysis are hallmarks of hepatocyte failure in advance liver disease. J Hepatol. 2014;60(6):1203–1211.
- Lunt SY, Vander Heiden MG. Aerobic glycolysis: meeting the metabolic requirements of cell proliferation. Annu Rev Cell Dev Biol. 2011;27:441–464.
- Vander Heiden MG, Cantley LC, Thompson CB. Understanding the warburg effect: the metabolic requirements of cell proliferation. Science. 2009;324(5930):1029–1033.
- Satyanarayana G, Turaga RC, Sharma M, et al. Pyruvate kinase M2 regulates fibrosis development and progression by controlling glycine auxotrophy in myofibroblasts. Theranostics. 2021;11(19):9331–9341.
- McPhail MJW, Shawcross DL, Lewis MR, et al. Multivariate metabotyping of plasma predicts survival in patients with decompensated cirrhosis. J Hepatol. 2016;64(5):1058–1067.
- Nie Y, Liu LX, Chen T, et al. Serum lactate level predicts 6-months mortality in patients with hepatitis B virus-related decompensated cirrhosis: a retrospective study. Epidemiol Infect. 2021;149:e26.
- Kottmann RM, Trawick E, Judge JL, et al. Pharmacologic inhibition of lactate production prevents myofibroblast differentiation. Am J Physiol Lung Cell Mol Physiol. 2015;309(11):L1305–12.
- Trivedi P, Wang S, Friedman SL. The power of Plasticity-Metabolic regulation of hepatic stellate cells. Cell Metab. 2021;33(2):242–257.
- Zheng D, Jiang Y, Qu C, et al. Pyruvate kinase M2 tetramerization protects against hepatic stellate cell activation and liver fibrosis. Am J Pathol. 2020;190(11):2267–2281.
- Liu J, Jiang S, Zhao Y, et al. Geranylgeranyl diphosphate synthase (GGPPS) regulates non-alcoholic fatty liver disease (NAFLD)-fibrosis progression by determining hepatic glucose/fatty acid preference under high-fat diet conditions. J Pathol. 2018;246(3):277–288.
- Yang L, Roh YS, Song J, et al. Transforming growth factor beta signaling in hepatocytes participates in steatohepatitis through regulation of cell death and lipid metabolism in mice. Hepatology. 2014;59(2):483–495.
- Zhang J, Muise ES, Han S, et al. Molecular profiling reveals a common metabolic signature of tissue fibrosis. Cell Rep Med. 2020;1(4):100056.
- Kuchay MS, Choudhary NS, Mishra SK. Pathophysiological mechanisms underlying MAFLD. Diabetes Metab Syndr. 2020;14(6):1875–1887.
- Bobowski-Gerard M, Zummo FP, Staels B, et al. Retinoids issued from hepatic stellate cell lipid droplet loss as potential signaling molecules orchestrating a multicellular liver injury response. Cells. 2018;7(9):137.
- Hernández-Gea V, Ghiassi-Nejad Z, Rozenfeld R, et al. Autophagy releases lipid that promotes fibrogenesis by activated hepatic stellate cells in mice and in human tissues. Gastroenterology. 2012;142(4):938–946.
- Ye P, Xiang M, Liao H, et al. Dual-specificity phosphatase 9 protects against nonalcoholic fatty liver disease in mice through ASK1 suppression. Hepatology. 2019;69(1):76–93.
- Fang W, Deng Z, Benadjaoud F, et al. Cathepsin B deficiency ameliorates liver lipid deposition, inflammatory cell infiltration, and fibrosis after diet-induced nonalcoholic steatohepatitis. Transl Res. 2020;222:28–40.
- Gluais-Dagorn P, Foretz M, Steinberg GR, et al. Direct AMPK activation corrects NASH in rodents through metabolic effects and direct action on inflammation and fibrogenesis. Hepatol Commun. 2022;6(1):101–119.
- Hu J, Du H, Yuan Y, et al. MFG-E8 knockout aggravated nonalcoholic steatohepatitis by promoting the activation of TLR4/NF-κB signaling in mice. Mediators Inflamm. 2022;2022:5791915.
- Aragón-Herrera A, Otero-Santiago M, Anido-Varela L, et al. The treatment with the SGLT2 inhibitor empagliflozin modifies the hepatic metabolome of male zucker diabetic fatty rats towards a protective profile. Front Pharmacol. 2022;13:827033.
- Wu KT, Kuo PL, Su SB, et al. Nonalcoholic fatty liver disease severity is associated with the ratios of total cholesterol and triglycerides to high-density lipoprotein cholesterol. J Clin Lipidol. 2016;10(2):420–425.e1.
- Wang G, Li Z, Li H, et al. Metabolic profile changes of CCl4-Liver fibrosis and inhibitory effects of Jiaqi Ganxian granule. Molecules. 2016;21(6):698.
- Liu Y, Binz J, Numerick MJ, et al. Hepatoprotection by the farnesoid X receptor agonist GW4064 in rat models of intra- and extrahepatic cholestasis. J Clin Invest. 2003;112(11):1678–1687.
- Li J, Kuruba R, Wilson A, et al. Inhibition of endothelin-1-mediated contraction of hepatic stellate cells by FXR ligand. PLoS One. 2010;5(11):e13955.
- Yan T, Luo Y, Yan N, et al. Intestinal peroxisome proliferator-activated receptor α-fatty acid-binding protein 1 axis modulates nonalcoholic steatohepatitis. Hepatology. 2022;2022:hep.32538.
- Samah M, El-Aidy Ael R, Tawfik MK, et al. Evaluation of the antifibrotic effect of fenofibrate and rosiglitazone on bleomycin-induced pulmonary fibrosis in rats. Eur J Pharmacol. 2012;689(1–3):186–193.
- Oruqaj G, Karnati S, Vijayan V, et al. Compromised peroxisomes in idiopathic pulmonary fibrosis, a vicious cycle inducing a higher fibrotic response via TGF-β signaling. Proc Natl Acad Sci USA. 2015;112(16):E2048–57.
- Kim K, Bae GD, Lee M, et al. Allomyrina dichotoma larva extract ameliorates the hepatic insulin resistance of high-fat diet-induced diabetic mice. Nutrients. 2019;11(7):1522.
- Yu M, Zhu Y, Cong Q, et al. Metabonomics research progress on liver diseases. Can J Gastroenterol Hepatol. 2017;2017:8467192.
- Enomoto H, Sakai Y, Aizawa N, et al. Association of amino acid imbalance with the severity of liver fibrosis and esophageal varices. Ann Hepatol. 2013;12(3):471–478.
- Gaggini M, Carli F, Rosso C, et al. Altered amino acid concentrations in NAFLD: impact of obesity and insulin resistance. Hepatology. 2018;67(1):145–158.
- Du K, Hyun J, Premont RT, et al. Hedgehog-YAP signaling pathway regulates glutaminolysis to control activation of hepatic stellate cells. Gastroenterology. 2018;154(5):1465–1479.e13.
- Li J, Ghazwani M, Liu K, et al. Regulation of hepatic stellate cell proliferation and activation by glutamine metabolism. PLoS One. 2017;12(8):e0182679.
- Huang T, Terrell JA, Chung JH, et al. Electrospun microfibers modulate intracellular amino acids in liver cells via integrin β1. Bioengineering. 2021;8(7):88.
- Chakravarthy MV, Neutel J, Confer S, et al. Safety, tolerability, and physiological effects of AXA1665, a novel composition of amino acids, in subjects with child-Pugh a and B cirrhosis. Clin Transl Gastroenterol. 2020;11(8):e00222.
- Holeček M. Branched-Chain amino acids and branched-chain Keto acids in hyperammonemic states: metabolism and as supplements. Metabolites. 2020;10(8):324.
- Zorov DB, Juhaszova M, Sollott SJ. Mitochondrial reactive oxygen species (ROS) and ROS-induced ROS release. Physiol Rev. 2014;94(3):909–950.
- Krähenbühl S, Krähenbühl-Glauser S, Stucki J, et al. Stereological and functional analysis of liver mitochondria from rats with secondary biliary cirrhosis: impaired mitochondrial metabolism and increased mitochondrial content per hepatocyte. Hepatology. 1992;15(6):1167–1172.
- Di Ciaula A, Passarella S, Shanmugam H, et al. Nonalcoholic Fatty Liver Disease (NAFLD). mitochondria as players and targets of therapies? Int J Mol Sci. 2021;22(10):5375.
- Zhao Y, Wang Z, Feng D, et al. p66Shc contributes to liver fibrosis through the regulation of mitochondrial reactive oxygen species. Theranostics. 2019;9(5):1510–1522.
- Low TY, Leow CK, Salto-Tellez M, et al. A proteomic analysis of thioacetamide-induced hepatotoxicity and cirrhosis in rat livers. Proteomics. 2004;4(12):3960–3974.
- Diamond DL, Jacobs JM, Paeper B, et al. Proteomic profiling of human liver biopsies: hepatitis C virus-induced fibrosis and mitochondrial dysfunction. Hepatology. 2007;46(3):649–657.
- Li Z, Li Y, Zhang HX, et al. Mitochondria-mediated pathogenesis and therapeutics for non-alcoholic fatty liver disease. Mol Nutr Food Res. 2019;63(16):e1900043.
- Durand M, Coué M, Croyal M, et al. Changes in key mitochondrial lipids accompany mitochondrial dysfunction and oxidative stress in NAFLD. Oxid Med Cell Longev. 2021;2021:9986299.
- Fernández-Tussy P, Fernández-Ramos D, Lopitz-Otsoa F, et al. miR-873-5p targets mitochondrial GNMT-Complex II interface contributing to non-alcoholic fatty liver disease. Mol Metab. 2019;29:40–54.
- Chella Krishnan K, Floyd RR, Sabir S, et al. Liver pyruvate kinase promotes NAFLD/NASH in both mice and humans in a Sex-Specific manner. Cell Mol Gastroenterol Hepatol. 2021;11(2):389–406.
- Pusec CM, De Jesus A, Khan MW, et al. Hepatic HKDC1 expression contributes to liver metabolism. Endocrinology. 2019;160(2):313–330.
- Kubes P, Jenne C. Immune responses in the liver. Annu Rev Immunol. 2018;36:247–277.
- Liaskou E, Wilson DV, Oo YH. Innate immune cells in liver inflammation. Mediators Inflamm. 2012;2012:949157.
- O’Rourke JM, Sagar VM, Shah T, et al. Carcinogenesis on the background of liver fibrosis: implications for the management of hepatocellular cancer. World J Gastroenterol. 2018;24(39):4436–4447.
- Krenkel O, Tacke F. Liver macrophages in tissue homeostasis and disease. Nat Rev Immunol. 2017;17(5):306–321.
- Aydın MM, Akçalı KC. Liver fibrosis. Turk J Gastroenterol. 2018;29(1):14–21.
- Roehlen N, Crouchet E, Baumert TF. Liver fibrosis: mechanistic concepts and therapeutic perspectives. Cells. 2020;9(4):875.
- Seki E, Schwabe RF. Hepatic inflammation and fibrosis: functional links and key pathways. Hepatology. 2015;61(3):1066–1079.
- Shi J, Zhao J, Zhang X, et al. Activated hepatic stellate cells impair NK cell anti-fibrosis capacity through a TGF-β-dependent emperipolesis in HBV cirrhotic patients. Sci Rep. 2017;7:44544.
- Jeong WI, Gao B. Innate immunity and alcoholic liver fibrosis. J Gastroenterol Hepatol. 2008;23(Suppl 1):S112–S8.
- Jeong WI, Park O, Suh YG, et al. Suppression of innate immunity (natural killer cell/interferon-γ) in the advanced stages of liver fibrosis in mice. Hepatology. 2011;53(4):1342–1351.
- Gao B, Radaeva S. Natural killer and natural killer T cells in liver fibrosis. Biochim Biophys Acta. 2013;1832(7):1061–1069.
- Hammoutene A, Rautou PE. Role of liver sinusoidal endothelial cells in non-alcoholic fatty liver disease. J Hepatol. 2019;70(6):1278–1291.
- Kumar S, Duan Q, Wu R, et al. Pathophysiological communication between hepatocytes and non-parenchymal cells in liver injury from NAFLD to liver fibrosis. Adv Drug Deliv Rev. 2021;176:113869.
- Matsuda M, Seki E. The liver fibrosis niche: Novel insights into the interplay between fibrosis-composing mesenchymal cells, immune cells, endothelial cells, and extracellular matrix. Food Chem Toxicol. 2020;143:111556.
- Chen RJ, Wu HH, Wang YJ. Strategies to prevent and reverse liver fibrosis in humans and laboratory animals. Arch Toxicol. 2015;89(10):1727–1750.
- Mack M. Inflammation and fibrosis. Matrix Biol. 2018;68–69:106–121.
- McAnulty RJ. Fibroblasts and myofibroblasts: their source, function and role in disease. Int J Biochem Cell Biol. 2007;39(4):666–671.
- Stone RC, Pastar I, Ojeh N, et al. Epithelial-mesenchymal transition in tissue repair and fibrosis. Cell Tissue Res. 2016;365(3):495–506.
- Lua I, Li Y, Zagory JA, et al. Characterization of hepatic stellate cells, portal fibroblasts, and mesothelial cells in normal and fibrotic livers. J Hepatol. 2016;64(5):1137–1146.
- Iwaisako K, Jiang C, Zhang M, et al. Origin of myofibroblasts in the fibrotic liver in mice. Proc Natl Acad Sci USA. 2014;111(32):E3297–305.
- Mederacke I, Hsu CC, Troeger JS, et al. Fate tracing reveals hepatic stellate cells as dominant contributors to liver fibrosis independent of its aetiology. Nat Commun. 2013;4:2823.
- Kisseleva T, Uchinami H, Feirt N, et al. Bone marrow-derived fibrocytes participate in pathogenesis of liver fibrosis. J Hepatol. 2006;45(3):429–438.
- Pellicoro A, Ramachandran P, Iredale JP, et al. Liver fibrosis and repair: immune regulation of wound healing in a solid organ. Nat Rev Immunol. 2014;14(3):181–194.
- Tanwar S, Rhodes F, Srivastava A, et al. Inflammation and fibrosis in chronic liver diseases including non-alcoholic fatty liver disease and hepatitis C. World J Gastroenterol. 2020;26(2):109–133.
- Godwin JW, Pinto AR, Rosenthal NA. Chasing the recipe for a pro-regenerative immune system. Semin Cell Dev Biol. 2017;61:71–79.
- Elsherif SA, Alm AS. Role of macrophages in liver cirrhosis: fibrogenesis and resolution. Anat Cell Biol. 2022;55(1):14–19.
- Fallowfield JA, Mizuno M, Kendall TJ, et al. Scar-associated macrophages are a major source of hepatic matrix metalloproteinase-13 and facilitate the resolution of murine hepatic fibrosis. J Immunol. 2007;178(8):5288–5295.
- Nilsson J, Hörnberg M, Schmidt-Christensen A, et al. NKT cells promote both type 1 and type 2 inflammatory responses in a mouse model of liver fibrosis. Sci Rep. 2020;10(1):21778.
- Wang H, Yin S. Natural killer T cells in liver injury, inflammation and cancer. Expert Rev Gastroenterol Hepatol. 2015;9(8):1077–1085.
- Mitra A, Satelli A, Yan J, et al. IL-30 (IL27p28) attenuates liver fibrosis through inducing NKG2D-rae1 interaction between NKT and activated hepatic stellate cells in mice. Hepatology. 2014;60(6):2027–2039.
- Poisson J, Lemoinne S, Boulanger C, et al. Liver sinusoidal endothelial cells: physiology and role in liver diseases. J Hepatol. 2017;66(1):212–227.
- Lafoz E, Ruart M, Anton A, et al. The endothelium as a driver of liver fibrosis and regeneration. Cells. 2020;9(4):929.
- Iwakiri Y, Shah V, Rockey DC. Vascular pathobiology in chronic liver disease and cirrhosis – current status and future directions. J Hepatol. 2014;61(4):912–924.
- Pi X, Xie L, Patterson C. Emerging roles of vascular endothelium in metabolic homeostasis. Circ Res. 2018;123(4):477–494.
- Elvevold K, Smedsrød B, Martinez I. The liver sinusoidal endothelial cell: a cell type of controversial and confusing identity. Am J Physiol Gastrointest Liver Physiol. 2008;294(2):G391–400.
- Braet F, Wisse E. Structural and functional aspects of liver sinusoidal endothelial cell fenestrae: a review. Comp Hepatol. 2002;1(1):1.
- DeLeve LD. Liver sinusoidal endothelial cells in hepatic fibrosis. Hepatology. 2015;61(5):1740–1746.
- Huang C, Ogawa R. The vascular involvement in soft tissue Fibrosis-lessons learned from pathological scarring. Int J Mol Sci. 2020;21(7):2542.
- Deleve LD, Wang X, Guo Y. Sinusoidal endothelial cells prevent rat stellate cell activation and promote reversion to quiescence. Hepatology. 2008;48(3):920–930.
- Gupta TK, Toruner M, Chung MK, et al. Endothelial dysfunction and decreased production of nitric oxide in the intrahepatic microcirculation of cirrhotic rats. Hepatology. 1998;28(4):926–931.
- Matei V, Rodríguez-Vilarrupla A, Deulofeu R, et al. The eNOS cofactor tetrahydrobiopterin improves endothelial dysfunction in livers of rats with CCl4 cirrhosis. Hepatology. 2006;44(1):44–52.
- Fleischer JR, Jodszuweit CA, Ghadimi M, et al. Vascular heterogeneity with a special focus on the hepatic microenvironment. Front Physiol. 2020;11(591901):591901.
- Brusilovskaya K, Königshofer P, Schwabl P, et al. Vascular targets for the treatment of portal hypertension. Semin Liver Dis. 2019;39(4):483–501.
- Povero D, Panera N, Eguchi A, et al. Lipid-induced hepatocyte-derived extracellular vesicles regulate hepatic stellate cell via microRNAs targeting PPAR-γ. Cell Mol Gastroenterol Hepatol. 2015;1(6):646–663.e4.
- Tugues S, Fernandez-Varo G, Muñoz-Luque J, et al. Antiangiogenic treatment with sunitinib ameliorates inflammatory infiltrate, fibrosis, and portal pressure in cirrhotic rats. Hepatology. 2007;46(6):1919–1926.
- Friedman SL. Evolving challenges in hepatic fibrosis. Nat Rev Gastroenterol Hepatol. 2010;7(8):425–436.
- Ding BS, Cao Z, Lis R, et al. Divergent angiocrine signals from vascular niche balance liver regeneration and fibrosis. Nature. 2014;505(7481):97–102.
- Fernández M, Semela D, Bruix J, et al. Angiogenesis in liver disease. J Hepatol. 2009;50(3):604–620.
- Ogawa R, Akaishi S. Endothelial dysfunction may play a key role in keloid and hypertrophic scar pathogenesis – Keloids and hypertrophic scars may be vascular disorders. Med Hypotheses. 2016;96:51–60.
- Taura K, De Minicis S, Seki E, et al. Hepatic stellate cells secrete angiopoietin 1 that induces angiogenesis in liver fibrosis. Gastroenterology. 2008;135(5):1729–1738.
- Cong M, Liu T, Wang P, et al. Antifibrotic effects of a recombinant adeno-associated virus carrying small interfering RNA targeting TIMP-1 in rat liver fibrosis. Am J Pathol. 2013;182(5):1607–1616.
- Kagan HM, Li W. Lysyl oxidase: properties, specificity, and biological roles inside and outside of the cell. J Cell Biochem. 2003;88(4):660–672.
- Georges PC, Hui JJ, Gombos Z, et al. Increased stiffness of the rat liver precedes matrix deposition: implications for fibrosis. Am J Physiol Gastrointest Liver Physiol. 2007;293(6):G1147–54.
- Perepelyuk M, Terajima M, Wang AY, et al. Hepatic stellate cells and portal fibroblasts are the major cellular sources of collagens and lysyl oxidases in normal liver and early after injury. Am J Physiol Gastrointest Liver Physiol. 2013;304(6):G605–14.
- Liu SB, Ikenaga N, Peng ZW, et al. Lysyl oxidase activity contributes to collagen stabilization during liver fibrosis progression and limits spontaneous fibrosis reversal in mice. Faseb J. 2016;30(4):1599–1609.
- Ikenaga N, Peng ZW, Vaid KA, et al. Selective targeting of lysyl oxidase-like 2 (LOXL2) suppresses hepatic fibrosis progression and accelerates its reversal. Gut. 2017;66(9):1697–1708.
- Chen G, Xia B, Fu Q, et al. Matrix mechanics as regulatory factors and therapeutic targets in hepatic fibrosis. Int J Biol Sci. 2019;15(12):2509–2521.
- Iredale JP, Thompson A, Henderson NC. Extracellular matrix degradation in liver fibrosis: biochemistry and regulation. Biochim Biophys Acta. 2013;1832(7):876–883.
- Massey VL, Dolin CE, Poole LG, et al. The hepatic "matrisome" responds dynamically to injury: characterization of transitional changes to the extracellular matrix in mice. Hepatology. 2017;65(3):969–982.
- Akbari Dilmaghnai N, Shoorei H, Sharifi G, et al. Non-coding RNAs modulate function of extracellular matrix proteins. Biomed Pharmacother. 2021;136:111240.
- Popov Y, Patsenker E, Stickel F, et al. Integrin alphavbeta6 is a marker of the progression of biliary and portal liver fibrosis and a novel target for antifibrotic therapies. J Hepatol. 2008;48(3):453–464.
- Conroy KP, Kitto LJ, Henderson NC. αv integrins: key regulators of tissue fibrosis. Cell Tissue Res. 2016;365(3):511–519.
- Song S, Shackel NA, Wang XM, et al. Discoidin domain receptor 1: isoform expression and potential functions in cirrhotic human liver. Am J Pathol. 2011;178(3):1134–1144.
- Karsdal MA, Manon-Jensen T, Genovese F, et al. Novel insights into the function and dynamics of extracellular matrix in liver fibrosis. Am J Physiol Gastrointest Liver Physiol. 2015;308(10):G807–30.
- Schuppan D, Schmid M, Somasundaram R, et al. Collagens in the liver extracellular matrix bind hepatocyte growth factor. Gastroenterology. 1998;114(1):139–152.
- Natarajan V, Harris EN, Kidambi S. SECs (sinusoidal endothelial cells), liver microenvironment, and fibrosis. Biomed Res Int. 2017;2017:4097205.
- Caliari SR, Perepelyuk M, Soulas EM, et al. Gradually softening hydrogels for modeling hepatic stellate cell behavior during fibrosis regression. Integr Biol. 2016;8(6):720–728.
- Olsen AL, Bloomer SA, Chan EP, et al. Hepatic stellate cells require a stiff environment for myofibroblastic differentiation. Am J Physiol Gastrointest Liver Physiol. 2011;301(1):G110–8.
- Liu L, You Z, Yu H, et al. Mechanotransduction-modulated fibrotic microniches reveal the contribution of angiogenesis in liver fibrosis. Nat Mater. 2017;16(12):1252–1261.
- El-Safy S, Tammam SN, Abdel-Halim M, et al. Collagenase loaded chitosan nanoparticles for digestion of the collagenous scar in liver fibrosis: the effect of chitosan intrinsic collagen binding on the success of targeting. Eur J Pharm Biopharm. 2020;148:54–66.
- Thomas JA, Pope C, Wojtacha D, et al. Macrophage therapy for murine liver fibrosis recruits host effector cells improving fibrosis, regeneration, and function. Hepatology. 2011;53(6):2003–2015.
- Barry-Hamilton V, Spangler R, Marshall D, et al. Allosteric inhibition of lysyl oxidase-like-2 impedes the development of a pathologic microenvironment. Nat Med. 2010;16(9):1009–1017.
- Mannaerts I, Leite SB, Verhulst S, et al. The Hippo pathway effector Yap controls mouse hepatic stellate cell activation. J Hepatol. 2015;63(3):679–688.
- Murata T, Arii S, Nakamura T, et al. Inhibitory effect of Y-27632, a ROCK inhibitor, on progression of rat liver fibrosis in association with inactivation of hepatic stellate cells. J Hepatol. 2001;35(4):474–481.
- Marcellin P, Gane E, Buti M, et al. Regression of cirrhosis during treatment with tenofovir disoproxil fumarate for chronic hepatitis B: a 5-year open-label follow-up study. Lancet. 2013;381(9865):468–475.
- Ebrahimi H, Naderian M, Sohrabpour AA. New concepts on reversibility and targeting of liver fibrosis; a review article. Middle East J Dig Dis. 2018;10(3):133–148.
- Glass LM, Dickson RC, Anderson JC, et al. Total body weight loss of ≥ 10% is associated with improved hepatic fibrosis in patients with nonalcoholic steatohepatitis. Dig Dis Sci. 2015;60(4):1024–1030.
- Sun YM, Chen SY, You H. Regression of liver fibrosis: evidence and challenges. Chin Med J. 2020;133(14):1696–1702.
- Abu Dayyeh BK, Yang M, Dienstag JL, et al. The effects of angiotensin blocking agents on the progression of liver fibrosis in the HALT-C trial cohort. Dig Dis Sci. 2011;56(2):564–568.
- McHutchison J, Goodman Z, Patel K, et al. Farglitazar lacks antifibrotic activity in patients with chronic hepatitis C infection. Gastroenterology. 2010;138(4):1365–1373.
- Liu L, Yannam GR, Nishikawa T, et al. The microenvironment in hepatocyte regeneration and function in rats with advanced cirrhosis. Hepatology. 2012;55(5):1529–1539.
- Kisseleva T, Cong M, Paik Y, et al. Myofibroblasts revert to an inactive phenotype during regression of liver fibrosis. Proc Natl Acad Sci USA. 2012;109(24):9448–9453.
- Troeger JS, Mederacke I, Gwak GY, et al. Deactivation of hepatic stellate cells during liver fibrosis resolution in mice. Gastroenterology. 2012;143(4):1073–1083.e22.